
Abstract
Recently, there has been an increased focus on the immune checkpoint protein PD-1 and its ligand PD-L1 due to the discovery that blocking the PD-1/PD-L1 pathway with monoclonal antibodies elicits striking clinical results in many different malignancies. We have described naturally occurring PD-L1-specific T cells that recognize both PD-L1-expressing immune cells and malignant cells. Thus, PD-L1-specific T cells have the ability to modulate adaptive immune reactions by reacting to regulatory cells. Thus, utilization of PD-L1-derived T cell epitopes may represent an attractive vaccination strategy for targeting the tumor microenvironment and for boosting the clinical effects of additional anticancer immunotherapy. This review summarizes present information about PD-L1 as a T cell antigen, depicts the initial findings about the function of PD-L1-specific T cells in the adjustment of immune responses, and discusses future opportunities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antigen presentation on the cell surface, which is mediated by HLA molecules, is not sufficient to initiate an efficient T cell response. Accordingly, TCR co-stimulatory pathways are crucial for maintaining immune system homeostasis by regulating T cell activation. After recognition of an antigen presented in the context of an HLA molecule, cellular components rearrange to form distinctive immunological synapses upon immune cell polarization. The CD28 family of receptors, which includes CD28, CTLA-4, ICOS, and PD-1, comprises key elements of the immunological synapse. When these receptors interact with their corresponding ligands, they generate potent co-stimulatory or inhibitory signals [1]. Notably, the receptors that generate inhibitory signals prevent T cell-mediated damage to self-tissue by inhibiting the T cell response.
Importantly, tumor cells can engage these T cell pathways by expressing ligands for the inhibitory receptors on the cell surface. The first of these receptors to be successfully targeted by therapeutic monoclonal antibodies was CTLA-4 or (CD152). CTLA-4 is upregulated after T cell stimulation via the TCR. CTLA-4 binds to B7 with a higher affinity than CD28, inhibiting T cell priming. Work in animal models shows that blocking CTLA-4 can shift the immune system balance toward T cell activation and, consequently, exert anticancer effects. These effects have been confirmed in human clinical trials, and, in 2011, the anti-CTLA-4 antibody ipilimumab was the first-in-class therapeutic monoclonal antibody to be approved by the FDA for the treatment of metastatic melanoma (MM) on the basis of a phase III trial showing improved survival [2].
PD-1 is a central regulatory surface protein that delivers inhibitory signals to maintain the functional silence of T cells against their cognate antigens. The PD-1 receptor was identified in 1992 as a protein that was upregulated during apoptosis in lymphocytes [3]. PD-1 is expressed on monocytes, DCs, T cells, B cells, and NK cells. Persistent expression of PD-1 is a marker for T cell exhaustion, as recently reviewed by Wherry [4]. The PD-1 ligand PD-L1 (B7-H1) was discovered in 1999 and is a 290 amino acid transmembrane protein encoded by the CD274 gene [5]. The extracellular portion of PD-L1 comprises IgV- and IgC-like domains, while the intracellular part comprises a 30 amino acid tail. PD-L1 is expressed on non-hematopoietic cells as well as on antigen-presenting cells and on placental cells that are located in an inflammatory microenvironment [6]. PD-L1 is upregulated in a JAK-/STAT-dependent manner by type I and type II IFNs via IFN regulatory factor-1 (IRF-1) [6, 7].
In general, interactions between PD-L1 and PD-1 regulate the induction and maintenance of peripheral T cell tolerance throughout regular immune responses [5]. The interactions between PD-1 and PD-L1 negatively regulate T cell proliferation and cytokine production. Thus, PD-L1 is a critical negative regulator of self-reactive T cells during both the induction and effector phases of the immune response. PD-L1 acts as an inhibitor in multiple ways. For example, in addition to being a ligand for PD-1, PD-L1 binds B7-1 (CD80) preventing B7-1 co-stimulation [8]. Ligation of PD-L1 results in IL-10 production and may augment the apoptosis of activated T cells [9]. In addition, PD-L1 plays a critical role in the conversion of naïve T cells to regulatory T cells (Tregs) [7].
