
Abstract
The bone marrow has been long known to host a unique environment amenable to colonization by metastasizing tumor cells. Yet, the underlying molecular interactions within this specialized microenvironment which give rise to the high incidence of bone metastasis in breast and prostate cancer patients have long remained uncharacterized. With the recent description of the bone metastatic “niche,” considerable focus has been placed on understanding how the bone stroma contributes to each step of metastasis. Discoveries within this field have demonstrated that when cancer cells home to the niche in which hematopoietic and mesenchymal stem/progenitor cells normally reside, a bidirectional crosstalk emerges between the tumor cells and the bone metastatic stroma. This communication modulates every step of cancer cell metastasis to the bone, including the initial homing and seeding, formation of micrometastases, outgrowth of macrometastases, and the maintenance of long-term dormancy of disseminated tumor cells in the bone. In clinical practice, targeting the bone metastatic niche is evolving into a promising avenue for the prevention of bone metastatic relapse, therapeutic resistance, and other aspects of cancer progression. Here, we review the current knowledge concerning the role of the bone metastatic niche in bone metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The bone is an organ frequently infiltrated by the metastatic spread of solid tumors [1–4]. An estimated 350,000 people in the USA die each year with bone metastasis as 65–80 % of patients with metastatic breast or prostate cancer present skeletal complications, while lower rates are observed in patients with lung, kidney, thyroid, or other cancers [1–3, 5]. Despite the recent approval of several bone-specific agents to alleviate skeletal-related complications, bone metastasis remains largely incurable and such treatments are usually palliative in nature [5–7]. This status may soon change as significant advances in bone metastasis research in recent years have revealed an intricate interaction between the metastatic tumor cells and the resident bone microenvironment essential for the development of osteolytic or osteoblastic bone lesions [1–3, 5]. Collectively, the unique combination of cell types, connective tissues, and signaling molecules has been named the “bone metastatic niche.”
Anatomically, the bone areas most frequently colonized by disseminated tumor cells (DTCs) are the axial skeleton, including the spine, ribs, and pelvic bones [8]. This pattern correlates with areas of red marrow in fully mature adults, indicating that active hematopoietic processes are available to provide ample cells, extracellular matrix, and nutrition to metastatic tumor cells at these sites [9]. These bone stromal cells, such as osteoblasts, osteoclasts, mesenchymal stem/stromal cells (MSCs), endothelial cells, macrophages, neutrophils, lymphocytes, and hematopoietic stem/progenitor cells (HSPCs), have been shown to either expedite or impede the progression of cancer cell metastases [10, 11]. Furthermore, a series of trophic factors, cytokines, and chemokines serve as bone stroma-derived mediators that play critical roles in building the specialized bone metastatic niche. Of these known regulators, CX-chemokine ligand 12 (CXCL12), integrins, osteopontin (OPN), vascular cell adhesion molecule-1 (VCAM-1), transforming growth factor beta (TGF-β), Jagged 1, and the receptor activator of nuclear factor kappa-b ligand (RANKL) display the greatest influence in specifying the metastatic niche. Taken together, these bone marrow (BM) niche cells and factors constitute a finely organized network that promotes DTC homing, seeding, hibernation, and proliferation, while facilitating the progressive breakdown of normal hematopoiesis and osteogenesis [12–14]. These tumor-stroma interactions not only serve as a quintessential proof for Stephen Paget’s visionary “seed and soil” hypothesis of cancer metastasis [15] but also led to the development of effective therapeutic agents, such as osteoclast-targeting bisphosphonates and the RANKL-neutralizing antibody (Denosumab) for controlling cancer-induced bone complications[1, 5]. In the future, a more comprehensive understanding of the bone metastatic niche will facilitate the development of novel therapeutic strategies for preventing or curing otherwise fatal bone complications.
The normal bone niche and homeostasis
The bone is the principal site that houses hematopoiesis and osteogenesis in healthy individuals. Accumulating evidence has shown that there is an intricately-organized microenvironment inside the bone that regulates the dynamic balance among various stem cells, progenitor cells, mature immune cells, and supporting stromal cells; this has been named the bone niche [12, 16]. In recent years, a remarkable progress has been made in the identification and characterization of the key cells and molecules regulating hematopoiesis and osteogenesis in the bone microenvironment. One emerging concept describes the existence of two primary niches in the BM: the osteoblastic niche and the perivascular niche. These contrasting niches house two types of adult stem cells and their progeny: hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs) [16–18]. HSCs differentiate to form the entire immune system and are the most well-defined stem cells, whereas the latter, the MSCs, have been recently revealed as a new niche component in the BM serving to differentiate into the mesenchymal lineage cells, which include osteoblasts, adipocytes, chondrocytes, fibroblasts, and other stromal cells. These two cell lineages are intertwined with each other in the bone niche to sustain normal bone homeostasis. Compromising the normal bone niche and homeostasis not only leads to bone complications such as osteopetrosis, osteoporosis, osteoarthritis and malignancy but also induces a series of broad disorders affecting most of the organs in the body [19]. While this new definition of the normal bone niche is still under development, a better understanding will help to understand how the niche functions under pathological conditions and may contribute to curative approaches for metastatic bone diseases.
