
Abstract
Erdheim-Chester disease (ECD) is a rare clonal myeloid neoplasm typically affecting adults over 50 years old, with bone lesions in almost all patients. The prognosis is poor in most cases if left untreated. Clinical manifestations are not specific, which hinders early diagnosis. The disease has distinct radiological features. However, three-phase bone scintigraphy exhibits the most typical pattern of all imaging modalities, which is the prominent strikingly symmetrical radiotracer uptake in the distal ends of the femurs and proximal and distal ends of the tibiae, sparing the epiphyses. We report a case of a 54-year-old female patient, presenting with atypical persistent knee joint pain. After an MRI scan, she underwent a three-phase bone scan, revealing the characteristic pattern, thus indicating a possible ECD diagnosis, which was eventually confirmed in biopsy material. Novel aspects of the pathophysiology and treatment of the disease, as well as a differential diagnosis from the perspective of an MSK radiologist and nuclear medicine physician, are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first two cases of Erdheim Chester disease (ECD) were described in 1930 by William Chester, an American post-doctoral student, and the Viennese pathologist Jakob Erdheim. The disease was initially termed “lipoid granulomatosis,” and it was only in 1972 that the name “Erdheim Chester disease” was coined by Dr. Ronald Jaffe [1, 2]. Many things have changed since then regarding the classification, diagnostic criteria, our knowledge about the pathophysiology and genetics of the disease, and above all the treatment and prognosis. However, nuclear medicine undoubtedly continues to play an indispensable role in the diagnosis of this rare entity.
Case report
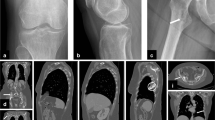
We report a case of a 54-year-old female patient with persistent atypical knee joint pain. The patient was initially treated with non-steroidal anti-inflammatory drugs without resolution of her symptoms. Knee joint radiography revealed diffuse sclerosing lesions in the metaphyses of the tibia and femurs (Fig. 1). Magnetic resonance imaging (MRI) revealed bone marrow edema, with a “geographical” pattern, including the femoral metaphysis and the lower part of the diaphysis, as well as the tibial metaphysis and upper part of the diaphysis, with heterogeneous low signal in T1 and high signal in STIR and proton density sequences. The findings were nonspecific and suggested a potential bone marrow disorder or bone infarct-like lesion (Fig. 2).

Mediolateral radiographs of the right (R) and left (L) knee. Diffuse sclerosing lesions (arrows) in the tibial and femoral metaphyses, and the distal femoral and proximal tibial diaphyses

T1 (a), STIR (b), and proton density (PD) (c) weighted coronal MRI images of the left knee: geographical pattern with low signal (a) and high signal appearance (b, c) of the femoral and tibial bone marrow (arrows). Note that the epiphysis is spared (arrowheads indicating the epiphyseal line). The scan was performed in a GE 1.5 T, 8-channel MRI system
Continuing the diagnostic workup, the patient underwent a three-phase whole-body bone scan to differentiate and clarify the MRI findings. The dynamic and blood pool phase showed symmetrically increased blood flow and blood pool in the upper parts of the tibial diaphyses as well as in the femoral metaphyses (Fig. 3A). Delayed images revealed symmetrically increased uptake in the femoral metaphyses and the upper and lower parts of the tibial diaphyses and proximal metaphyses, without involvement of the epiphyses (Figs. 3B, 3C and 4). The findings were strongly suggestive of Erdheim Chester disease and further work-up was recommended.

A Blood pool images of the three-phase bone scan showing symmetrically increased blood flow in the upper parts of the tibial diaphyses and femoral metaphyses. B, C Delayed images showing symmetrically increased uptake in the femoral metaphyses and the upper and lower parts of the tibial diaphyses and proximal metaphyses, sparing the epiphyses

Delayed whole-body image of the three-phase bone scan
Eventually, the patient underwent bone marrow biopsy of the involved femur which revealed extensive fibrosis of the bone marrow cavity, with the ample presence of foamy histiocytes, findings suggestive of histiocytosis. Subsequent molecular analysis revealed BRAFV600E mutation and the diagnosis of ECD was confirmed. The patient was initially treated with corticosteroids, and as soon as the BRAFV600E mutation was confirmed, they were replaced by vemurafenib. To date, about 10 months after induction of vemurafenib, the patient’s symptoms have significantly improved, without any reported adverse effects.
