
Abstract
Programmed death receptor ligand 1 (PD-L1, also called B7-H1) is a recently described B7 family member. In contrast to B7-1 and B7-2, PD-L1 does not interact with either CD28 or CTLA-4. To date, one specific receptor has been identified that can be ligated by PD-L1. This receptor, programmed death receptor 1 (PD-1), has been shown to negatively regulate T-cell receptor (TCR) signaling. Upon ligating its receptor, PD-L1 has been reported to decrease TCR-mediated proliferation and cytokine production. PD-1 gene–deficient mice developed autoimmune diseases, which early led to the hypothesis of PD-L1 regulating peripheral tolerance. In contrast to normal tissues, which show minimal surface expression of PD-L1 protein, PD-L1 expression was found to be abundant on many murine and human cancers and could be further up-regulated upon IFN-γ stimulation. Thus, PD-L1 might play an important role in tumor immune evasion. This review discusses the currently available data concerning negative T-cell regulation via PD-1, the blockade of PD-L1/PD-1 interactions, and the implications for adoptive T-cell therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Conventional therapies (surgery, chemotherapy, and radiation) for many metastatic solid tumors have been relatively disappointing, and survival rates of these patients have not been substantially improved during the last decade [1]. Therefore, the search for alternative therapeutic approaches has been intense. One attractive concept has been to modulate the immune system in a way that enables it to recognize and kill tumor cells [40, 71]. Initial work has shown that most human tumors express tumor-associated antigens (TAAs) that can be recognized by T cells and thus are potentially capable of inducing immune responses [8]. Early phase clinical trials have been initiated by vaccinating cancer patients with TAA or professional antigen-presenting cells pulsed with TAA [18, 25, 39, 52]. Induction of tumor antigen–specific CD8+ T cells has been achieved in many of these trials [38, 52]. Adoptive transfer of tumor antigen–specific T cells into patients also has been pursued and has revealed homing of the expanded cytotoxic T lymphocytes (CTLs) to tumor sites [43]. However, despite tumor infiltration of immune effector cells, tumor growth was seldom controlled. It is well known that the tumor microenvironment can protect tumor cells from immune destruction [26, 65]. Soluble factors, as well as membrane-bound molecules including transforming growth factor β (TGF-β), interleukin (IL)–10, prostaglandin E2, FAS, CTLA-4 ligands, tumor necrosis factor–related apoptosis-inducing ligand (TRAIL), and programmed death receptor ligand 1 (PD-L1, also B7-H1), have been found to be expressed by tumors and have been postulated to mediate immune evasion [21, 33, 66]. Blocking of such negative immune regulatory signals on tumor cells has given rise to hope that their manipulation may lead to enhanced tumor-specific CD8+ T-cell immunity in vivo (Fig. 1).

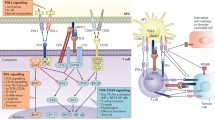
Schematic description of the requirements for an CD8+ T-cell–mediated antitumor immune response. Naïve tumor-specific CD8+ T cells can be activated directly from tumor cells or indirectly from APCs presenting TAA. In a second step, differentiation toward a type I phenotype is optimal. This step is favored by IL-12 and IFN-γ and suppressed by IL-4 or IL-13. Furthermore, sufficient expansion of tumor-specific T cells is important and may require IL-2. Activation-induced nonresponsiveness (AINR), anergy induction, and T-cell deletion may interfere with sufficient expansion and in vivo survival. Tumor penetration is influenced by chemokines and adhesion molecules. Furthermore, the interaction between tumor-specific T cells and the tumor cell is improved by high MHC expression, high antigen densitiy on the tumor, and B7 expression of the tumor. Immune escape might be mediated via TGF-β, MHC loss, or engagement of Fas, PD-1, or CTLA-4 on the T cell. Finally, long-term responses require the development of a memory immune response.
