
Abstract
The yeast Dekkera bruxellensis is well-known for its adaptation to industrial ethanol fermentation processes, which can be further improved if nitrate is present in the substrate. To date, the assimilation of nitrate has been considered inefficient because of the apparent energy cost imposed on cell metabolism. Recent research, however, has shown that nitrate promotes growth rate and ethanol yield when oxygen is absent from the environment. Given this, the present work aimed to identify the biological mechanisms behind this physiological behaviour. Proteomic analyses comparing four contrasting growth conditions gave some clues on how nitrate could be used as primary nitrogen source by D. bruxellensis GDB 248 (URM 8346) cells in anaerobiosis. The superior anaerobic growth in nitrate seems to be a consequence of increased cell metabolism (glycolytic pathway, production of ATP and NADPH and anaplerotic reactions providing metabolic intermediates) regulated by balanced activation of TORC1 and NCR de-repression mechanisms. On the other hand, the poor growth observed in aerobiosis is likely due to an oxidative stress triggered by nitrate when oxygen is present. These results represent a milestone regarding the knowledge about nitrate metabolism and might be explored for future use of D. bruxellensis as an industrial yeast.
Key points
• Nitrate can be regarded as preferential nitrogen source for D. bruxellensis.
• Oxidative stress limits the growth of D. bruxellensis in nitrate in aerobiosis.
• Nitrate is a nutrient for novel industrial bioprocesses using D. bruxellensis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The yeast Dekkera bruxellensis (the teleomorph of Brettanomyces bruxellensis) is known for its introduction and maintenance in industrial fermentative processes for the production of ethanol fuel. Regarded initially as a contaminant, D. bruxellensis, in recent studies, has revealed a set of physiological characteristics that indicate its biotechnological potential (Blomqvist et al. 2010; Leite et al. 2013; Parente et al. 2015; Reis et al. 2016; De Barros Pita et al. 2019). A striking feature in the yeast D. bruxellensis is its ability to assimilate nitrate, a source of nitrogen present in sugarcane juice and which is not used by Saccharomyces cerevisiae (de Barros Pita et al. 2011; de Barros Pita et al. 2013; Pereira et al. 2012, 2014). Our recent results have shown for the first time that D. bruxellensis strains isolated from sugarcane juice fermentation can grow anaerobically with nitrate as the sole source of nitrogen (N), producing ethanol with yields similar to S. cerevisiae (Peña-Moreno et al. 2019). This is a particular feature of bioethanol strains since their wine counterparts can only grow in anaerobiosis in the presence of nitrate if at least traces of ammonium or amino acids are present in the medium (Galafassi et al. 2013; Tiukova et al. 2013). Unlike what has been observed in aerobiosis, the presence of nitrate in the substrate nitrate increases both cell growth and ethanol production (Peña-Moreno et al. 2019).
The limited capacity to grow in nitrate has been related to the energy cost of the assimilatory reactions, where 4 mols of reduced nicotinamide dinucleotide (phosphate) (NAD(P)H) are necessary for the conversion of one mol of nitrate to ammonium (Siverio 2002; Galafassi et al. 2013). To fulfil this requirement, D. bruxellensis cells produce acetate by the so-called pyruvate dehydrogenase bypass (PDH-bypass) pathway, from which the pyruvate is converted to acetaldehyde by pyruvate decarboxylase and this molecule further converted to acetate by NADP+-dependent acetaldehyde dehydrogenase (ACDH) with NADPH production (Van Rossum et al. 2016). This last reaction competes with NADH-dependent alcohol dehydrogenase (ADH) in such a way that the more acetate produced, the less ethanol is generated (de Barros Pita et al. 2013; Leite et al. 2013). This yeast is more accurately classified as acetogenic due the constitutive production of acetate even in ammonium (Leite et al. 2013; Teles et al. 2018). This physiological characteristic seems to be the cause of the Custers effect, defined as negative effect on the glycolytic pathway caused by the reduced NADH re-oxidation and shortage of NAD+ cofactor by the ACDH/ADH competition (van Dijken and Scheffers 1986). The proteome analysis of D. bruxellensis cells cultivated in aerobiosis revealed some aspects of the yeast metabolism in nitrate as N source compared to cultivation in ammonium (Neto et al. 2014). The results in nitrate showed the overproduction of proteins involved in the pentose phosphate pathway PPP) as the provider of NADPH required for nitrate assimilation, as well as proteins of the tricarboxylic acid (TCA) cycle as provider of anabolic metabolites and adenosine triphosphate (ATP) biosynthetic pathway as energy provider (Neto et al. 2014). These results were in agreement with gene expression profiles studied under the same conditions (de Barros Pita et al. 2013).