PD-L1 and cancer
It is clear that the immune system can recognize and kill malignant cells in patients with cancer. However, the immunosuppressive tumor microenvironment results in vast immune dysregulation, eventually leading to an insufficient immune response and the out-of-control growth of cancer cells. Notably, cancer cells can directly suppress anticancer immune mechanisms. In addition, cancer cells attract and/or convert immune cells to generate and maintain an immune-suppressive microenvironment. PD-1 and its ligands play central roles in the creation of an immune inhibitory tumor microenvironment that protects cancer cells from immune cell-mediated cell death [10–13]. Thus, PD-L1 helps protect malignant cells from immune destruction and, notably, is expressed by cancer cells in many different malignancies [14–22]. PD-L1 was first depicted as a marker of tumor aggressiveness in renal cell carcinoma [23]. PD-L1 expression on tumor cells correlates with increased tumor aggressiveness and with a poor prognosis in a number of solid cancers, including pancreatic cancer and ovarian cancer [24–26]. Additionally, PD-1 expression by TILs is a negative prognostic factor in several cancers [27–30].
Surface expression of PD-L1 has been described not only in solid tumors but also in several hematological cancers [17, 19, 21, 22]. PD-L1 is expressed both on malignant cells and on infiltrating immune cells in subsets of aggressive B cell lymphomas [31]. In myeloma, PD-L1 upregulation on malignant cells induces T cell apoptosis and tumor-specific T cell anergy, and it enhances the aggressive characteristics of myeloma cells [21]. In multiple myeloma, myeloma cells that overexpress PD-L1 inhibit the generation of CTLs in vitro [32, 33]. In addition, co-culture of CD4+ T cells with myeloma cells results in the generation of Tregs in a contact-dependent manner. These Tregs have a suppressive phenotype and show increased PD-1 expression compared with naturally occurring Tregs. Furthermore, the PD-1/PD-L1 pathway not only promotes the progression of myeloma indirectly by leading to immune control failure; in addition, bone marrow stromal cells induce myeloma cells to express PD-L1, which results in increased tumor cell proliferation and reduced susceptibility to anti-myeloma chemotherapy. Accordingly, clinical progression is observed in patients that have myeloma cells that express high levels of PD-L1.
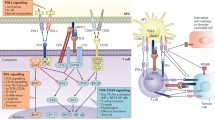
Multiple studies of anti-PD-1 and anti-PD-L1 blockade report the subsequent restoration of T cell effector function and proliferation as well as increased infiltration of tumors by CTLs. This alters the CTL/Treg ratio and ultimately results in the death of tumor cells [25, 34]. The blockade of either PD-1 or PD-L1 by monoclonal antibodies has produced outstanding clinical responses [35, 36], and the Food and Drug Administration (FDA) recently approved the anti-PD-1 antibodies pembrolizumab and nivolumab in September and December of 2014, respectively. Blocking the PD-1 pathway shows great clinical promise, and there is high commercial interest and intense competition among drug companies to develop agents that target PD-1 or PD-L1. Anti-PD-1 antibodies block PD-1:PD-L1 and PD-1:PD-L2 interactions, whereas anti-PD-L1 antibodies block PD-1:PD-L1 and PD-L1:CD80 interactions. This distinction results in slightly different modes of action and in different adverse events and response patterns. Another example in which the PD-1 pathway is targeted is the use of a recombinant B7-DC-Fc fusion protein that has a unique mode of action. Specifically, this fusion protein depletes T cells that express high levels of PD-1, thus allowing a more vigorous anticancer response [37]. Interestingly, it was also shown recently that the immune system itself has an anticancer mechanism that works via PD-L1-specific effector T cells (Fig. 1) [38, 39].