Besides the well-described B7-1/B7-2/CD28/CTLA-4 pathway, a growing number of B7 family ligands and their receptors have been identified that have been shown to modulate TCR signals [10, 11]. This review focuses on the recently described B7 family member B7-H1 (PD-L1) interacting with programmed death gene 1 (PD-1). This pathway can have negative regulatory functions and might play an important role in immune evasion from tumor-specific T cells.
Expression and function of PD-1
Programmed death receptor 1 is a 55-kDa type I transmembrane receptor that was initially identified in a murine T-cell hybridoma undergoing activation-induced cell death (AICD) [31]. PD-1 is a member of the Ig superfamily that contains a single Ig V-like domain in its extracellular region but lacks the MYPPPY motif, a sequence critical for CTLA-4 and CD28 binding to B7-1 and B7-2 [2]. The cytoplasmatic tail of PD-1 contains two tyrosines within a YXXL/I motif that is hypothesized to be a common feature of an immunoreceptor tyrosine-based inhibitory motif (ITIM) and thought to mediate negative regulatory signals [15, 31, 62, 73]. Recent data imply the C-terminal tyrosine to be found within an immunoreceptor tyrosine switch motif (ITSM) [80]. Coligation of PD-1 and BCR leads to a rapid phosphorylation of Src homology region 2 containing tyrosine phosphatase-2 (SHP-2), resulting in dephosphorylation of key signal transducers of BCR signaling, such as Syk and Igα/β. Downstream signaling events such as activation of PI3 K, PLCγ2, and ERK, and elevation of intracellular Ca2+ are consequently inhibited. However, SHP-2 recruitment to the C-terminal phosphotyrosine and the N-terminal tyrosine residue is almost dispensable for the inhibitory effects of PD-1, raising questions about the precise biochemical mechanism of this effect [50]. Moreover, detailed studies of the biochemical nature of PD-1 signaling in T cells are lacking.
Programmed death receptor 1 has been found to be expressed on thymocytes at transition from DN (CD4−CD8−) to DP (CD4+CD8+) stage [6, 45] and on mature T and B cells following activation [2, 7, 10]. PD-1 was also found to be expressed on macrophages [45]. Studies in PD-1–deficient mice indicate that PD-1 serves as a negative regulator for immune responses. Absence of PD-1 in mice leads to glomerulonephritis, arthritis, and cardiomyopathy [46–48].
Two ligands for PD-1 have been identified: PD-L1 (B7-H1) and PD-L2 (B7-DC) [24, 35]. Interaction of PD-1 with PD-L1 or PD-L2 has been described to negatively regulate cytokine production and proliferation of T cells [9, 12, 24, 59]. However, some other reports identified costimulatory functions of these ligands possibly mediated via an unidentified receptor different from PD-1 [16, 69, 70, 74, 79].
Expression of PD-L1 and PD-L2
Expression of PD-L1 and PD-L2 transcripts has been described in a wide range of murine and human tissues including placenta, heart, pancreas, spleen, lymph node, and thymus [24, 35, 45, 51], while it was absent in brain and kidney tissue. Interestingly, surface expression of the proteins encoded by these genes has been shown to be more limited, suggesting the possibility of post-transcriptional regulation.
Programmed death receptor ligand 1 and PD-L2 surface expression has been intensively studied on antigen-presenting cells (APCs): resting B cells and monocytes express neither PD-L1 nor PD-L2; surface expression of PD-L1 was detected upon activation on macrophages and B lymphocytes [14, 37, 72]; whereas PD-L2 was only expressed on inflammatory macrophages [37]. Dendritic cells (DCs) have been shown to express PD-L1 and PD-L2 upon stimulation [14, 78]. The data so far published concerning the surface expression of PD-L1 and PD-L2 on immature DCs are conflicting [14, 59, 78].
In addition to the receptor PD-1, T cells also can express the corresponding ligands, particularly following TCR stimulation even in the absence of CD28 costimulation. PD-L2 is not expressed in naïve T cells, and the current results concerning a possible up-regulation upon stimulation are controversial [7, 9, 32, 37, 78]. Expression of PD-1 and PD-L1 on activated T cells has given rise to the hypothesis that T:T interactions via PD-1/PD-L1 might limit TCR signaling after activation, although there is little direct evidence for this phenomenon.