Our recent data on the high growth rate of D. bruxellensis in nitrate when oxygen is completely absent raised questions about the real extension of the energetic cost for nitrate assimilation (Peña-Moreno et al. 2019). Another relevant issue in this matter is the paradox created when considering the previous classification of nitrate as a non-preferential or even poorly assimilable N source in the presence of oxygen (Cajueiro et al. 2017; Parente et al. 2017). Therefore, the aim of the present work was to identify genetic and metabolic factors leading to the distinct physiological profiles of D. bruxellensis observed in nitrate or ammonium, depending on the oxygen availability. In this study, we performed differential proteomics in nitrate-based media both in aerobiosis and anaerobiosis. In addition, we compared the proteomic profiles of D. bruxellensis cells in anaerobiosis when ammonium or nitrate was used as the N source, in a feasible attempt to resemble the industrial environment. Overall, the results uncovered some relevant physiological aspects of nitrate assimilation in anaerobiosis and also revealed the major regulatory metabolism behind the cellular conversion of nitrate to ammonium in D. bruxellensis in the presence or absence of oxygen.
Material and methods
Yeast strain and preculture conditions
D. bruxellensis GDB 248 (URM 8346) was used in the present work (Peña-Moreno et al. 2019). This strain is deposited at the Department of Mycology Culture Collection (URM-Recife), Federal University of Pernambuco, which is part of the World Directory of Collections of Culture of Microorganisms (WFCC) under the registration number 604, and can be released for research purpose upon request. The cells were maintained in a solid yeast extract–peptone–dextrose (YPD) medium (10 g L−1 yeast extract, 20 g L−1 glucose, 20 g L−1 peptone and 20 g L−1 agar). For the pre-inoculum, cells were cultivated in flasks containing 50 mL liquid YPD and remained at 30 °C for 48 h in a rotatory shaker at 160 rpm. Then, the cells were centrifuged and re-suspended in liquid synthetic defined (SD) medium (1.7 g L−1 yeast nitrogen base without amino acids and ammonium sulphate, 20 g L−1 glucose, 5 g L−1 ammonium sulphate) at 30 °C and 160 rpm for 24 h. These were the seed cells used in the experiments under different growth conditions.
Media composition and cultivation procedures
Microfermentation experiments used specific SD media containing sucrose at 110 mmol L−1 (20 g L−1) as the carbon source and ammonium sulphate or sodium nitrate at 75 mmol of nitrogen L−1 as the nitrogen (N) source. Yeast seed cells were used to inoculate 1.5 mL of specific SD in FlowerPlates™ (m2p Labs, Baesweiler, Germany) for initial absorbance of 0.1 units at 600 nm (A600nm). For the aerobic assays, the yeast cells were cultivated in a BioLector NA device (m2p Labs, Baesweiler, Germany) at 30 °C, with 85% humidity and at 800 rpm (equivalent to 180 rpm in rotatory shaker), with a constant flux of sterile air. For anaerobic growth, specific SD media were supplemented with Tween 80 to 420 mg L−1 and ergosterol to 10 mg L−1 to meet the lipid requirement in anaerobiosis (Jouhten et al. 2008; Blomqvist et al. 2012; Peña-Moreno et al. 2019). Cultivations were performed under the same conditions as aerobiosis in the BioLector NA device, with the air replaced by ultrapure gaseous nitrogen (O2 < 1 ppm). The complete absence of oxygen was verified by the failure of the S. cerevisiae JP1 strain to grow in the SD-ammonium medium containing glycerol as the carbon source (data not shown), which is strictly metabolised by respiration. Automatic readings of light scattering were recorded every 30 min for 24 h and converted to optical density values (A600) by means of a calibration curve. Data were recovered in .xls format and processed in Excel® 2010 (Microsoft, Redmond, Washington, USA) to generate growth curves. Specific growth rates were calculated from the linear portion of the exponential phase as previously reported (Peña-Moreno et al. 2019). All experiments were performed in biological triplicate. For protein extraction, experiments were repeated and samples were collected from ten wells when the cultures reached around 0.5 A600 nm, then pooled to increase the number of cells, centrifuged and the cell sediment was frozen in liquid nitrogen.