PD-L1-specific T cells target immune regulatory cells as well as cancer cells. Cancer cells (purple) as well as other regulatory immune cells [e.g., tumor-associated dendritic cells (TADC) (dark red) and MDSC (light red)] express checkpoint inhibitors (e.g., PD-L1), inhibitory cytokines as well as metabolic enzymes that restrain the antitumor activity of anti-tumor-specific T cells (green) in the tumor microenvironment. Specific T cells recognizing HLA-restricted PD-L1-derived epitopes (yellow), which are generated from intracellular degraded PD-L1, are able to eliminate (red arrows) regulatory immune cells as well as cancer cells. Hence, the activation of PD-L1-specific T cells by vaccination may directly target immune inhibitory pathways in the tumor microenvironment, modulate immune regulation, and potentially alter tolerance to tumor antigens. The addition of PD-L1 epitopes to therapeutic cancer vaccines would thus be a simple and highly synergistic means to increase the outcome
PD-L1-specific T cells
Our group was the first to describe spontaneous CD8+ and CD4+ T cell reactivity against PD-L1 in the peripheral blood of both patients with various cancers and healthy donors. These PD-L1-specific CD8+ T cells release IFN-γ and TNF-α. Notably, a few individuals in whom we were able to measure specific T cell responses directly ex vivo had a relatively high number of PD-L1-specific T cells. With very few exceptions, it is not feasible to evaluate tumor-associated antigen-specific T cells either by tetramer staining or by ELISPOT in PBMCs ex vivo without in vitro peptide stimulation [40]. We verified that the PD-L1-specific T cells in PBMCs were cytolytic effector cells using the Granzyme B ELISPOT assay. In addition, we generated PD-L1-specific T cell cultures by re-stimulating PBMCs with the PD-L1 peptide in vitro and showed that the subsequent T cell lines were PD-L1 specific. We further established that the PD-L1-specific CD8+ T cells were cytolytic effector cells that recognize and kill PD-L1-expressing melanoma cells as well as cutaneous T cell lymphoma cells. Recently, Minami et al. [41] described HLA-A24-restricted PD-L1-specific T cells that could lyse PD-L1+ HLA-A24+ renal cell carcinoma cells. In addition to recognizing tumor cells, PD-L1-specific CTLs can recognize and kill normal immune cells in a PD-L1-dependent manner. Thus, using siRNA transfection to knockdown PD-L1 protects DCs from death due to PD-L1-specific T cells [38].
Cross-presentation is defined as the processing of exogenous antigens into the HLA class I pathway [42]. We showed that long peptides (20 amino acids) derived from PD-L1 are readily cross-presented by B cells and T2 cells in the absence of antigen-presenting cells such as DCs or macrophages. This result is interesting in light of the observation that patients with renal cell carcinoma produce soluble PD-L1 that retains its immune-suppressive activity [43]. The ability of T2 cells to process the long PD-L1 peptide and, to some extent, to process the full-length recombinant PD-L1 protein demonstrates the transporter associated with antigen processing (TAP)-independent nature of the cross-presentation. Non-professional APCs were shown previously to cross-present HLA class I-restricted epitopes in a similar TAP-independent way, e.g., from exogenous NY-ESO polypeptides [44].
We additionally described that by reacting to PD-L1-expressing cells, PD-L1-specific T cells directly and indirectly augment other T cell responses [45, 46]. First, since the PD-L1/PD-1 pathway is important for the regulation of both viral and anticancer CTL responses, we considered using PD-L1-specific CTLs to influence anti-viral immunity. Indeed, in culture the addition of PD-L1-specific CTLs 1 week after virus epitope stimulation resulted in an vast increase in the number of virus-specific CD8+ T cells [45]. A similar increase in virus-specific T cells was observed in cultures after co-stimulation with the PD-L1 peptide epitope compared to cultures that were co-stimulated with an irrelevant epitope from HIV-1 [46]. Hence, PD-L1-specific CTLs may efficiently augment the effector phase of the immune response by suppressing PD-L1-expressing regulatory cells that restrain PD-1-expressing effector T cells. Second, we began investigating the possibility of influencing the immunogenicity of a DC-based vaccine using co-stimulation with two PD-L1-derived epitopes. We stimulated PBMCs from DC-vaccinated MM patients with the DC-based vaccine used in the clinical study either with or without the PD-L1-derived peptide epitopes. We observed a significant increase in the number of vaccine-reactive T cells in cultures that were co-stimulated with the PD-L1 peptide epitope compared to cultures co-stimulated with an irrelevant HIV epitope (unpublished observation). Thus, boosting PD-L1-specific T cells may directly modulate the immunogenicity of a DC-based vaccine (Fig. 2). If these findings translate to the clinic, co-vaccination with PD-L1 epitopes may be useful for boosting the immunogenicity of the vaccine. Thus, adding PD-L1 epitopes to cancer vaccines may be an easy and attractive way to increase vaccine efficiency.