In addition to TCR or BCR signals in lymphocytes, IFN-γ was shown to influence PD-L1 expression predominantly on nonlymphoid tissues. These data are in line with results showing that the PD-L1 promoter region contains several IFN-γ–responsive elements [17, 41]. Human and murine endothelial cells (ECs) constitutively express PD-L1, and in vitro treatment with IFN-γ, but not LPS or TNF-α, results in up-regulation of PD-L1. Human ECs have also been shown to up-regulate PD-L1 upon TNF-α stimulation [23, 34, 41]. Thus, IFN-γ–mediated up-regulation of PD-L1 on endothelial cells in vivo might play a significant role in regulation of T-cell function at peripheral sites and mediate peripheral tolerance. Regulation of tolerance might also be reflected by the high PD-L1 expression at the human maternal–fetal interface [53]. Several studies have suggested an immunosuppressive role of IFN-γ in autoimmune settings. Consistent with these findings, PD-L1 expression was found to be up-regulated at sites of inflammation in experimental autoimmune encephalomyelitis [56] and in muscle biopsies from patients with inflammatory myopathies [76]. In the latter publication the authors showed that IFN-γ leads to increased PD-L1 expression on human muscle cells, resulting in decreased cytokine production of allogenic CD4+ and CD8+ T cells. Thus, the beneficial effect of IFN-γ reported in some autoimmune settings might be partly contributed to induction of PD-L1 ligand expression at the sites of inflammation and subsequent down-regulation of immune responses by PD-1 engagement [10].
In addition to these normal nonhematologic tissues, PD-L1 also has been found to be expressed on a broad range of cancers. PD-L1 expression has been detected by immunohistochemical stainings on the surface of human cancers of larynx, lung, stomach, colon, breast, cervix, ovary, renal cell, bladder, liver, glioma, and melanoma [9, 17, 77]. In addition, PD-L1 was found to be expressed on various human tumor cell lines including lung, colon, breast, placenta, melanoma, and glioma cancer cell lines [7, 17, 35, 77]. Similar to endothelial cells, murine and human tumor cells up-regulate PD-L1 upon IFN-γ stimulation [7, 17, 77]. These observations have led to the hypothesis that tumors might escape from the host immune system by negative attenuation of tumor-specific T-cell responses via PD-L1/PD-1 interactions.
Immunological functions of PD-L1
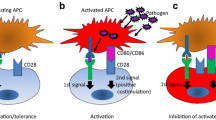
Identification of PD-L1 as ligand for PD-1 led rapidly to the hypothesis that PD-L1 might negatively regulate immune responses [24]. Neither PD-L1 nor PD-L2 bound to CD28, CTLA-4, or ICOS, and reciprocally, soluble forms of B7-1 and B7-2 did not bind PD-1 [10]. Cross-linking PD-1 by either PD-L1-Ig or PD-L2-Ig resulted in decreased proliferation, and decreased IFN-γ, IL-10, IL-4, and IL-2 secretion from anti-CD3-stimulated T cells [24, 35], without an increase in cell death [35; and H. Wiendl, personal communication], indicating that the nomenclature for this receptor and its ligands is perhaps misleading. In vitro studies using T cells from PD-1–deficient mice support the negative functional consequences of PD-1/PD-L1 interactions. Proliferation upon anti-CD3 mAb stimulation of wild-type, but not PD-1–deficient, T cells was inhibited by PD-L1-Ig [24]. Further studies using blocking antibodies to PD-L1 performed on human endothelial cells [41, 55], DCs [9, 14, 54, 59], liver nonparenchymal cells [33], and glioma cells [77] support negative regulation of T-cell activation via PD-1/PD-L1 interaction. In vivo studies on PD-1 gene–deficient mice or blocking PD-L1 with mAb showed that PD-1 can serve as a negative regulator of autoimmune responses in vivo, supporting further the negative regulatory function of PD-1/PD-L1 interactions [3, 46–48, 57]. PD-L1-Ig promoted in vivo cardiac allograft survival, protection from chronic rejection [51], and long-term pancreas islet allograft survival [27].