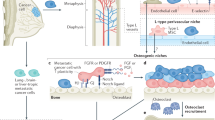
The osteoblastic niche is better described in terms of both composition and function. Localized at the inner surface of the bone cavity, this niche has been previously revealed to be the primary site to accommodate quiescent HSCs [20, 21]. Casting doubt on this old theory, the latest studies have suggested that HSCs are mostly localized in the perivascular niche where endothelial cells, CXCL12-abundant reticular (CAR) cells, and MSCs regulate the HSCs through a series of growth factors, cytokines, and chemokines such as stem cell factor (SCF), CXCL-12, and angiopoietin-1 [17, 22–24]. In particular, BM-MSCs, marked by expression of platelet-derived growth factor receptor α (PDGFRα) [25], CD51 [26], nestin [18], CD146, Mx-1 [27], leptin receptor (Lepr) [22, 23], and Prx [24], are capable of generating osteoprogenitor cells to form the osteoblastic niche and releasing CXCL-12, SCF, and other factors to maintain HSC self-renewal in the perivascular niche (Fig. 1). As a consequence of such exquisitely regulated machinery in the bone microenvironment, the hierarchical lineages of HSCs, such as myeloid cells, B lymphocytes, osteoclasts, as well as those derived from MSCs including osteoblasts and adipocytes, are precisely maintained to fulfill the functions of hematopoiesis, osteoclastogenesis, and osteogenesis. In addition to maintaining healthy local bone development, the bone niche continuously exports immune cells and tissue progenitor cells into circulation to constitute the peripheral immune system and sustain tissue repair and regeneration [28–30].

The normal bone niche and homeostasis. There are two major identified BM niches supporting hematopoiesis and osteogenesis: osteoblastic niche and perivascular niche. Within these niches, two types of adult stem cells HSCs and MSCs along with a hierarchy of their differentiated progenitor cells and mature lineage cells contribute to sustain a healthy bone microenvironment. As well, HSCs and MSCs play crucial roles to maintain the systemic homeostasis via exporting immune cells and tissue progenitor and stromal cells. Albeit still under debate, recent studies implicated that quiescent HSCs may mainly reside in the perivascular niche, whereas early B-cells occupy the osteoblastic niche. Further identification of the HSC niches and their functions will shed light on exploring the less-defined bone metastatic niche. CAR CXCL12-abundant reticular cells, MSC mesenchymal stem cells, HSC hematopoietic stem cells, CLP common lymphoid progenitors
Bone marrow contributes to distinct steps of cancer progression by exporting tumor-modulating stromal cells
Many functions of the normal bone niche are hijacked to support the growth of both primary and metastatic cancers. In particular, the tumor microenvironment (TME) has attracted attention of researchers since numerous studies have outlined key roles of the TME in the growth, spread, and therapeutic resistance of primary tumors [31, 32]. In direct contrast to tumor cells, these stromal cells in the TME make for an attractive target in cancer therapeutics as they are genetically stable. However, research efforts have focused on the cells within primary tumor sites with the assumption that associated stromal cells are also derived from the local region. In fact, characterization of TME-stromal cells at the cellular and molecular levels shows that many of them are primarily, if not exclusively, originated from the BM. This has been shown for myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), MSCs, and endothelial progenitor cells (EPCs) [33]. Carcinoma-associated fibroblasts (CAFs), another key tumor stromal population, have long been thought to derive from tumor resident tissue fibroblasts [34]. However, recent data demonstrated that CAFs can also originate from BM precursors such as BM-MSCs [35, 36]. The BM-derived CAFs, uniquely expressing alpha-smooth muscle actin (α-SMA) and certain pro-invasive molecules such as matrix metalloproteinase 13 (MMP13), represent 20–40 % of the total CAFs varying among distinct tumor types [37–40]. Along with the BM-derived cells, some well-defined tumor-regulating growth factors and cytokines are also largely derived from BM stromal cells, including vascular endothelial growth factor (VEGF), CXCL-12, TGF-β, etc [41]. Therefore, the impact of the BM microenvironment on tumorigenesis before metastasis into the bone niche is likely underestimated.
Supporting this notion, there is increasing evidence that BM-resident precursor cells and mature lineage cells could be actively recruited by tumor cells to migrate into either primary tumor sites or the distant organs in which the tumor cells metastasize, such as the lung and brain [41]. Once BM-derived cells arrive at their new niche, they quickly adapt and cooperate with the local stromal cells to motivate further cancer progression [14, 42]. In a study specifically exploring the role of BM in the crosstalk among tumors with differentially aggressive capabilities, it was shown that the more vigorous primary tumors could instigate the outgrowth of otherwise-indolent tumors through activation and mobilization of BM stromal cells to the secondary tumors [43]. Therefore, through releasing abundant tumor-modulating stromal cells, the BM microenvironment serves as a key site fostering the various steps of cancer progression.