Erdheim Chester disease: a brief review
Erdheim-Chester disease (ECD) is a rare clonal myeloid neoplasm characterized by tissue infiltration by foamy histiocytes that harbor mutations of the RAS-RAF-MEK-ERK kinase pathway [3]. According to the 2020 WHO classification of soft tissue and bone tumors, Langerhans cell histiocytosis (LCH), Erdheim Chester disease (ECD), and Rosai-Dorfman-Destombes disease (RDD) are now categorized as hematopoietic neoplasms of bone [4]. To date, only a few thousand cases have been described worldwide. However, the number of ECD diagnoses has increased exponentially during the last two decades, owing to increased awareness and diagnostic capabilities [3]. Epidemiologic data suggest a male preponderance, with a mean age of 48–56 years [5]. ECD manifestations range from localized lesions to a multisystem disease, which can prove fatal if left untreated [6]; thus, early diagnosis is crucial. Diagnosis of ECD is multimodal and requires awareness and integration of clinical information, imaging, and pathology studies [7].
Clinical manifestations
Most cases of ECD follow a progressive course, with lesions that tend to accumulate in the affected sites [3]. The pathogenesis of organ dysfunction seems to be mediated by local effects of the histiocytic infiltrate, as well as by chronic systemic inflammation [7]. Bone manifestations are the most frequent, with long-bone osteosclerosis observed in 80–95% of patients [3]. The most prevalent presenting symptom is bone pain [7]. Endocrine manifestations are among the most common. Diabetes insipidus (DI) is observed in 25% of patients and is often the first sign of ECD [3]. Ophthalmologic manifestations occur in almost a third of ECD cases, usually in the form of exophthalmos, which may be bilateral, and ophthalmoplegia [8]. ECD should be strongly suspected in patients presenting with DI of unknown etiology, bone pain, and painless bilateral exophthalmos, which compose the typical clinical triad of the disease.
Cardiovascular involvement is common in ECD and is usually associated with a poorer prognosis [7]. The most described feature is a sheathing of the aorta, with peri-aortic infiltration, also known as “coated aorta.” Retroperitoneal fibrosis (RPF) is observed in a third of patients and may be complicated by proximal obstruction of the ureters and bilateral hydronephrosis [9]. Renal artery stenosis has also been documented [10]. Nephrological manifestations may occur. Abdominal CT scans reveal infiltration of the peri-renal fat, with the typical pattern of “hairy kidneys” described in more than half of the patients [10]. Pulmonary parenchymal and pleural infiltration may present with an interstitial lung disease-like pattern [6]. Cutaneous manifestations classically affect the eyelids and periorbital tissues, presenting with palpebral xanthelasmas [8]. CNS involvement has been identified as an independent predictor of death. The most frequent signs are cerebellar and pyramidal syndromes [3]. Meningeal involvement can also occur [5].
Laboratory and histological findings and molecular analysis
There are no specific laboratory abnormalities. Elevation of erythrocyte sedimentation rate and C-reactive protein [7] as well as elevated levels of many inflammatory cytokines, such as IFN-a, IL-6, and IL-12, and chemokines [11], are observed in the majority of ECD patients, indicating a strong local and systemic inflammatory response.
Βiopsy is strongly recommended, not only to confirm the diagnosis but also to identify the mutational background of each patient, which may lead to the use of targeted therapy [6]. The gold standard is the detection of BRAFV600E or another MAPK pathway mutation [3]. Several malignancies such as melanoma and thyroid carcinoma harbor the BRAFV600E mutation, which is an activating mutation of the BRAF proto-oncogene [12]. The mutation is detected in more than half (54%) of the patients with ECD and 38% of patients with LCH, but not in any other type of histiocytosis [13, 14]. It is a strong independent determinant of cardiac and aortic infiltration and also CNS involvement [3].
Imaging
ECD has distinct radiological features. The osseous abnormalities may appear as diffuse osteosclerotic lesions in the diaphyseal and metaphyseal regions of the long bones by plain film or CT, and concomitant narrowing of the marrow cavity, seen clearly on CT [7].
MRI imaging shows a patchy “geographical” pattern with low signal on T1 and high signal on T2 fat suppression weighted images. The affected areas are usually significantly enhanced after gadolinium injection [15]. The epiphyses are generally spared and this can also be appreciated on MRI as seen in this case (Fig. 2). These features are important for an MSK radiologist to suspect ECD in the first place and to differentiate it from other bone disorders, as described below.