However, some evidence has been generated that PD-L1 and PD-L2 may provide costimulatory signals in certain contexts. One group reported PD-L1-Ig to stimulate T-cell proliferation and IL-10 production [16, 70]. In addition, local expression of PD-L1 promoted organ-specific autoimmunity and transplant rejection in a pancreatic islet model [69]. These results might be explained by the possibility of PD-1 mediating either positive or negative signals, resulting from signals via its hypothesized cytoplasmatic ITSM motif, which has recently been described as being capable of providing both signals [60, 64]. Another possibility might be a different positioning of PD-1 toward the TCR. A recent study has indicated that PD-1 negative regulatory signals only occur when PD-1 is in close proximity with the TCR [5]. Most likely, however, is the hypothesis that PD-L1–mediated positive costimulatory effects are independent of PD-1 and occur through an unidentified second receptor. Recent binding studies and in vitro studies using PD-1–deficient T cells suggest the existence of an alternative receptor, distinct from PD-1, that could mediate the costimulatory effects from PD-L1/L2 [61, 74]. Therefore, one might envision that the outcome of an immune response upon PD-L1 costimulation will depend on the expression kinetics of the hypothesized costimulatory receptor and PD-1, or on a balance of signaling between the two receptors, as postulated for CD28 and CTLA-4 [58]. The fact that PD-L1/PD-1 engagement can attenuate ICOS-positive costimulatory signals [5], that its negative signaling however can be negated by CD28 ligation or IL-2 [12], and the identification of a further negative regulatory receptor, BTLA [11, 75], indicates that the overall result from negative and positive costimulation will depend on the relative expression of the individual molecules. This orchestra of B7 family costimulatory molecules and their interactions in modulating the overall outcome of the immune response is illustrated in Fig. 2.

The growing number of B7 superfamily members shaping TCR signaling. CD28 and CTLA-4 have MYPPPY motifs that are essential to bind B7-1 (CD80) and B7-2 (CD86). B7-1 and B7-2 provide important costimulatory signals via an interaction with the constitutively expressed CD28 and provide a negative signal via CTLA-4 (CD152), which is induced upon T-cell activation. ICOS (H4) does not show any binding to B7 and seems to mediate costimulatory effects on recently activated and effector T cells binding to its ligand, ICOS-L (B7 h, B7-H2). B7-H3 is constitutively expressed on IFN-γ–treated DCs and binds to an unidentified receptor on activated, but not on resting, T cells. B7-H3 costimulates proliferation of CD4+ and CD8+ T cells. PD-L1 (B7-H1) negatively regulates proliferation and cytokine production of T and B cells that express PD-1 upon activation. A secondary receptor mediating costimulatory signals from PD-L1 has been postulated, but not yet identified. This receptor possibly interacts preferentially with PD-L2 (B7-DC). B7x (B7-H4) has been reported to interact with BTLA, which is expressed on activated T and B cells, inhibiting lymphocyte activation. In contrast to the APC shown here, most tumor cells lack B-7 costimulatory molecules and express strongly PD-L1, either constitutively or upon IFN-γ. This expression pattern might shift the costimulatory balance of the TCR signal toward inhibition of tumor-specific T cells when interacting with the tumor cell.