Growth conditions and physiological the comparisons
Four growth conditions were tested in the present work. In condition #1, yeast cells were cultivated in an SD medium containing nitrate as the N source in anaerobiosis (NO3−/N2); in condition #2, yeast cells were cultivated in an SD medium containing nitrate as the N source in aerobiosis (NO3−/O2); in condition #3, yeast cells were cultivated in an SD medium containing ammonium as the N source in anaerobiosis (NH4+/N2); and in condition #4, the yeast cells were cultivated in an SD medium containing ammonium as the N source in aerobiosis (NH4+/O2). This last condition was only used as a control quality parameter to evaluate the growth profile (Cajueiro et al. 2017). From these conditions, two physiological comparisons were performed. In comparison [A], the protein profile of condition #1 was compared to condition #2 to describe how cells respond to the availability of oxygen when nitrate is used as the N source. In comparison [B], the protein profile of condition #1 was compared to condition #3 to describe how the cells respond to different N sources in the absence of oxygen.
Protein extraction
Cells collected in the mid log phase under each of the four growth conditions were thawed on ice and proteins were extracted as previously described (Neto et al. 2014). Cell sediments were washed with 2 mL of sterile deionised water and re-suspended in 200 μL of sterile deionised water. Then, 200 μL of 600 mM NaOH was added and the tubes were incubated for 5 min at room temperature. Cells were recovered by centrifugation (5 min, 10,000 rpm at 4 °C), re-suspended with 200 μL SDS sample buffer (60 mM Tris-HCl pH 6.8, 5% glycerol, 4% β-mercaptoethanol, 2% SDS, 0.0025% bromophenol blue) and lysed by boiling for 3 min. The lysates were centrifuged and the cell-free extracts transferred to new tubes. The sediments were re-suspended with 200 μL SDS sample buffer and glass beads (425–600 μm Ø, Sigma-Aldrich, St. Louis, MO, USA) were added to complete the buffer volume. The tubes were submitted to five cycles of 30 s in vortex and 30 s in an ice bath to break down the remaining intact cells. After further centrifugation, the second cell-free extract was pooled with the first cell-free extract and kept at 4 °C for protein extraction (Neto et al. 2014).
Afterwards, cell-free extracts were mixed with 1 mL extracting solution (100 mM ammonium acetate in methanol), incubated at − 20 °C for 30 min and centrifuged (20 min, 17,000 rpm, 4 °C). One millilitre of cold 80% acetone was slowly added to protein precipitates, and the tubes carefully inverted and centrifuged again. This procedure was repeated twice and then repeated once again with 1 mL cold 70% ethanol. After centrifugation, the sediments were dried at room temperature for 15 min, solubilised with 200 μL solubilising buffer (8 M urea and 2 M thiourea) and stored at − 80 °C. Total proteins were quantified by the Bradford method (Bradford 1976) using Coomassie brilliant blue G-250. Bovine serum albumin (BSA) was used for the establishment of the standard curve.
To verify the integrity of the extracted proteins, electrophoresis was performed in a 12% SDS-PAGE in Tris-glycine buffer at 150 V and 17 mA. A 14–97-kDa molecular weight marker (GE Healthcare, Chicago, USA) was used to estimate protein size. The gels were stained with 0.08% w/v Coomassie brilliant blue G-250 and later digitised on an ImageScanner III transparency scanner (GE Healthcare, Chicago, USA).