Bone scintigraphy exhibits the most typical pattern of all imaging modalities. The hallmark of ECD imaging is the prominent, strikingly symmetrical radiotracer uptake in all three phases of a bone scan, especially in the lower extremities, more specifically in the distal ends of the femurs and proximal and distal ends of the tibiae, sparing the epiphyses [3, 16]. This pattern is clearly depicted in our case (Fig. 4). The FDG-PET scan shows a similar uptake pattern in the long bones but can simultaneously and globally depict the extent and activity of ECD lesions, including sites of extraosseous involvement [17]. Other conditions that may exhibit symmetrical bone radiopharmaceutical uptake include systemic mastocytosis, progressive diaphyseal dysplasia (Engelmann-Camurati disease), G-6PD deficiency, Gaucher disease, multifocal osteonecrosis, usually steroid-induced, and cinacalcet treatment [18]. However, the typical ECD bone scan pattern (namely the strikingly symmetrical uptake at the femoral and tibial metaphyses with epiphyseal sparing) rarely or never presents with the aforementioned conditions.
Differential diagnosis
Bone metastases
The bone area most frequently colonized by disseminated tumor cells is the axial skeleton [19]. Radiographically, metastases can be lytic, sclerotic, or mixed, depending on the primary tumor, while typical ECD lesions are sclerotic and involve the metaphyses of long bones.
Langerhans cell histiocytosis
LCH can affect one or multiple systems. Single-system LCH is predominantly skeletal (skull, femur, vertebrae, pelvis, ribs, and mandible) with a predilection for the axial skeleton. Patients are usually young children with multiple or sequential destructive bone lesions, often with adjacent soft tissue masses. Lesions can be radiolucent on plain film and appear as intramedullary lesions with cortical erosion on MRI [4]. Bone scintigraphy may be a complementary technique, mainly for whole-body investigation [20].
Rosai-Dorfman-Destombes disease
RDD usually presents as painless cervical lymphadenopathy [21], with bone involvement in up to 10% of patients. Intraosseous RDD most commonly involves the craniofacial skeleton and metaphysis of long bones and usually affects a single bone [4], in the form of a well-defined lytic, septated mass with occasional periosteal reaction [22]. Purely sclerotic lesions—typically seen in ECD—are rare in RDD [21].
Solitary plasmacytoma of bone (SPB)
Less than 5% of patients with plasma cell dyscrasia present with a single bone lesion without evidence of systemic disease [23, 24]. The axial skeleton is mainly affected. Radiographically, SPB manifests as a well-circumscribed geographical lytic bone lesion without an identifiable matrix [4]. The lesions are isointense on T1 and hyperintense on T2-weighted images and contrast enhanced. [25]. Upon detection of multiple vertebral lesions, positivity on FDG-PET, or bone marrow disease, some patients will be upstaged for multiple myeloma [23].
Multiple myeloma
Up to 90% of myeloma patients develop osteolytic lesions, predominantly in the axial skeleton (over half of which affect the vertebrae) as well as the proximal areas of the arms and legs. Lytic lesions on plain film are typically punched-out with no surrounding reactive sclerosis—usually in the flat bones of the skull and pelvis. In long bones, appearances range from endosteal scalloping to large destructive lesions, more easily appreciated on CT. [26]. Whole-body FDG-PET imaging can be useful in clarifying disease classification [25]. Bone scans are often negative in myeloma patients with purely lytic lesions [26].
POEMS
Polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS) syndrome is a rare plasmacyte-associated disease, with typical osteosclerotic high-density lesions, or less frequent osteolytic or mixed lesions with a soap-bubble appearance [27, 28].
Primary non-hodgkin lymphoma of bone
This uncommon, extranodal lymphoma manifests as a localized solitary lesion of the medullary cavity. The tumor favors persistent bone marrow formation sites, such as the femur, pelvis, tibia, fibula, and humerus [29]. Imaging frequently shows a large, lytic, and destructive tumor with possible cortical erosion and soft tissue extension, with often moth-eaten or permeative borders [4, 29]. On FDG-PET it usually appears as a focal hypermetabolic lesion [29].
Takayasu arteritis
Thickening of the aorta wall may be observed in Takayasu arteritis. However, in this case, the entire wall (adventitia, media, and intima) is affected, whereas in ECD only the adventitial and periadventitial periaortic spaces, but not the wall itself, are involved [30].
Idiopathic retroperitoneal fibrosis (RPF)
Idiopathic RPF is not circumferential, meaning that it spares the posterior wall of the aorta, whereas in ECD, fibrosis encircles the entire aorta [30]. In ECD the distal ureters appear to be spared [9], and the inferior vena cava is usually not affected [30]. In contrast, both of the above may be affected in idiopathic RPF.