PD-L1 and tumor evasion
Programmed death receptor ligand 1 was originally identified from a homology search of the human cDNA expressed sequence tag (EST) database using published human B7-1 and B7-2 amino acid sequences. Two overlapping ESTs with homology to B7-1 and B7-2 were found to be expressed in an EST clone of human ovary tumor [16], suggesting the expression of PD-L1 by human cancers. Initial functional experiments suggested that PD-L1 costimulates T cells to induce IL-10 production [16]. In contrast, a second group showed that PD-L1 decreased T-cell proliferation, and IFN-γ and IL-10 secretion [24]. PD-L1 expression was found to be expressed at mRNA levels on a wide range of tumor cell lines, but surface expression using mAbs showed only a small fraction of human tumor cell lines to express PD-L1. However, the majority of murine and human tumor lines can be up-regulated to express PD-L1 upon treatment with IFN-γ [7, 17, 68]. Immunhistochemistry analysis demonstrated PD-L1 reactivity in a majority of freshly isolated human carcinomas [17, 68, 77], indicating that inflammatory cytokines or other in vivo signals may be actively inducing the expression of this molecule in vivo. Broad expression of PD-L1 on human cancer tissues led to the hypothesis of PD-L1/PD-1 interactions mediating immune evasion from tumor-specific T cells [17, 33]. To address this question, several groups have implemented experiments using human tumor cell lines in vitro or murine tumors in vitro and in vivo.
Transfection of murine mastocytoma P815 with PD-L1 led to decreased lysis of the tumor cells when cocultured with a tumor-specific CTL clone. Lysis was restored when anti-PD-L1 mAb was added [33]. Endogenous PD-L1 surface expression on IFN-γ–treated murine melanoma was also able to decrease lysis by wild-type CTLs compared with PD-1–deficient CTLs [7]. Transfection of PD-L1 into a PD-L1–negative human melanoma line led to an increased percentage of apoptotic T cells, [17]. The latter data have suggested that PD-L1 mediates a decreased proliferation upon TCR stimulation [12, 14] rather than apoptosis. Neutralizing mAbs against PD-L1 also have been shown to augment production of IFN-γ and IL-2 from tumor-specific CTLs when in vitro cocultured with PD-L1–expressing tumors or tumor-antigen loaded myeloid dendritic cells [7, 12, 33, 77].
In vivo studies revealed a positive effect on tumor growth control and survival of tumor-bearing animals when anti-PD-L1 mAb was administered [33, 68]. Interestingly, not only the acute growth of the tumors was influenced, but also the long-term survival of tumor-bearing animals, indicating that PD-L1 blockade might improve the development of a memory immune response.
There is some controversy whether PD-L1 transfection of murine tumors leads to decreased tumorigenicity in vivo. Whereas one group has shown that PD-L1 transfection of P815 mastocytoma resulted in increased tumor growth in syngeneic mice [33], another group found the growth to be unaltered compared with mock-transfected P815 cell line [17]. The latter group, however, has reported in the same publication that the rejection of a B7-expressing P815 mastocytoma line was negated when cotransfected with PD-L1. More congruent are the data from tumor mouse models utilizing PD-1–deficient animals. PD-L1–expressing myeloma cells grew only in wild-type animals, but not PD-1–deficient mice [33]. Similarly, PD-1–deficient CD8+ TCR transgenic T cells caused potent tumor rejection in an adoptive transfer model under conditions in which wild-type T cells as well as CTLA-4–deficient T cells failed [7]. At first glance these data appear to be contrary to previous data indicating a benefit in tumor rejection of CTLA-4 blockade [36]. However this discrepancy might be due to the focus on a monoclonal population of CD8+ T cells, in the absence of CD4+ cells. Previous data indicate that CTLA-4 primarily regulates CD4+ T-cell responses [4, 29, 63]. Furthermore, the ligands for CTLA-4, B7-1, and B7-2 would be expressed predominantly on APCs rather than on the tumor cells themselves. At the level of the APC, the role of PD-L1/PD-1 seems to be minor because CD28 costimulation by B7 overcomes its inhibitory effects [12]. Thus, PD-L1 could play a more critical role in suppressing the execution of T-cell effector function during the process of recognition of B7-negative tumor cells.