Two-dimensional electrophoresis
Isoelectric focusing (IEF) was carried out with 11 cm/pH 3–10 Immobiline Drystrip Gels (IPG drystrips) (GE Healthcare, Chicago, USA) for each of the three biological replicates from each treatment. Approximately 200 μg of protein were dissolved in rehydration solution composed of 7 M urea, 2 M thiourea, 2% w/v CHAPS detergent (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 19.4 mM dithiotreitol (DTT), IPG buffer pH 4–7 non-linear 1.0 % (GE Healthcare, Chicago, USA) and 0.005% bromophenol blue (w/v). The rehydration step was made in the IEF dispositive (IPG-Box, GE Healthcare, Chicago, USA) at room temperature for 18 h. Proper IEF was performed on Multiphor II Electrophoresis Unit (GE Healthcare, Chicago, USA) and conducted at 10 °C in three stages (i) 300 V–30 V h−1, (ii) 3500 V–2900 V h−1 and (iii) 3500 V–6170 V h−1, under constant current (2 mA) and power (5 W) conditions. After IEF, strips were stored at − 80 °C until the second dimension running (Neto et al. 2014). For this, the strips were stabilised in a solution containing 50 Mm Tris-HCI (pH 8.8), 6 M urea, 30% (v/v) glycerol, 2% (m/v) SDS, 1% DTT and 2.5% IAA (iodoacetic acid); the last two were added separately in two steps of 20 min under orbital rotation. In the second dimension, the proteins were resolved via 12.5% SDS-PAGE using a DGV-20 system (Biosystems, Curitiba, Paraná, Brazil) at 30 mA/gel for 15 min, 40 mA/gel for 30 min, and then 50 mA/gel for 3 h. After electrophoresis, the 2D gels were stained in 0.08% w/v Coomassie brilliant blue G-250. 2D gel images were obtained by digitalization in ImageScanner III (GE Healthcare, Chicago, USA) at 300 dpi, calibrated for optical density, with program LabScan 6.0 m (GE Healthcare, Chicago, USA).
Two-dimensional gel image analysis
Differentially accumulated proteins/peptides (DAPs) were selected through comparative analysis of 2D-PAGE (two-dimensional polyacrylamide gel electrophoresis) digital images by using the Image Master 2D Platinum v7.05 software (IMP, GE Healthcare, Chicago, USA), according to previous protocols (Pacheco et al. 2013; Neto et al. 2014). Three biological replicates were considered for each condition. Replicates presenting a correlation coefficient (R) equal to or higher than 0.7 were considered for analysis. Based on normalisation and statistical analyses performed in IMP, it was possible to verify the differential accumulation of spots, as well as exclusive spots in comparison [A] and comparison [B]. Differential spots that presented a percent volume (%vol) ratio ≥ 1.5 and ANOVA test value ≤ 0.05 were considered DAPs and selected for identification.
DAP identification by mass spectrometry and putative annotation
Selected spots were manually excised from gels and digested with trypsin (Neto et al. 2014). The resulting peptides were eluted in solution containing 5% trifluoroacetic acid and 50% acetonitrile, concentrated in Speed-vac (Eppendorf, Hamburg, Germany) for 30 min and stored at – 20 °C until mass spectrometry (MS) analysis. The mass spectra (MS) of the peptides originating from samples digestion were obtained in the Autoflex III MALDI-ToF/ToF analyser (Bruker Daltonics, Billerica, USA), using an α-cyano-4-hydroxycinnamic acid matrix. The equipment was calibrated using a peptide 158 mixture [M + H]+ ions for standard calibration. The resulting m/z ratio comprises the specific molecular mass of the sample peptides and is then named the peptide mass fingerprint (PMF method). Further, the acquisition of the MS/MS spectrum was achieved by collision energy of 1 kV, first in negative and then in positive ion mode using argon collision gas. The same precursor ions generated by the negative ion MS were analysed in negative and positive MS/MS. The peak processing of LC1 MALDI MS was controlled by the so-called peak selection method throughout the work. Peak selection was conducted automatically to analyse MS spectra and acquire MS/ MS data for selected peaks. Peak lists files were generated using the Flex Analysis 3.4 software (Bruker Daltonics, Billerica, USA).