Co-stimulation with PD-L1 epitopes boosts the immunogenicity of a DC-based vaccine. PBMC (numerous colors) was stimulated with an autologous DC-based vaccine (blue) in the presence of IL-2. Subsequently, DC-reactive T cells (green) expand, and this is augmented when PD-L1-specific T cells are activated by co-stimulation with PD-L1-derived epitopes (yellow) assessed in cultures co-stimulated with an HIV control epitope (red)
It should be noted that the function and effects of PD-L1-specific CTLs may vary according to the microenvironment and the condition of the immune response. The major role of the PD-1 pathway is thought to be its involvement in regulating effector T cell responses to control tissue damage rather than its actions at the initial T cell activation stage [9]. Hence, the occurrence of PD-L1-specific CTLs during the activation phase of an immune response may not enlarge this response. In fact, adding PD-L1-specific CTLs simultaneously with virus antigen stimulation somewhat decreases the number of virus-specific T cells [45], possibly due to the expression of PD-L1 on APCs or on resting T cells.
Owing to the vital functions of PD-L1 in immune regulation, it may seem surprising that there is a natural specific T cell response against PD-L1. However, Yu and colleagues recently described that clonal deletion in the thymus prunes the T cell repertoire, but it does not eliminate self-reactive T cell clones [47]. The authors proposed that a complete deletion of self-reacting T cells would create holes in the immune repertoire that could be exploited by infectious pathogens. Hence, self-peptide-specific CD8+ T cells are present at levels similar to those specific for non-self-antigens in the blood of healthy humans. These self-reactive T cells are substantially anergic compared to non-self-specific T cells; however, they can be activated by strong activation signals. PD-L1 is highly expressed during inflammation and/or stress in professional antigen-presenting cells. Self-reactive T cells that recognize PD-L1 may therefore be activated by the strong activation signals of their cognate targets.
Conclusions and perspectives
Regulatory feedback mechanisms are essential for limiting the strength and magnitude of immune responses that might otherwise harm their host [48, 49]. However, immune evasion is detrimental in the framework of cancer immunotherapy. Thus, it may be very beneficial to target one or more immunosuppressive pathways in combination with anticancer immunotherapy. Immune regulatory cells suppress anticancer immunity in many different ways: by checkpoint inhibitors like PD-L1 and PD-L2, expressing cytokines like TGF-β and IL-10, via metabolic enzymes like tryptophan 2,3-dioxygenase (TDO) and IDO [50] and via arginase, as well as by inducible nitric oxide synthases (iNOS) and adenosine [51, 52]. In addition, regulatory cells release chemokines like CCL22 that attract additional immune regulatory cells. Several different therapeutic strategies are being utilized to target immunosuppression in cancer, including blocking inhibitory pathways such as the PD-1/PD-L1 pathway. In practice, antibodies that target the PD-L1 checkpoint have been shown to elicit impressive, dynamic, and durable tumor regression. We suggest the use of specific T cells as yet another approach to target immune suppression. This review describes naturally occurring specific T cells that recognize PD-L1 in immune-suppressive cells and in malignant cells. A major difference between targeting PD-L1 with monoclonal antibodies versus utilizing PD-L1-specific T cells is that in addition to decreasing the immunoregulatory effects of PD-L1, the PD-L1-specific T cells also inhibit other routes of immune suppression that are mediated by PD-L1+ target cells. Accordingly, a vaccine targeting PD-L1 should attract PD-L1-specific pro-inflammatory T cells to the tumor microenvironment. PD-L1-specific T cells may directly support anticancer immunity by killing target cells and indirectly support it by releasing pro-inflammatory cytokines in the microenvironment to boost additional anticancer immunity. Thus, a PD-L1-based vaccine should be viewed as complementing rather than competing with other forms of immunotherapy. Vaccine-activated PD-L1-specific T cells may, for example, be further boosted by PD-L1 blockade, since PD-L1 mAbs target the same cells as vaccine-induced T cells; this therapeutic strategy will therefore make cells more vulnerable targets (Fig. 3).