The bone niche and bone metastasis
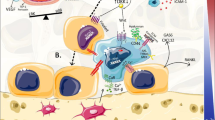
Bone metastasis is a frequent occurrence in late stages of solid tumors. The interaction between tumor cells and the bone microenvironment is critical in supporting the metastatic cascade, from the initial survival and seeding of DTCs, activation of indolent micrometastases, and expansion of osteolytic or osteoblastic bone lesions [1, 2, 5]. This mechanism does not appear to be niche-specific as both the osteoblastic and the perivascular niches have been reported to impact metastatic survival and tumor cell proliferation during bone metastasis. While direct competition for the osteoblastic niche has been observed between HSCs and metastatic cancer cells [44], the perivascular niche has also been characterized as an alternate site for bone metastatic colonization and accommodation of dormant cancer cells [10, 45–47]. The roles of bone metastatic niche in directing successive steps of bone metastasis of solid tumors are described below (Fig. 2).

Bone metastatic niche in distinct steps of bone metastasis. Bone niche cells including vascular cells, BM-MSCs and osteoblasts, recruit CXCR4+ DTCs to home to the bone via releasing CXCL12. The DTCs then seed onto either vascular niche or osteoblastic niche by stroma-mediated adhesion. These DTCs either enter a dormant status or develop into osteolytic or osteoblastic macrometastasis. Abundant studies have illustrated a “vicious cycle” in tumor-stroma interaction toward formation of the macrometastasis at relatively late stages of bone metastasis. Endocrine factors especially the sex steroids serve to impede such an osteolytic process by induction of osteoclast apoptosis or inhibition of osteoclast activation. In contrast to the macrometastasis-supporting niche, it remains largely uncharacterized for the early metastatic niche as well as the niche governing tumor cell dormancy. ECM extracellular matrix, DTC disseminated tumor cells, MSC mesenchymal stem cells, HSC hematopoietic stem cells, LOX lysyl oxidase, MMP matrix metalloproteinase, PTHrP parathyroid hormone-related protein, IGF insulin-like growth factor
DTCs homing to the bone
The first step of metastasis requires the escape of metastasized tumor cells from the primary tumor into circulation. This invasion and intravasation are driven by tumor cell-intrinsic mechanisms, their associated stroma, and the local extracellular matrix (ECM) surrounding the primary tumor [48, 49]. Regulation of bone metastasis by early events at the primary tumor sites has been highlighted by a recent study showing that the CAF-enriched primary tumor stroma could mimic the CXCL12-rich bone metastatic niche and therefore pre-select the cancer cells that are primed for metastasis to bone [50].
When tumor cells successfully escape the primary tumors and disseminate into the periphery, only a very small percentage (0.02–0.1 %) survive during circulation and homing to the secondary organs [51]. One essential pathway governing this tumor cell homing to the bone is the CXCL12–CXC-chemokine receptor 4 (CXCR4) signaling axis. CXCL12 is predominantly produced by a diversity of BM stromal cells including BM-MSCs, endothelial cells, CAR cells, and osteoblasts. This pathway has been extensively studied for its role in controlling both HSC and leukemia cell trafficking through BM [24, 52, 53]. By using mice with conditional deletion of Cxcl12 in candidate bone niche cells, recent studies have mapped the CXCL12-expressing bone stromal cells critical for HSC and leukemic cell maintenance in the bone. The results from these studies underlined a more important contribution of the perivascular niche than the osteoblastic niche in supporting quiescent HSCs [22, 24, 52]. Solid tumor cells employ the same homing mechanism by overexpressing CXCL12 receptors CXCR4 and CXCR7, which induce chemotaxis along CXCL12 gradients to home to the bone [54, 55]. In clinical cancer patients, CXCR4 expression predicts early metastatic relapse in breast, prostate, colorectal, and other types of cancer [55, 56]. As such, targeting the CXCL12-CXCR4 (CXCR7) axis is emerging as a promising therapy for prevention and treatment of bone metastatic diseases [56]. In addition to this major signaling pathway, other chemokines and cytokines have also been reported to mediate the DTC homing to the bone, such as CCL-2 and CCL-22 [57, 58]. In contrast to homing mechanisms, certain growth factors such as granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF), utilized for HSC mobilization out of BM, have been shown to similarly mobilize prostate cancer cells into the peripheral circulation [44, 59, 60].
BM hematopoietic progenitor cells contribute to the “pre-metastatic” niche formation at secondary metastatic sites such as the lung in animal models [42]. However, the “pre-metastatic” niche at the bone during primary tumor progression is less studied, although a few reports showed the evidence of bone pre-metastatic conditioning by tumor cell-secreted proteins and exosomes [61, 62]. A lack of experimental evidence is likely due to limited spontaneous bone metastasis models available. Future efforts might be made to explore whether a receptive or resistant bone niche could be driven by primary tumor growth, the cellular and molecular mechanisms governing such niche formation, as well as their clinical relevance.
Seeding and formation of micrometastases
When DTCs arrive at the bone marrow niche, most are rapidly cleared by resident immunosurveillance while few survive after a firm adhesion to the BM stroma [63, 64]. Cell-cell adhesion is a critical step during the initial seeding of DTCs to the bone metastatic niche; multiple adhesion molecules have been implicated in this initial step. The expression of integrin αvβ3 on metastatic tumor cells primarily promotes their adherence to ECM components such as osteopontin, fibronectin, vitronectin, and thrombospondin [65]. On the other hand, cancer cells also express α4β1 integrin to engage to intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) expressed by BM stromal cells and vascular cells [65]. These integrin-mediated cell-cell adhesions have been described in prostate, breast, lung, and other cancer types, to provide survival, proliferation, and osteoclast activation signals for developing the DTCs into overt macrometastases [66, 67]. As a result, integrins have become another attractive target for the treatment of skeletal metastasis of solid tumors in pre-clinical studies and clinical trials [65].