Protein identification
The identification of the spectra was performed through the MASCOT online software (http://www.matrixscience.com) (Lisacek 2006), initially with the public access version, based on the peptide mass fingerprinting method (PMF) for MS spectra and MS/MS ion search method for MS/MS spectra, against the sub-banks D. bruxellensis and S. cerevisiae from NCBI protein and SwissProt databases. Subsequently, additional identification was conducted through a private version of the MASCOT program, kindly made available for access in collaboration with the Centre for Advanced Proteomics of the University of Washington (Seattle, Washington, USA (http://proteomicsresource.washington.edu/), against the sub-database D. bruxellensis. The experimental error used for mass values was as follows: (i) to MS spectra analysis: up to 1.2 Da of tolerance; (ii) to MS/MS spectra: 100 to 200 ppm and 0.2 to 1.2 Da (for parental ion) and 100 to 200 ppm and 0.2 to 0.6 Da (for ion fragment). Another search parameter was set in both methods as follows: carbamidomethylation of cysteines as the fixed modification and methionine oxidation as the variable modifications, allowing up to one missed cleavage. Protein identification was performed using the Mascot search probability-based molecular weight search (MOWSE) score. Only the identifications with calculated probability score equal or greater than the cut-off value were considered significant. The score equals − 10.log (P), where P is the probability that the similarity found is random.
Additional searches were performed with the SearchGUI v.3.2.20 software (Vaudel et al. 2011) using xml files obtained after analysis via MALDI-ToF/ToF that were individually converted to mzXML format through the FlexAnalysis software tool (Bruker Daltonics, Billerica, Massachusetts, USA), and submitted to presumptive identification (Carmona et al. 2016), with results visualized using the program 1PeptideShaker v.1.16.15 (de Groot et al. 2007). The following parameters were used to search the SearchGUI: tolerance m/z of the parental ion: 0.25 Da; ion fragments m/z tolerance: 0.25 Da; charge of the parental ion: 1–1; isotope: 0–1; maximum missed cleavages: 1; fixed modifications: carbamidomethylation (C); variable modifications: oxidation (M). The other parameters remained in the default software mode. Those proteins identified with the SwissProt database were compared by BLASTp against the NCBI database for B. bruxellensis AW.
Ontology and protein–protein interaction (PPI) analysis
Initially, the Uniprot ID of previously identified sequences from B. bruxellensis database were aligned through BLASTP (https://blast.ncbi.nlm.nih.gov) to find the protein accession statistical significance of matches against the S. cerevisiae proteome. These accesses served to identify the names of correspondents for each protein identified using UniProt (http://www.uniprot.org/) and reconfirmed in the S. cerevisiae database (http://yeastgenome.com/). For gene ontology, the Yeastmine software (https://yeastmine.yeastgenome.org) was used considering the biological process at each of the four conditions. The Holm–Bonferroni correction test was used and a maximum p value of 0.5. The most prominent gene clusters were included in our study. The proteome of S. cerevisiae in the STRING database v. 11.0 (https://string-db.org/) was used as reference for the identification of D. bruxellensis proteins (cut-off of confidence: 0.4) using the following prediction methods: conserved neighbourhood, co-occurrence, fusion, co-expression, experiments, databases and text mining. Hence, the predicted PPI analyses containing the ratio data of the treatments were transported via StringApp to Cytoscape (version: 3.8.0) (Szklarczyk et al. 2017) for interactome drawings. Each treatment was identified by three different colours and each protein mapped was differentiated by node size gradient.
Gene expression
RT-qPCR experiments were performed to transcriptionally validate the results of the proteomic. Aliquots of frozen cells collected from each of the four growth conditions were used for primer design and calibration, RNA extraction, cDNA synthesis and reference gene normalisation followed the procedures described specifically for D. bruxellensis (de Barros Pita et al. 2012). Target genes were those reported in our recent work (Peña-Moreno et al. 2019). Amplification reactions were carried out by using the SYBR Green PCR Master Mix kit in ABI Prism 7300 detection system (Applied Biosystems, Foster City, CA, USA) (de Barros Pita et al. 2012; Peña-Moreno et al. 2019). For relative expression analyses, condition #2 (NO3/O2) was used as a reference for the comparison [A] and condition #4 (NH4/N2) was the reference for the comparison [B].
Results
Absence of oxygen speeds up D. bruxellensis growth in nitrate
It has been widely reported that D. bruxellensis grows slowly when nitrate is available in the medium as the N source, which led us to place this nutrient as non-preferential for the yeast metabolism (Jouhten et al. 2008; de Barros Pita et al. 2011; de Barros Pita et al. 2013; Parente et al. 2017). However, nitrate has been proposed as an important adaptive factor for this yeast in industrial fermentation processes, co-assimilated with ammonium with sugar cane juice as the substrate (de Barros Pita et al. 2011). Recently, we showed that yeast growth rate in nitrate is practically the same as that in ammonium, when oxygen is absent (Peña-Moreno et al. 2019). So, we concluded that the assimilatory pathway for nitrate is very efficient and that this N source could be re-classified as preferential under the anaerobic growth condition. This raised the question of what are the biological constraints that slow yeast growth in nitrate when oxygen is present? And what are the metabolic traits that promote accelerated cell growth in anaerobiosis when oxygen is absent?