A PD-L1 vaccine and checkpoint inhibitors are complimentary. a PD-L1-expressing regulatory immune cells (red) degrade intracellular PD-L1 into peptides (yellow) that are subsequently processed into peptides and presented on the cell surface by HLA molecules, where they are recognized by PD-L1-specific T cells (green). Hence, PD-L1-specific T cells can promote local immune suppression by the secretion of effector cytokines or by killing regulatory immune cells directly (red arrow), thereby influencing general immune reactions. Similarly, they can eliminate PD-L1-expressing malignant cells, b PD-1-positive, PD-L1-specific T cells are themselves hampered by the suppressive effects of PD-L1 expression on their targets and c PD-L1-specific T cells may thus be further boosted by PD-L1 blockade, since PD-L1 mAbs target the same cells as vaccine-induced T cells; this therapeutic strategy will therefore make cells more vulnerable targets. Thus, a PD-L1-based vaccine should be viewed as complementing rather than competing with checkpoint inhibitors
Cancer vaccines represent a promising way to eliminate minimal residual disease without inducing significant toxicity and secondary malignancies. However, so far they have been largely failed to demonstrate a significant improvement in patient outcome [53]. This probably reflects the ability of malignant cells to suppress the function of the induced immune cells. The addition of PD-L1 epitope-based therapy to current cancer vaccine strategies would be easy to implement and is likely to be highly beneficial. It should be noted that the loss of PD-L1 expression in cells during vaccination therapy might result in immune escape, i.e., it might protect target cells from immune-mediated killing by vaccine-activated T cells. However, this should reduce local immune suppression, thereby permitting circulating effector T cells to function or to become activated. PD-L1 may thus serve as a widely accessible target for immunotherapeutic strategies that has an entirely different function and expression pattern than previously described antigens.
In conclusion, these findings justify clinical testing to evaluate the efficacy of PD-L1-based vaccination. We plan to conduct the first PD-L1 vaccine study in humans at Herlev Hospital (Denmark) in which PD-L1 epitopes will be administered to patients with MM. The vaccine will consist of two PD-L1-derived peptides [54]. Long-peptide vaccines that combine MHC class I and II TAA epitopes can efficiently potentiate broad T cell effector function and long-term immunity [55]. The phase I/II trial will explore the safety and toxicity (primary objective) of vaccinating MM patients with two PD-L1 epitopes. The secondary objectives include (a) induction of PD-L1-specific immune responses and (b) obtaining clinical response. To summarize, a PD-L1-based cancer vaccine represents a completely novel immuno-oncological therapeutic approach. PD-L1-specific T cells is a fascinating example of the immune system’s ability to effect adaptive immune reactions by directly acting on the immune-suppressive mechanisms of cancerous cells.