In addition to integrins, annexin II, a protein mediating the adhesion of HSCs to osteoblasts, has also been reported to play a role in prostate cancer cell seeding on the bone niche [68]. In a recent study specifically exploring the early bone metastatic niche, it was shown that breast cancer DTCs prefer to seed on the osteogenic niche through heterotypic adherens junctions (hAJs) between E-cadherin in tumor cells and N-cadherin in osteogenic cells. Upon hAJ interaction, such adhesion further activates mTOR and AKT signaling to promote the DTC proliferation to form micrometastases [69].
As these adhesion and seeding mechanisms are also employed by the bone niche to regulate HSC cell homing and retention in the BM, it has been hypothesized that DTCs may compete with the HSC cells for the limited number of niche sites. Indeed, in an experimental bone metastasis model of prostate cancer, it was shown that prostate cancer cells use the CXCL12-CXCR4 axis to home to the osteoblastic niche, where they compete with the HSC cells for niche support [44]. Supporting this notion, in human cancer patients the prevalence of DTCs in the BM were found to be independent of the primary tumor size, possibly due to the limited niche sites available in the bone [70]. Recent advances in the recognition of specific BM niches accommodating quiescent HSCs may provide insights to uncover the less-studied bone metastatic niche. Furthermore, stresses that perturb HSCs may in turn be exploited to treat bone metastasis [17, 18, 22–24].
Metastatic outgrowth in bone
Bone tissue constantly undergoes dynamic remodeling mediated by the balanced activity of osteoclasts and osteoblasts [13, 71, 72]. When metastatic cancer cells finally settle down in the bone niche by overtaking the host stroma, they often exploit the normal bone homeostatic process and tip the equilibrium toward either hyperactive bone lysis or bone growth to facilitate the formation of bone metastasis. The proclivity of breast cancer in forming osteolytic bone metastasis has been frequently cited as the classic example of “seed and soil” interactions between tumor and stroma in metastasis [2, 15]. A “vicious cycle” of molecular crosstalk between tumor cells and the bone metastatic niche often takes place in osteolytic bone metastasis [2]. In this process, tumor cells produced a variety of cytokines and growth factors such as tumor necrosis factor alpha (TNF-α), IL11, VCAM-1, MMP1, Jagged 1, and parathyroid hormone-related protein (PTHrP), which either directly stimulate the osteoclast maturation or indirectly promote osteoclast differentiation through stimulating the BM osteoblasts to produce IL-6 and RANKL [2]. With the osteoclast activation, TGF-β and insulin-like growth factor-1 (IGF1) released by bone matrix resorption in turn promote the survival and proliferation of cancer cells, thus forming an amplifying loop to efficiently drive osteolytic metastasis [1, 2, 14]. The enzyme lysyl oxidase (LOX) secreted by primary tumor cells, previously shown essential for creating a permissive lung metastatic niche [73], is recently reported as an activator of osteoclasts during osteolytic bone metastasis [74]. In addition to these molecules, microRNAs have emerged as new players in mediating the interplay between osteoclasts and the tumor cells, adding another layer to the “vicious cycle” of osteolytic metastasis [75].
In sex steroid (androgen, estrogen, and progesterone) responsive cancers such as prostate, breast, and ovarian cancers, the host endocrine changes caused by disease progression or hormone deprivation therapy can also promote the osteolytic bone metastasis [76]. Both clinical and pre-clinical evidence showed that reduction of the systemic sex steroid levels by castration or ovariectomy contributed to accelerated osteolytic bone metastases in multiple tumor models [77–79]. These effects involve a hormone-bone niche crosstalk as estrogen, androgen, vitamin D, and other endocrine factors can directly or indirectly promote osteoclast apoptosis and simultaneously prolong the osteoblast lifespan [80–83]. As a consequence, hormone deprivation will induce a significant bone resorption leading to enhanced osteolytic lesion formation during bone metastasis. Currently, prevention of bone loss by inhibiting osteoclasts is a commonly applied strategy in clinical treatment of bone metastasis during hormone deprivation therapy [84, 85].
In contrast to the more frequent osteolytic metastasis leading to the aberrant bone resorption witnessed in hormone independent breast cancers and multiple myeloma, osteoblastic lesions found in prostate or hormone-dependent breast cancers involve increased osteoblast differentiation with uncontrolled bone formation [6]. In osteoblastic metastasis, WNT signaling originated from tumor cells is essential to direct osteoblast differentiation by activating transcription factor Runt-related transcription factor 2 (RUNX2) [6, 86]. Additionally, other factors such as fibroblast growth factor (FGF), bone morphogenetic proteins (BMPs), and IGFs can also be secreted by tumor cells to stimulate osteoblast differentiation and activity. Accompanied with the aberrant bone formation, abnormal osteolysis take place owing to the osteoblast-derived RANKL [87]. These relatively late stages of the bone metastasis occur primarily at the tumor-bone interface within the osteoblastic niche and both the osteoclasts and osteoblasts function as major players. Approved clinical therapies targeting bone metastasis, such as bisphosphonates (inhibitors of osteoclasts) and anti-RANKL antibody denosumab, are directed at this step and are therefore only palliative in nature [88].