Implications of PD-L1 for tumor immunotherapy
Adoptive T-cell therapy approaches have gained increased attention for clinical applications [71]. Several steps, including sufficient activation of tumor-specific T cells, the induction of a type I T-cell phenotype, T-cell expansion, tumor infiltration, and development of T-cell memory are thought to be required for an effective antitumor immune response (Fig. 1). PD-L1 has been shown to be expressed on dendritic cells as well as on tumor cells, mainly in response to IFN-γ. Because a goal of many immunotherapy protocols is to induce a type I immune response [52, 67], PD-L1 may play an important role in tumors resisting the effector phase of IFN-γ–producing antitumor T cells. Of note, PD-L1 expression was also found on naïve cells and up-regulated on activated T cells [37]. So far there is no evidence for a T cell:T cell interaction via PD-L1/PD-1 during stimulation of naïve T cells in the presence of B7 costimulus and/or IL-2 [7, 12].
A major challenge for adoptive T-cell therapies is to ensure a long-term survival of the transferred T cells. Despite efficient approaches for rapid expansion of tumor-specific T cells in vitro [20, 38, 40, 49], the data regarding long-term survival of tumor-specific T cells after transfer into patients are controversial. Whereas some groups have found short half-life of T cells after transfer, other groups have shown long-term persistence [22, 43, 44]. Only one group so far has clearly shown a significant expansion of the transferred tumor-specific T-cell populations in vivo [18]. In mouse tumor experiments the blockade of PD-L1 resulted in an increased expansion of tumor-specific T cells and relatively decreased numbers of apoptotic T cells early after transfer [14, 17]. Therefore, the blockade of PD-L1 during T-cell transfer might increase the numbers of surviving cells. Whether this effect might be synergistic with, or overcome the need for, additional IL-2 needs to be evaluated.
Clinical responses have been found to correlate with CD8+ lymphocyte infiltration of the carcinomas and with CD8+ T cells producing IFN-γ [28, 30]. Interestingly, in vitro studies have revealed increased IFN-γ production by CD8+ T cells upon stimulation with tumor cells when PD-L1 was blocked [7, 33]. Thus, the blockade of negative regulation via PD-L1/PD-1 might improve the induction of type I immune responses.
Despite the presence of tumor-specific T cells within the tumors [42], tumor cell lysis is often prevented, and the clinical outcome of adoptive T-cell therapies is often disappointing [13, 20]. It is thought that the tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes [26, 65]. The blockade of PD-L1 on the tumor cells might improve stimulation of T cells after infiltration and thus allow for improved lysis of target cells in vivo [7, 68].
Interfering with the interaction of PD-L1, either on antigen-presenting dendritic cells or the tumor cells themselves, with its receptor PD-1 on T cells, either adoptively transferred or induced by vaccination, might potentiate antitumor T-cell effector function in vivo. Translating these ideas to therapy approaches for human cancer patients should be a high priority in future studies.
References
Abbott A (2002) Cancer research: on the offensive. Nature 416:470–474
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T (1996) Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 8:765–772
Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ et al (2003) The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 198:63–69
Bachmann MF, Waterhouse P, Speiser DE, McKall-Faienza K, Mak TW, Ohashi PS (1998) Normal responsiveness of CTLA-4-deficient anti-viral cytotoxic T cells. J Immunol 160:95–100
Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, Khan N, Veldman G, Jacobs KA, Valge-Archer VE et al (2003) Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol 170:711–718
Blank C, Brown I, Marks R, Nishimura H, Honjo T, Gajewski TF (2003) Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J Immunol 171:4574–4581
Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF (2004) PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 64:1140–1145
Boon T, Gajewski TF, Coulie PG (1995) From defined human tumor antigens to effective immunization? Immunol Today 16:334–336
Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ (2003) Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 170:1257–1266
Carreno BM, Collins M (2002) The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol 20:29–53
Carreno BM, Collins M (2003) BTLA: a new inhibitory receptor with a B7-like ligand. Trends Immunol 24:524–527
Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM (2002) PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol 32:634–643
Cohen PA, Peng L, Kjaergaard J, Plautz GE, Finke JH, Koski GK, Czerniecki BJ, Shu S (2001) T-cell adoptive therapy of tumors: mechanisms of improved therapeutic performance. Crit Rev Immunol 21:215–248