Abbreviations
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- FDA:
-
Food and Drug Administration
- iNOS:
-
Inducible nitric oxide synthases
- IRF-1:
-
IFN regulatory factor-1
- MM:
-
Metastatic melanoma
- TADC:
-
Tumor-associated dendritic cells
- TAP:
-
Transporter associated with antigen processing
- TDO:
-
Tryptophan 2,3-dioxygenase
- Tregs:
-
Regulatory T cells
References
Hornig N, Reinhardt K, Kermer V, Kontermann RE, Muller D (2013) Evaluating combinations of costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. Cancer Immunol Immunother 62:1369–1380. doi:10.1007/s00262-013-1441-7
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. doi:10.1056/NEJMoa1003466
Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11:3887–3895
Wherry EJ (2011) T cell exhaustion. Nat Immunol 12:492–499
Dong H, Zhu G, Tamada K, Chen L (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5:1365–1369
Seo SK, Seo DI, Park WS, Jung WK, Lee DS, Park SG, Choi JS, Kang MS, Choi YH, Choi I, Yu BC, Choi IW (2014) Attenuation of IFN-gamma-induced B7-H1 expression by 15-deoxy-delta(12,14)-prostaglandin J2 via downregulation of the Jak/STAT/IRF-1 signaling pathway. Life Sci 112:82–89. doi:10.1016/j.lfs.2014.07.021
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206:3015–3029. doi:10.1084/jem.20090847
Haile ST, Bosch JJ, Agu NI, Zeender AM, Somasundaram P, Srivastava MK, Britting S, Wolf JB, Ksander BR, Ostrand-Rosenberg S (2011) Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J Immunol 186:6822–6829. doi:10.4049/jimmunol.1003682
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264. doi:10.1038/nrc3239
Ostrand-Rosenberg S, Horn LA, Alvarez JA (2015) Novel strategies for inhibiting PD-1 pathway-mediated immune suppression while simultaneously delivering activating signals to tumor-reactive T cells. Cancer Immunol Immunother 64:1287–1293. doi:10.1007/s00262-015-1677-5
Durgan K, Ali M, Warner P, Latchman YE (2011) Targeting NKT cells and PD-L1 pathway results in augmented anti-tumor responses in a melanoma model. Cancer Immunol Immunother 60:547–558. doi:10.1007/s00262-010-0963-5
Blank C, Mackensen A (2007) Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 56:739–745
Blank C, Gajewski TF, Mackensen A (2005) Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 54:307–314
Kozako T, Yoshimitsu M, Fujiwara H, Masamoto I, Horai S, White Y, Akimoto M, Suzuki S, Matsushita K, Uozumi K, Tei C, Arima N (2009) PD-1/PD-L1 expression in human T-cell leukemia virus type 1 carriers and adult T-cell leukemia/lymphoma patients. Leukemia 23:375–382. doi:10.1038/leu.2008.272
Atanackovic D, Luetkens T, Kroger N (2014) Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia 28(5):993–1000. doi:10.1038/leu.2013.310
Yang H, Bueso-Ramos C, Dinardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, Parmar S, Cortes J, Kantarjian H, Garcia-Manero G (2014) Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 28:1280–1288. doi:10.1038/leu.2013.355
Krejsgaard T, Odum N, Geisler C, Wasik MA, Woetmann A (2012) Regulatory T cells and immunodeficiency in mycosis fungoides and Sezary syndrome. Leukemia 26:424–432. doi:10.1038/leu.2011.237
Kollgaard T, Petersen SL, Hadrup SR, Masmas TN, Seremet T, Andersen MH, Madsen HO, Vindelov L, Thor SP (2005) Evidence for involvement of clonally expanded CD8+ T cells in anticancer immune responses in CLL patients following nonmyeloablative conditioning and hematopoietic cell transplantation. Leukemia 19:2273–2280
Ame-Thomas P, Le PJ, Yssel H, Caron G, Pangault C, Jean R, Martin N, Marafioti T, Gaulard P, Lamy T, Fest T, Semana G, Tarte K (2012) Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia 26:1053–1063. doi:10.1038/leu.2011.301
van de Donk NW, Kamps S, Mutis T, Lokhorst HM (2012) Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma. Leukemia 26:199–213. doi:10.1038/leu.2011.214
Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, Hyodo H, Shinya E, Takahashi H, Dong H, Tamada K, Chen L, Dan K, Ogata K (2013) Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 27:464–472. doi:10.1038/leu.2012.213
Greaves P, Gribben JG (2013) The role of B7 family molecules in hematologic malignancy. Blood 121:734–744. doi:10.1182/blood-2012-10-385591
Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, Zincke H, Blute ML, Strome SE, Leibovich BC, Kwon ED (2004) Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA 101:17174–17179
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N, Honjo T, Fujii S (2007) Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA 104:3360–3365
Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, Nakajima Y (2007) Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 13:2151–2157
Maine CJ, Aziz NH, Chatterjee J, Hayford C, Brewig N, Whilding L, George AJ, Ghaem-Maghami S (2014) Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol Immunother 63:215–224. doi:10.1007/s00262-013-1503-x
Thompson RH, Dong H, Lohse CM, Leibovich BC, Blute ML, Cheville JC, Kwon ED (2007) PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res 13:1757–1761
Hsu MC, Hsiao JR, Chang KC, Wu YH, Su IJ, Jin YT, Chang Y (2010) Increase of programmed death-1-expressing intratumoral CD8 T cells predicts a poor prognosis for nasopharyngeal carcinoma. Mod Pathol 23:1393–1403. doi:10.1038/modpathol.2010.130
Krambeck AE, Dong H, Thompson RH, Kuntz SM, Lohse CM, Leibovich BC, Blute ML, Sebo TJ, Cheville JC, Parker AS, Kwon ED (2007) Survivin and b7-h1 are collaborative predictors of survival and represent potential therapeutic targets for patients with renal cell carcinoma. Clin Cancer Res 13:1749–1756
Sun S, Fei X, Mao Y, Wang X, Garfield DH, Huang O, Wang J, Yuan F, Sun L, Yu Q, Jin X, Wang J, Shen K (2014) PD-1(+) immune cell infiltration inversely correlates with survival of operable breast cancer patients. Cancer Immunol Immunother 63:395–406. doi:10.1007/s00262-014-1519-x
Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, Yu H, Fletcher C, Freeman GJ, Shipp MA, Rodig SJ (2013) PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res 19:3462–3473. doi:10.1158/1078-0432.CCR-13-0855
Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, Saudemont A, Quesnel B (2007) Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110:296–304
Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, Zhang J, Yu J, Smith MK, Greenfield CN, Porcu P, Devine SM, Rotem-Yehudar R, Lozanski G, Byrd JC, Caligiuri MA (2010) The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 116:2286–2294. doi:10.1182/blood-2010-02-271874
Peng W, Lizee G, Hwu P (2013) Blockade of the PD-1 pathway enhances the efficacy of adoptive cell therapy against cancer. Oncoimmunology 2(2):e22691
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366(26):2455–2465. doi:10.1056/NEJMoa1200694
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2453. doi:10.1056/NEJMoa1200690
Mkrtichyan M, Najjar YG, Raulfs EC, Liu L, Langerman S, Guittard G, Ozbun L, Khleif SN (2012) B7-DC-Ig enhances vaccine effect by a novel mechanism dependent on PD-1 expression level on T cell subsets. J Immunol 189:2338–2347. doi:10.4049/jimmunol.1103085
Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH (2013) HLA-restricted cytotoxic T cells that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res 73(6):1764–1776. doi:10.1158/0008-5472.CAN-12-3507
Munir S, Andersen GH, Woetmann A, Odum N, Becker JC, Andersen MH (2013) Cutaneous T cell lymphoma cells are targets for immune checkpoint ligand PD-L1-specific, cytotoxic T cells. Leukemia 27:2251–2253. doi:10.1038/leu.2013
Keilholz U, Weber J, Finke JH, Gabrilovich DI, Kast WM, Disis ML, Kirkwood JM, Scheibenbogen C, Schlom J, Maino VC, Lyerly HK, Lee PP, Storkus W, Marincola F, Worobec A, Atkins MB (2002) Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. J Immunother 25:97–138