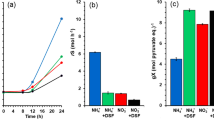
To address these questions, we performed growth experiments under the four different conditions. Based the growth curves (Fig. 1), specific growth rates were calculated as 0.21 h−1, 0.20 h−1 and 0.22 h−1 for conditions #1 (NO3−/N2), #3 (NH4+/N2) and #4 (NH4+/O2), respectively. This confirmed that nitrate was as efficiently assimilated by the yeast cells as ammonium in the absence of oxygen. Given these conditions, the cells grew at the same rate as that calculated for those in the ammonium-based medium in the presence of oxygen, used as reference condition. Noteworthy, these high growth rate values were achieved in the medium containing sucrose as the sole carbon (C) source. It has already been reported that growth rate of D. bruxellensis in sucrose is higher than in glucose (Leite et al. 2013; da Silva et al. 2019). In the present work, sucrose was chosen as the C source because it is the major sugar constituent of sugar cane juice, used for industrial fermentation for bioethanol. Sucrose consumption rates (mmol L−1 h−1) were calculated as 4.36 (± 0.65) for condition #1, 3.58 (± 0.43) for condition #3 and 4.17 (± 0.45) for condition #4. These results showed that the source of N, either ammonium or nitrate, did not affect the way the cells consumed sucrose from the medium when in absence of oxygen. Under all these three conditions, the yeast cells fermented sucrose to ethanol, even though at different rates. In contrast, a growth rate of 0.13 h−1 was calculated for condition #2 (NO3−/O2), confirming that the nitrate had restricted cell growth in the presence of oxygen. Under the condition of aerobic nitrate cultivation #2, the sugar assimilation rate was calculated as 2.49 mmol L−1 h−1 (± 0.31), 38% lower than that calculated for anaerobic nitrate cultivation condition #1. This value was close to that observed for C-limited aerobic chemostat cultivation of D. bruxellensis in sucrose and ammonium running at dilution rate of 0.1 h−1 (Leite et al 2013). That chemostat cultivation has a common feature with the present condition #2 in that both are strictly respiratory conditions. Based on the physiological features described above, the protein profile of this yeast was investigated in order to identify potential functional biomarkers that could explain this behaviour.

Growth profile of Dekkera bruxellensis GDB 248 cultivated in the presence (open symbols) or absence (solid symbols) of oxygen in synthetic defined medium containing sucrose as carbon source and ammonium (diamond) or nitrate (square) as a nitrogen source. The results represent the average of two independent experiments
Overall proteomic analysis
Proteomic analysis was undertaken for two different comparisons to find the panel of proteins that might help in the understanding of nitrate metabolism by the yeast D. bruxellensis. To normalise the analyses, proteins were extracted when each yeast culture had reached the same point in the growth curve (0.5 A600nm), assuming they were at the same physiological stage at the exponential growth phase (Fig. 2a). Moreover, as stated above, the sucrose assimilation rate was very similar among the conditions. A total of 12 protein extracts were used for 2D electrophoresis comparative analysis to identify differential accumulated proteins (DAPs) (Fig. 2a). A total of 344 spots were identified in the comparison [A] (condition #1 vs. condition #2) (Fig. 2b) and 357 spots in the comparison [B] (condition #1 vs. condition #3) (Fig. 2c). Subsequently, DAP spots were assigned as (i) exclusive if they were detected only under a specific growth condition or (ii) prevalent when its relative amount in one condition was ≥ 1.5 higher than in the other growth condition, with a 99.99% confidence level (p value ≤ 0.0001). Consequently, 95 and 91 DAPs were validated in comparisons [A] and [B], respectively (a complete list of proteins and their characteristics is available in the supplementary Tables S1 to S4). DAPs were submitted to GO analysis for the identification of the major biological processes as well as the cellular mechanisms that might be more active in each of the growth conditions (Fig. 3).