Treatment
Most patients require treatment, except for some selected cases of asymptomatic minimal-burden disease, which can be monitored closely [6]. In the past, many therapeutic regimens have been used [3, 6, 31]. The therapeutic landscape of ECD has changed dramatically since the discovery of BRAF and MAPK mutations, which paved the way for the use of targeted therapies, with BRAF and MEK inhibitors. BRAF inhibitors, such as vemurafenib, are known to improve the survival of patients with melanoma [3]. In ECD patients carrying the BRAFV600E mutation, BRAF inhibitor therapy has been shown to achieve robust and durable responses, with minimal and dose-limited toxicity [6, 32]. After the introduction of new targeted therapies, the 5-year survival rate has significantly increased, from 43% in a study from 1996 [33] to 83% in recent studies [3, 34].
Conclusion
ECD can now be defined as a clonal myeloid neoplasm due to MAPK pathway mutations. An inflammatory milieu is important in the pathogenesis and clinical manifestations of the disease [7]. ECD is difficult to diagnose, mainly owing to its variable manifestations, generally nonspecific findings, and rarity. Multidisciplinary collaboration is often needed, as diagnosis and treatment of the disease present unique challenges and require the implementation of clinical and pathological findings, molecular analysis, and imaging. Nonetheless, along with radiology, nuclear medicine undoubtedly continues to play an indispensable role in the diagnosis of ECD. In light of new and effective therapies, increased awareness of the disease is of critical importance, for clinicians, MSK radiologists, and nuclear medicine physicians alike.
References
Chester W. Über Lipoidgranulomatose. Virchows Arch Pathol Anat Physiol Klin Med [Internet]. 1930;279(2):561–602. Available from: https://doi.org/10.1007/BF01942684
Jaffe HL. Lipid (Cholesterol) granulomatosis. Metabolic, degenerative, and inflammatory disease of bones and joints. 1972;535–41.
Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease [Internet]. 2020. Available from: http://ashpublications.org/blood/article-pdf/135/16/1311/1724169/bloodbld2019002766c.pdf. Accessed 6 Sept 2023
The WHO Classification of Tumours Editorial Board 2020 WHO Classification of Tumours Soft Tissue and Bone Tumours 3 5 IARC Press Lyon 337 499
Papo M, Emile JF, Maciel TT, Bay P, Baber A, Hermine O, et al. Erdheim-Chester disease: a concise review, vol. 21. Current Rheumatology Reports: Springer; 2019.
Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood [Internet]. 2020 May 28;135(22):1929–45. Available from: https://doi.org/10.1182/blood.2019003507
Starkebaum G, Hendrie P. Erdheim–Chester disease. Vol. 34, Best Practice and Research: Clinical Rheumatology. Bailliere Tindall Ltd; 2020.
Kanakis M, Petrou P, Lourida G, Georgalas I. Erdheim-Chester disease: a comprehensive review from the ophthalmologic perspective. Vol. 67, Survey of Ophthalmology. Elsevier Inc.; 2022. p. 388–410.
Yelfimov DA, Lightner DJ, Tollefson MK. Urologic manifestations of Erdheim-Chester disease. Urology. 2014;84(1):218–21.
Estrada-Veras JI, O’Brien KJ, Boyd LC, Dave RH, Durham BH, Xi L, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1(6):357–66.
Arnaud L, Gorochov G, Charlotte F, Lvovschi V, Parizot C, Larsen M, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood [Internet]. 2011 Mar 10;117(10):2783–90. Available from: https://doi.org/10.1182/blood-2010-10-313510
Croce L, Coperchini F, Magri F, Chiovato L, Rotondi M. The multifaceted anti-cancer effects of BRAF-inhibitors the wild type BRAF gene. Oncotarget [Internet]. 2019 [cited 2023 Aug 29];10(61):6623–40. Available from: www.oncotarget.com
Haroche J, Charlotte F, Arnaud L, Von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700–3.
Emile JF, Diamond EL, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood [Internet]. 2014;124(19):3016–9. Available from: https://www.sciencedirect.com/science/article/pii/S0006497120354161. Accessed 6 Sept 2023
Sun Z, Shen C. Differential diagnosis of rare diseases involving bilateral lower extremities with similar 99mTc-MDP bone scan patterns: analysis of a case series. Diagnostics (Basel). 2022 Apr 6;12(4).