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC et al (2003) Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med 9:562–567
D’Ambrosio D, Fong DC, Cambier JC (1996) The SHIP phosphatase becomes associated with Fc gammaRIIB1 and is tyrosine phosphorylated during ‘negative’ signaling. Immunol Lett 54:77–82
Dong H, Zhu G, Tamada K, Chen L (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5:1365–1369
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K et al (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM et al (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854
Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA et al (2002) A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother 25:243–251
Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA (2003) Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–342
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998
Economou JS, Belldegrun AS, Glaspy J, Toloza EM, Figlin R, Hobbs J, Meldon N, Kaboo R, Tso CL, Miller A et al (1996) In vivo trafficking of adoptively transferred interleukin-2 expanded tumor-infiltrating lymphocytes and peripheral blood lymphocytes: results of a double gene marking trial. J Clin Invest 97:515–521
Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP (2002) Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 9:133–145
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192:1027–1034
Gajewski TF, Fallarino F, Ashikari A, Sherman M (2001) Immunization of HLA-A2+ melanoma patients with MAGE-3 or MelanA peptide-pulsed autologous peripheral blood mononuclear cells plus recombinant human interleukin 12. Clin Cancer Res 7:895s–901s
Ganss R, Hanahan D (1998) Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res 58:4673–4681
Gao W, Demirci G, Strom TB, Li XC (2003) Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation 76:994–999
Harlin H, Artz AS, Mahowald M, Rini BI, Zimmerman T, Vogelzang NJ, Gajewski TF (2004) Clinical responses following nonmyeloablative allogeneic stem cell transplantation for renal cell carcinoma are associated with expansion of CD8+ IFN-gamma-producing T cells. Bone Marrow Transplant 33:491–497
Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H (1998) The central role of CD4(+) T cells in the antitumor immune response. J Exp Med 188:2357–2368
Ikeguchi M, Oi K, Hirooka Y, Kaibara N (2004) CD8+ lymphocyte infiltration and apoptosis in hepatocellular carcinoma. Eur J Surg Oncol 30:53–57
Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11:3887–3895
Ishida M, Iwai Y, Tanaka Y, Okazaki T, Freeman GJ, Minato N, Honjo T (2002) Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett 84:57–62
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 99:12293–12297
Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T (2003) PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med 198:39–50
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R et al (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2:261–268
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736
Loke P, Allison JP (2003) PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A 100:5336–5341
Mackensen A, Wittnebel S, Veelken H, Noppen C, Spagnoli GC, Lindermann A (1999) Induction and large-scale expansion of CD8+ tumor specific cytotoxic T lymphocytes from peripheral blood lymphocytes by in vitro stimulation with CD80-transfected autologous melanoma cells. Eur Cytokine Netw 10:329–336
Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S (2000) Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74:181–273
Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH (2002) Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol 20:143–148
Mazanet MM, Hughes CC (2002) B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol 169:3581–3588
Meidenbauer N, Andreesen R, Mackensen A (2001) Dendritic cells for specific cancer immunotherapy. Biol Chem 382:507–520
Meidenbauer N, Marienhagen J, Laumer M, Vogl S, Heymann J, Andreesen R, Mackensen A (2003) Survival and tumor localization of adoptively transferred Melan-A-specific T cells in melanoma patients. J Immunol 170:2161–2169
Mitchell MS, Darrah D, Yeung D, Halpern S, Wallace A, Voland J, Jones V, Kan-Mitchell J (2002) Phase I trial of adoptive immunotherapy with cytolytic T lymphocytes immunized against a tyrosinase epitope. J Clin Oncol 20:1075–1086
Nishimura H, Honjo T (2001) PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol 22:265–268
Nishimura H, Minato N, Nakano T, Honjo T (1998) Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol 10:1563–1572
Nishimura H, Nose M, Hiai H, Minato N, Honjo T (1999) Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11:141–151
Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291:319–322
Oelke M, Moehrle U, Chen JL, Behringer D, Cerundolo V, Lindemann A, Mackensen A (2000) Generation and purification of CD8+ melan-A-specific cytotoxic T lymphocytes for adoptive transfer in tumor immunotherapy. Clin Cancer Res 6:1997–2005