Minami T, Minami T, Shimizu N, Yamamoto Y, De VM, Nozawa M, Yoshimura K, Harashima N, Harada M, Uemura H (2015) Identification of Programmed death ligand 1-derived peptides capable of inducing cancer-reactive cytotoxic T lymphocytes from HLA-A24+ patients with renal cell carcinoma. J Immunother 38:285–291. doi:10.1097/CJI.0000000000000090
Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR (1997) Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med 186:65–70
Frigola X, Inman BA, Lohse CM, Krco CJ, Cheville JC, Thompson RH, Leibovich B, Blute ML, Dong H, Kwon ED (2011) Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res 17:1915–1923. doi:10.1158/1078-0432.CCR-10-0250
Gnjatic S, Atanackovic D, Matsuo M, Jager E, Lee SY, Valmori D, Chen YT, Ritter G, Knuth A, Old LJ (2003) Cross-presentation of HLA class I epitopes from exogenous NY-ESO-1 polypeptides by nonprofessional APCs. J Immunol 170:1191–1196
Ahmad SM, Larsen SK, Svane IM, Andersen MH (2014) Harnessing PD-L1-specific cytotoxic T cells for anti-leukemia immunotherapy to defeat mechanisms of immune escape mediated by the PD-1 pathway. Leukemia 28:236–238. doi:10.1038/leu.2013.261
Ahmad SM, Svane IM, Andersen MH (2014) The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J 4:e230–e233. doi:10.1038/bcj.2014.50
Yu W, Jiang N, Ebert PJ, Kidd BA, Muller S, Lund PJ, Juang J, Adachi K, Tse T, Birnbaum ME, Newell EW, Wilson DM, Grotenbreg GM, Valitutti S, Quake SR, Davis MM (2015) Clonal deletion prunes but does not eliminate self-specific alphabeta CD8(+) T lymphocytes. Immunity 42(5):929–941. doi:10.1016/j.immuni.2015.05.001
Hurwitz AA, Watkins SK (2012) Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer Immunol Immunother 61:289–293. doi:10.1007/s00262-011-1181-5
Ostrand-Rosenberg S, Sinha P, Chornoguz O, Ecker C (2012) Regulating the suppressors: apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol Immunother 61:1319–1325. doi:10.1007/s00262-012-1269-6
Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ (2014) Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother 63:721–735. doi:10.1007/s00262-014-1549-4
Andersen MH (2015) Immune regulation by self-recognition: novel possibilities for anticancer immunotherapy. J Natl Cancer Inst. doi:10.1093/jnci/djv154
Becker JC, Thor SP, Andersen MH (2014) Self-reactive T cells: suppressing the suppressors. Cancer Immunol Immunother 63:313–319. doi:10.1007/s00262-013-1512-9
Rhee F (2007) Idiotype vaccination strategies in myeloma: how to overcome a dysfunctional immune system. Clin Cancer Res 13:1353–1355
Munir S, Andersen GH, Svane IM, Andersen MH (2013) The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology 2(4):e23991
Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, Wafelman AR, Oostendorp J, Fleuren GJ, van der Burg SH, Melief CJ (2009) Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 361(19):1838–1847. doi:10.1056/NEJMoa0810097
Acknowledgments
This work was supported by the Danish Cancer Society, the Danish Council for Independent Research, Lundbeck Foundation, Toyota Foundation, and Herlev Hospital. The funders did not have a role in the writing of the article or the decision to submit the article for publication
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Mads Hald Andersen is an author of three filed patent applications based on the use of PD-L1 vaccination. The rights of the patent applications have been transferred to Copenhagen University Hospital, Herlev/The Capital Region of Denmark, according to the Danish Law of Public Inventions at Public Research Institutions. All other authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Ahmad, S.M., Borch, T.H., Hansen, M. et al. PD-L1-specific T cells. Cancer Immunol Immunother 65, 797–804 (2016). https://doi.org/10.1007/s00262-015-1783-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1783-4