Resnick D, Greenway G, Genant H, Brower A, Haghighi P, Emmett M. Erdheim-Chester disease. Radiology. 1982;142(2):289–95.
Arnaud L, Malek Z, Archambaud F, Kas A, Toledano D, Drier A, et al. 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum. 2009;60(10):3128–38.
Lenz O, Sancassani R, Bottino C, Fornoni A. Reversible bone pain and symmetric bone scan uptake in a dialysis patient treated with cinacalcet: a case report. J Med Case Rep [Internet]. 2010;4(1):191. Available from: https://doi.org/10.1186/1752-1947-4-191
Ren G, Esposito M, Kang Y. Bone metastasis and the metastatic niche. J Mol Med (Berl). 2015;93(11):1203–12.
Khung S, Budzik JF, Amzallag-Bellenger E, Lambilliote A, Soto Ares G, Cotten A, et al. Skeletal involvement in Langerhans cell histiocytosis. Insights Imaging. 2013;4(5):569–79.
Mosheimer BA, Oppl B, Zandieh S, Fillitz M, Keil F, Klaushofer K, et al. Bone involvement in Rosai-Dorfman disease (RDD): a case report and systematic literature review. Curr Rheumatol Rep. 2017;19(5):29.
Demicco EG, Rosenberg AE, Björnsson J, Rybak LD, Unni KK, Nielsen GP. Primary Rosai-Dorfman disease of bone: a clinicopathologic study of 15 cases. Am J Surg Pathol. 2010;34(9):1324–33.
Kilciksiz S, Karakoyun-Celik O, Agaoglu FY, Haydaroglu A. A review for solitary plasmacytoma of bone and extramedullary plasmacytoma. Sci World J. 2012;2012:895765.
Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology. 2005;2005(1):373–6.
Durie BGM, Kyle RA, Belch A, Bensinger W, Blade J, Boccadoro M, et al. Myeloma management guidelines: a consensus report from the Scientific Advisors of the International Myeloma Foundation. Hematol J. 2003;4(6):379–98.
Dimopoulos M, Terpos E, Comenzo RL, Tosi P, Beksac M, Sezer O, et al. International myeloma working group consensus statement and guidelines regarding the current role of imaging techniques in the diagnosis and monitoring of multiple Myeloma. Leukemia. 2009;23(9):1545–56.
Shi XF, Hu SD, Li JM, Luo XF, Long ZB, Zhu Y, et al. Multimodal imaging and clinical characteristics of bone lesions in POEMS syndrome. Int J Clin Exp Med. 2015;8(5):7467–76.
Dispenzieri A. POEMS syndrome. Blood Rev. 2007;21(6):285–99.
Behzadi AH, Raza SI, Carrino JA, Kosmas C, Gholamrezanezhad A, Basques K, et al. Applications of PET/CT and PET/MR imaging in primary bone malignancies. PET Clin. 2018;13(4):623–34.
Haroche J, Amoura Z, Dion E, Wechsler B, Costedoat-Chalumeau N, Cacoub P, et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Vol. 83, Medicine. 2004. p. 371–92.
Haroche J, Cohen-Aubart F, Rollins BJ, Donadieu J, Charlotte F, Idbaih A, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol [Internet]. 2017 Feb 1;18(2):e113–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28214412. Accessed 6 Sept 2023
Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood [Internet]. 2013 Feb 28;121(9):1495–500. Available from: https://doi.org/10.1182/blood-2012-07-446286
Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, Remy M, et al. Erdheim-Chester disease clinical and radiologic characteristics of 59 cases. Medicine [Internet]. 1996;75(3). Available from: https://journals.lww.com/md-journal/fulltext/1996/05000/erdheim_chester_disease_clinical_and_radiologic.5.aspx. Accessed 6 Sept 2023
Cohen-Aubart F, Emile JF, Carrat F, Helias-Rodzewicz Z, Taly V, Charlotte F, et al. Phenotypes and survival in Erdheim-Chester disease: results from a 165-patient cohort. Am J Hematol [Internet]. 2018 May 1;93(5):E114–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29396850. Accessed 6 Sept 2023
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consent to participate
Informed consent was obtained from the subject described in this report.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bountas, D., Bountas, M., Exadactylou, P. et al. Erdheim-Chester disease and nuclear medicine imaging. A case report and brief review. Skeletal Radiol (2024). https://doi.org/10.1007/s00256-024-04718-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00256-024-04718-z