Tumor cell dormancy in bone
Both clinical observations and animal studies suggest that metastases arise from the DTCs distributed among different organs [89, 90]. Metastatic relapse can arise years after primary tumor removal since DTCs can remain dormant and resistant to conventional therapies until re-activation. In some cases, these dormant cancer cells may correspond to the recently recognized “cancer stem cells” which are also shown to be quiescent with the corresponding ability to evade various stresses [91]. Both the cell-intrinsic and stromal factors regulating eventual outgrowth of indolent DTCs still remain largely uncharacterized and present the most-promising treatment avenue for reducing bone metastasis.
Among the organs harboring dormant tumor cells, the bone contains a unique hypoxic microenvironment populated with various stromal cells replete with anti-apoptotic and survival signals, which are important for quiescent HSCs [90, 92]. In clinical cancer patients, the presence of DTCs or micrometastases in the BM is a significant indicator of poor prognosis in breast cancer, squamous cell carcinoma, and resected esophageal cancer [70, 93–95]. A valuable strategy for exploring the dormant BM niche for DTCs seeks to find the counterparts for maintaining HSC quiescence. With the recent progress in identifying the quiescent HSC niche, there are numerous research resources developed which may shed light on finding out the DTC dormant niche in the bone microenvironment [22–24]. Although still under debate, accumulating evidence revealed that quiescent HSCs predominantly reside within the perivascular niche of the BM in mouse models [17, 22, 25, 26]. Consistent with these findings, a recent study with breast cancer models indicated that dormant cancer cells preferentially reside in the bone vascular niche, where the tumor cells keep quiescent through their adhesion to endothelial-derived thrombospondin-1. This dormancy could be disrupted by sprouting neovasculature mediated by TGF-β1 and periostin produced by endothelial tip cells [47]. Such a switch between dormancy and outgrowth has also been reported in another breast cancer study showing upregulation of VCAM-1 on the dormant DTCs would stimulate them to develop into overt bone metastasis by interacting with its cognate receptor integrin α4β1 expressed on osteoclasts [66]. Moreover, certain molecules such as BMP7, TGFβ2, and growth arrest-specific protein 6 (GAS6), which regulate HSC quiescence, also appear to induce tumor dormancy in mouse models of prostate cancer and head and neck squamous cell carcinoma [96–98].
Despite these advances, many factors remain undescribed such as the physical location of the dormant bone niche, the cellular and molecular mechanisms balancing dormancy with re-activation, and the inability of many dormant DTCs to form bone metastasis. Generating a comprehensive understanding of the bone niche accommodating dormant DTCs will be an essential step toward developing clinical therapeutics to prevent tumor recurrence and metastasis. Directly targeting of the dormant tumor cells and the associated hypoxic niches represents a more effective treatment paradigm than waiting for the macrometastasis to emerge. Successful experience from treatments of leukemic stem cells and latent HIV may imply that disruption of the DTC niche could sensitize the dormant tumor cells to regular cytotoxic therapies [99, 100].
Bone metastatic niche alteration during cancer therapies
Conventional cancer therapies including chemotherapy and radiotherapy could cause a significant damage to the hematopoietic stem/progenitor cells and BM stromal cells [101, 102]. During treatment of hematological malignancies, several recent studies suggested that the cancer therapies could re-organize the already dysfunctional BM niches and confer a robust chemoresistance for invading leukemia cells or lymphoma cells in vivo [103, 104]. Additionally, numerous studies have shown that chemotherapies might induce an acquired chemoresistance through modulating the CXCL12-CXCR4 interaction between BM stroma and the blood cancer cells [55, 105, 106]. In solid tumors such as breast and prostate cancer, bone metastasis-targeting therapies have also been reported to disrupt normal BM hematopoiesis [107]. Yet, how cancer therapies modulate the BM niche to impact further metastatic progression of DTCs remains an unexplored field. Limited evidence from studies of ovarian cancer and rhabdomyosarcoma demonstrated that radiotherapy or the chemotherapeutic drug cyclophosphamide were capable of upregulating several chemokines and growth factors in the BM niche such as CXCL-12, VEGF, CCL-2 and hepatocyte growth factor (HGF), as well as bioactive lipids sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P). These therapy-induced factors may play roles in promoting tumor cell survival and proliferation [108–110]. Further investigation into therapeutic-induced bone metastatic niche alterations and their influence on bone metastatic relapse will not only deepen our understanding of microenvironmental control of cancer progression but more importantly, may lead to new clinical strategies for improving the efficacies of current cancer therapeutics.