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T (2001) PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A 98:13866–13871
Ozkaynak E, Wang L, Goodearl A, McDonald K, Qin S, O’Keefe T, Duong T, Smith T, Gutierrez-Ramos JC, Rottman JB et al (2002) Programmed death-1 targeting can promote allograft survival. J Immunol 169:6546–6553
Peterson AC, Harlin H, Gajewski TF (2003) Immunization with Melan-A peptide-pulsed peripheral blood mononuclear cells plus recombinant human interleukin-12 induces clinical activity and T-cell responses in advanced melanoma. J Clin Oncol 21:2342–2348
Petroff MG, Chen L, Phillips TA, Azzola D, Sedlmayr P, Hunt JS (2003) B7 family molecules are favorably positioned at the human maternal-fetal interface. Biol Reprod 68:1496–1504
Radhakrishnan S, Nguyen LT, Ciric B, Ure DR, Zhou B, Tamada K, Dong H, Tseng SY, Shin T, Pardoll DM et al (2003) Naturally occurring human IgM antibody that binds B7-DC and potentiates T cell stimulation by dendritic cells. J Immunol 170:1830–1838
Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ (2003) Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 33:3117–3126
Saint-Ruf C, Ungewiss K, Groettrup M, Bruno L, Fehling HJ, von Boehmer H (1994) Analysis and expression of a cloned pre-T cell receptor gene. Science 266:1208–1212
Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ (2003) Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med 198:71–78
Salomon B, Bluestone JA (2001) Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol 19:225–252
Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, Zlabinger G, Steinberger P, Pickl WF, Stockinger H et al (2003) B7-h1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol 170:3637–3644
Sharpe AH, Freeman GJ (2002) The B7-CD28 superfamily. Nat Rev Immunol 2:116–126
Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, Yagita H, Chen L, Powell J, Pardoll D, Housseau F (2003) Cooperative B7–1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med 198:31–38
Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T (1994) Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 23:704–706
Shrikant P, Khoruts A, Mescher MF (1999) CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity 11:483–493
Sidorenko SP, Clark EA (2003) The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol 4:19–24
Singh S, Ross SR, Acena M, Rowley DA, Schreiber H (1992) Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med 175:139–146
Smyth MJ, Godfrey DI, Trapani JA (2001) A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol 2:293–299
Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H (2002) Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity 17:737–747
Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W et al (2003) B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res 63:6501–6505
Subudhi SK, Zhou P, Yerian LM, Chin RK, Lo JC, Anders RA, Sun Y, Chen L, Wang Y, Alegre ML, Fu YX (2004) Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest 113:694–700
Tamura H, Dong H, Zhu G, Sica GL, Flies DB, Tamada K, Chen L (2001) B7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood 97:1809–1816
Thomas AK, June CH (2001) The promise of T-lymphocyte immunotherapy for the treatment of malignant disease. Cancer J 2[Suppl 7]:S67–S75
Trabattoni D, Saresella M, Biasin M, Boasso A, Piacentini L, Ferrante P, Dong H, Maserati R, Shearer GM, Chen L, Clerici M (2003) B7-H1 is upregulated in HIV infection and is a novel surrogate marker of disease progression. Blood 101:2514–2520
Vivier E, Daeron M (1997) Immunoreceptor tyrosine-based inhibition motifs. Immunol Today 18:286–291
Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L (2003) Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med 197:1083–1091
Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X et al (2003) BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 4:670–679
Wiendl H, Mitsdoerffer M, Schneider D, Chen L, Lochmuller H, Melms A, Weller M (2003) Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: a novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J 17:1892–1894
Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H (2003) Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res 63:7462–7467
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K et al (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169:5538–5545
Youngnak P, Kozono Y, Kozono H, Iwai H, Otsuki N, Jin H, Omura K, Yagita H, Pardoll DM, Chen L, Azuma M (2003) Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 307:672–677
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL (2004) SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 173:945–954
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Blank, C., Gajewski, T.F. & Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 54, 307–314 (2005). https://doi.org/10.1007/s00262-004-0593-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-004-0593-x