Perspectives
In the past decade, we have witnessed considerable progress in the understanding of the BM-HSC niche. While not complete, this may stimulate both the conceptual insight and technological development needed to probe the lesser-known bone metastatic niche. Given that clinical observations have revealed the bone as one of the major organs accommodating DTCs, a deeper exploration of the bone metastatic niche at structural, cellular, and molecular levels will be extremely useful for designing novel therapies to treat the incurable bone complications associated with cancer progression. These therapies will need to specifically target the mechanisms underlying tumor dormancy, metastatic relapse, and therapeutic resistance. A better understanding of the bone metastatic niche will also be instructional to developing early interventions before the formation of macrometastasis, as well as treatments specific to different stages of the bone metastasis. Of the several major weaknesses in our understanding of the bone niche mentioned here, many additional steps must be taken to further advance our understanding of the bone metastatic disease:
-
1.
Only a few spontaneous bone metastasis models have been established in either genetically engineered mice or orthotopic cancer models. Such spontaneous metastasis models more closely mimic the natural metastatic steps in human cancer patients. Although several intra-arterial tumor cell inoculation methods have been used to generate experimental bone metastasis at higher efficiency, such models may generate certain artificial interpretations due to omission of some key steps in cancer progression such as primary tumor growth and metastatic cell invasion and intravasation. It is also possible that different intravascular injections may alter the bone vascular niche. Development of improved mouse models of bone metastasis is needed to investigate different steps of bone metastasis development, particularly early steps of metastatic dissemination and seeding in the bone niche.
-
2.
Most of the bone metastatic niche studies are based on animal models, and it is not known how much similarity there is between animals and human patients regarding the histological structures, cellular components, and molecular pathways of the bone metastatic niche. The combination of human cancer patient specimen analysis with application of humanized animal model may improve this issue and facilitate the clinical translation.
-
3.
Although many signaling pathways have been recently identified to regulate metastatic tumor cells and the BM stromal cells during bone metastasis, many conclusions are drawn from the in vitro culturing systems. It is unclear to what extent the distinct bone stromal cells, especially osteoblasts, BM-MSCs, and endothelial cells in specific niches (perivascular niche vs osteoblastic niche) play a role in controlling each step of bone metastasis in vivo. Efforts might be made to define the functional contributions of the specific niche cells by applying genetically modified mice, lineage-specific tracing and ablating, intravital imaging, and other related techniques.
-
4.
It was recently shown that circadian rhythm and its associated sympathetic nervous system play a critical role in regulating HSC niche and functions [111, 112]. It might be interesting to further explore whether the nervous system and physiopathological stress also contribute to orchestrate the bone metastatic niche during bone metastasis [113].
-
5.
As mentioned above, the impact of cancer therapies on bone metastatic niche alterations and the influence of such changes on further metastatic progression remain largely undefined. This is an important question for cancer patients to investigate how different therapies functionally modify the bone metastatic niche into either a cancer-permissive or cancer-restrictive microenvironment, and the answers may help build the personalized cancer therapy.
References
Weilbaecher KN, Guise TA, McCauley LK (2011) Cancer to bone: a fatal attraction. Nat Rev Cancer 11:411–425
Ell B, Kang Y (2012) SnapShot: bone metastasis. Cell 151:690–690, e691
Mundy GR (2002) Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2:584–593
Pienta KJ, Robertson BA, Coffey DS, Taichman RS (2013) The cancer diaspora: metastasis beyond the seed and soil hypothesis. Clin Cancer Res Off J Am Assoc Cancer Res 19:5849–5855
Suva LJ, Washam C, Nicholas RW, Griffin RJ (2011) Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol 7:208–218
Sturge J, Caley MP, Waxman J (2011) Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol 8:357–368
Coleman RE (2012) Bone cancer in 2011: prevention and treatment of bone metastases. Nat Rev Clin Oncol 9:76–78
Hage WD, Aboulafia AJ, Aboulafia DM (2000) Incidence, location, and diagnostic evaluation of metastatic bone disease. Orthop Clin N Am 31:515–528, vii
Coleman RE (2006) Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res Off J Am Assoc Cancer Res 12:6243s–6249s
Kaplan RN, Psaila B, Lyden D (2006) Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev 25:521–529
Park SI, Soki FN, McCauley LK (2011) Roles of bone marrow cells in skeletal metastases: no longer bystanders. Cancer Microenviron Off J Int Cancer Microenviron Soc 4:237–246
Shen Y, Nilsson SK (2012) Bone, microenvironment and hematopoiesis. Curr Opin Hematol 19:250–255
Kingsley LA, Fournier PG, Chirgwin JM, Guise TA (2007) Molecular biology of bone metastasis. Mol Cancer Ther 6:2609–2617
Psaila B, Lyden D (2009) The metastatic niche: adapting the foreign soil. Nat Rev Cancer 9:285–293
Paget S (1989) The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 8:98–101
Frenette PS, Pinho S, Lucas D, Scheiermann C (2013) Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol 31:285–316
Morrison SJ, Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature 505:327–334
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466:829–834
Karsenty G, Ferron M (2012) The contribution of bone to whole-organism physiology. Nature 481:314–320
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ et al (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425:836–841
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR et al (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425:841–846
Ding L, Morrison SJ (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495:231–235
Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481:457–462
Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC (2013) CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495:227–230
Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-Hara C, Nagoshi N, Sunabori T et al (2009) Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206:2483–2496
Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, Frenette PS (2013) PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med 210:1351–1367
Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT (2012) Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10:259–272
Hess D, Li L, Martin M, Sakano S, Hill D, Strutt B, Thyssen S, Gray DA, Bhatia M (2003) Bone marrow-derived stem cells initiate pancreatic regeneration. Nat Biotechnol 21:763–770
Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM et al (2001) Bone marrow cells regenerate infarcted myocardium. Nature 410:701–705
Mercier FE, Ragu C, Scadden DT (2012) The bone marrow at the crossroads of blood and immunity. Nat Rev Immunol 12:49–60
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19:1423–1437
Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, Coussens LM, DeClerck YA (2012) Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res 72:2473–2480
Chantrain CF, Feron O, Marbaix E, DeClerck YA (2008) Bone marrow microenvironment and tumor progression. Cancer Microenviron Off J Int Cancer Microenviron Soc 1:23–35
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Ostman A, Augsten M (2009) Cancer-associated fibroblasts and tumor growth—bystanders turning into key players. Curr Opin Genet Dev 19:67–73
Shimoda M, Mellody KT, Orimo A (2010) Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol 21:19–25
Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA (2004) Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res 64:8492–8495
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS et al (2011) Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19:257–272
Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D (2008) Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 68:4331–4339
Lecomte J, Masset A, Blacher S, Maertens L, Gothot A, Delgaudine M, Bruyere F, Carnet O, Paupert J, Illemann M et al (2012) Bone marrow-derived myofibroblasts are the providers of pro-invasive matrix metalloproteinase 13 in primary tumor. Neoplasia 14:943–951
Casimiro S, Guise TA, Chirgwin J (2009) The critical role of the bone microenvironment in cancer metastases. Mol Cell Endocrinol 310:71–81
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827
McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, Reinhardt F, Harris LN, Hylander BL, Repasky EA et al (2008) Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133:994–1005
Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM et al (2011) Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Investig 121:1298–1312
Kuhn NZ, Tuan RS (2010) Regulation of stemness and stem cell niche of mesenchymal stem cells: implications in tumorigenesis and metastasis. J Cell Physiol 222:268–277
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA (2008) Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 322:1861–1865
Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY et al (2013) The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15:807–817
Zheng H, Kang Y (2014) Multilayer control of the EMT master regulators. Oncogene 33:1755–1763
Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG (2012) Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol 181:1895–1899
Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massague J (2013) Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154:1060–1073
Wan L, Pantel K, Kang Y (2013) Tumor metastasis: moving new biological insights into the clinic. Nat Med 19:1450–1464
Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA et al (2015) CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 27:755–768
Cojoc M, Peitzsch C, Trautmann F, Polishchuk L, Telegeev GD, Dubrovska A (2013) Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther 6:1347–1361
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Teicher BA, Fricker SP (2010) CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res Off J Am Assoc Cancer Res 16:2927–2931
Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK (2011) CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res Off J Am Assoc Cancer Res 17:2074–2080
Lu X, Kang Y (2009) Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 284:29087–29096
Nakamura ES, Koizumi K, Kobayashi M, Saitoh Y, Arita Y, Nakayama T, Sakurai H, Yoshie O, Saiki I (2006) RANKL-induced CCL22/macrophage-derived chemokine produced from osteoclasts potentially promotes the bone metastasis of lung cancer expressing its receptor CCR4. Clin Exp Metastasis 23:9–18
Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N et al (2002) G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol 3:687–694
Schroeder MA, DiPersio JF (2012) Mobilization of hematopoietic stem and leukemia cells. J Leukoc Biol 91:47–57
Kelly T, Suva LJ, Huang Y, Macleod V, Miao HQ, Walker RC, Sanderson RD (2005) Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res 65:5778–5784
Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C et al (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18:883–891
Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA et al (2012) Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 18:1224–1231
Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y et al (2010) Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Investig 120:2030–2039
Schneider JG, Amend SR, Weilbaecher KN (2011) Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone 48:54–65
Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, Yan J, Hua Y, Tiede BJ, Lu X et al (2011) VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 20:701–714
Zhao Y, Bachelier R, Treilleux I, Pujuguet P, Peyruchaud O, Baron R, Clement-Lacroix P, Clezardin P (2007) Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res 67:5821–5830
Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, Wang J, Lu G, Roodman GD, Loberg RD et al (2008) Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem 105:370–380
Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, Zhao H, Zhao Z, Du S, Tao J et al (2015) The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 27:193–210
Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G et al (2005) A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med 353:793–802
Sethi N, Dai X, Winter CG, Kang Y (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19:192–205
Esposito M, Kang Y (2014) Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther 141:222–233
Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ (2006) Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440:1222–1226
Cox TR, Rumney RM, Schoof EM, Perryman L, Hoye AM, Agrawal A, Bird D, Latif NA, Forrest H, Evans HR et al (2015) The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522:106–110
Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, Amadori D, Kang Y (2013) Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 24:542–556
Hofbauer LC, Rachner TD, Coleman RE, Jakob F (2014) Endocrine aspects of bone metastases. Lancet Diabetes Endocrinol 2:500–512
Ottewell PD, Wang N, Meek J, Fowles CA, Croucher PI, Eaton CL, Holen I (2014) Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr Relat Cancer 21:769–781
Ottewell PD, Wang N, Brown HK, Fowles CA, Croucher PI, Eaton CL, Holen I (2015) OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int J Cancer J Int Cancer 137:968–977
Shahinian VB, Kuo YF, Freeman JL, Goodwin JS (2005) Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 352:154–164
Clarke BL, Khosla S (2009) Androgens and bone. Steroids 74:296–305
Smith EP, Specker B, Korach KS (2010) Recent experimental and clinical findings in the skeleton associated with loss of estrogen hormone or estrogen receptor activity. J Steroid Biochem Mol Biol 118:264–272
Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y et al (2007) Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130:811–823
Takahashi N, Udagawa N, Suda T (2014) Vitamin D endocrine system and osteoclasts. BoneKEy Rep 3:495
Morrissey C, Roudier MP, Dowell A, True LD, Ketchanji M, Welty C, Corey E, Lange PH, Higano CS, Vessella RL (2013) Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration-resistant prostate cancer: results from the University of Washington Rapid Autopsy Series. J Bone Miner Res Off J Am Soc Bone Miner Res 28:333–340
Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H et al (2011) Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 377:813–822
Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET (2005) Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res 65:7554–7560
Patel LR, Camacho DF, Shiozawa Y, Pienta KJ, Taichman RS (2011) Mechanisms of cancer cell metastasis to the bone: a multistep process. Future Oncol 7:1285–1297
Clement-Demange L, Clezardin P (2015) Emerging therapies in bone metastasis. Curr Opin Pharmacol 22:79–86
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14:611–622
Kang Y, Pantel K (2013) Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23:573–581
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8:755–768
Anthony BA, Link DC (2014) Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol 35:32–37
Janni W, Vogl FD, Wiedswang G, Synnestvedt M, Fehm T, Juckstock J, Borgen E, Rack B, Braun S, Sommer H et al (2011) Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse--a European pooled analysis. Clin Cancer Res Off J Am Assoc Cancer Res 17:2967–2976
Grobe A, Blessmann M, Hanken H, Friedrich RE, Schon G, Wikner J, Effenberger KE, Kluwe L, Heiland M, Pantel K et al (2014) Prognostic relevance of circulating tumor cells in blood and disseminated tumor cells in bone marrow of patients with squamous cell carcinoma of the oral cavity. Clin Cancer Res Off J Am Assoc Cancer Res 20:425–433
Vashist YK, Effenberger KE, Vettorazzi E, Riethdorf S, Yekebas EF, Izbicki JR, Pantel K (2012) Disseminated tumor cells in bone marrow and the natural course of resected esophageal cancer. Ann Surg 255:1105–1112
Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, Schewe DM, Aguirre-Ghiso JA (2013) TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol 15:1351–1361
Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C et al (2011) Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 208:2641–2655
Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, Yumoto K, Berry JE, Shiozawa Y, Pienta KJ (2013) GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One 8:e61873
Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC et al (2012) Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485
Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A et al (2010) Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 28:275–280
Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, Tessarollo L, Frenette PS (2013) Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med 19:695–703
Shao L, Luo Y, Zhou D (2014) Hematopoietic stem cell injury induced by ionizing radiation. Antioxid Redox Signal 20:1447–1462
Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, Zhou XC, Cai YJ, Li ZQ, Zhang F et al (2014) Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25:778–793
Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA (2013) Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 121:4821–4831
Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, Walenkamp AM (2013) A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer 49:219–230
Sison EA, McIntyre E, Magoon D, Brown P (2013) Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res MCR 11:1004–1016
Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G (2008) Immunological aspects of cancer chemotherapy. Nat Rev Immunol 8:59–73
Park SI, Liao J, Berry JE, Li X, Koh AJ, Michalski ME, Eber MR, Soki FN, Sadler D, Sud S et al (2012) Cyclophosphamide creates a receptive microenvironment for prostate cancer skeletal metastasis. Cancer Res 72:2522–2532
Jankowski K, Kucia M, Wysoczynski M, Reca R, Zhao D, Trzyna E, Trent J, Peiper S, Zembala M, Ratajczak J et al (2003) Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res 63:7926–7935
Schneider G, Bryndza E, Abdel-Latif A, Ratajczak J, Maj M, Tarnowski M, Klyachkin YM, Houghton P, Morris AJ, Vater A et al (2013) Bioactive lipids S1P and C1P are prometastatic factors in human rhabdomyosarcoma, and their tissue levels increase in response to radio/chemotherapy. Mol Cancer Res MCR 11:793–807
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452:442–447
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124:407–421
Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner NL, Munoz SA, Zijlstra A, Yang X, Sterling JA et al (2012) Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol 10:e1001363
Acknowledgments
Research in our laboratory on bone metastasis was supported by grants from the Komen for the Cure (KG110464), Department of Defense (BC123187), the Breast Cancer Research Foundation, Brewster Foundation, and the National Institutes of Health (R01CA134519 and R01CA141062) to Y.K, an NIH K99 Award (K99CA188093) to G.R. and a pre-doctoral fellowship from the New Jersey Commission on Cancer Research to M.E.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ren, G., Esposito, M. & Kang, Y. Bone metastasis and the metastatic niche. J Mol Med 93, 1203–1212 (2015). https://doi.org/10.1007/s00109-015-1329-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-015-1329-4