Differential proteome analysis of D. bruxellensis GDB248 cells cultivated in ammonium or with nitrate as a nitrogen source and in the presence or absence of oxygen. a Protein profile of three independent cell extracts (R1 to R3) separated by 12% SDS-PAGE. The scheme of proteome comparison is shown. (M) Molecular weight marker is shown. b Isoelectrical focusing for the identification of differentially accumulated proteins (DAPs) in the comparison [A] between condition #1 nitrate/anaerobiosis (upper gel) and condition #2 nitrate/aerobiosis (lower gel). c Isoelectrical focusing for the identification of DAPs in the comparison [B] between condition #1 nitrate/anaerobiosis (upper gel) and condition #3 ammonium/anaerobiosis (lower gel) 2D-PAGE. The indicated spots were excised from the gels and peptides identified by PMF and MS/MS MALDI-ToF analyses. These pictures represented one of the three experiments performed for proteomic analysis. Peptide identity was statistically confirmed by the parameters described in the “Material and methods” Section. Each spot in triplicate was considered as positive for differentially accumulated protein when the variation coefficient of its quantification did not exceed 10% of the mean value

Gene ontology analysis of biological processes of differentially accumulated proteins in the proteome of Dekkera bruxellensis GDB248 cells in the comparison [A] (a) and the comparison [B] (b). Red bars represent condition #1 nitrate/anaerobiosis, green bars represent condition #2 nitrate/aerobiosis and blue bars represent condition #3 ammonium/anaerobioses
Increased central metabolism, protein synthesis and cell progression stimulate anaerobic growth in nitrate
In comparison [A], the peptides of D. bruxellensis cells, that had shown higher accumulation under condition #1 than under condition #2, represented the metabolic difference that allowed the yeast culture to growth 60% faster in anaerobiosis than in aerobiosis when nitrate was the N source in the medium (Fig. 1). These DAPs corresponded to central nitrogen metabolism, oxidative metabolism and biosynthesis of ATP, amino acid and purine nucleotides (Fig. 3a; Supplementary Table S1). Among these DAPs, the overproduction of three proteins of the central sugar metabolism (glucokinase/Glk, triophosphate isomerase/Tpi and pyruvate dehydrogenase component E1 beta subunit/Pdb1) and two proteins in central nitrogen metabolism (glutamate dehydrogenase/Gdh1 and glutamine synthetase/Gln1) can be highlighted (Supplementary Table S1). These pathways were connected by the overproduction of NADP+-dependent mitochondrial isocitrate dehydrogenase (Idh1p) responsible for the production of 2-oxoglutarate from the TCA cycle (Supplementary Table S1). This might have led to the higher flux through the central metabolism that resulted in increased levels of glucose and nitrate consumption (Peña-Moreno et al. 2019).
Greater production of DAPs involved in the amino acid biosynthesis, such as delta-1-pyrroline-5-carboxylase reductase (Pro3) and asparaginyl-tRNA synthetase (Ded81) involved in protein translation was found in condition #1. One higher weight isoform of Ded81p was found in condition #1 that could represent the highest phosphorylation level. Three lower weight isoforms were found in condition #2 which may represent less phosphorylated forms of the protein in the five phosphorylation sites identified. In addition, proteins involved in the regulatory processes of gene expression were identified (Fig. 3a). The polyadenylate-binding protein 2 (Pab1) that is part of the translation initiation was exclusively accumulated in anaerobic nitrate. The pre-mRNA SLT11 factor-splicing (Ecm2) involved in post-transcription modification, however, showed higher accumulation in aerobiosis (Supplementary Table S2). Transcription initiation factor IIb (Cti6) was present as high and low weighted isoforms in anaerobiosis and aerobiosis, respectively. Altogether, these results pointed at an incremented synthesis of protein in the cells cultivated under condition #1.
Regarding purine metabolism, the key enzyme adenylosuccinate lyase (Ade13) was exclusively found in condition #1 (Supplemental Table S1), indicating that de novo purine biosynthesis had been stimulated. The alpha (Atp1) and gamma (Atp3) subunits of ATP synthase F1 complex were accumulated in both condition #1 (Supplementary Table S1) and condition #2 (Supplementary Table S2), while the epsilon subunit (Atp15) exclusively accumulated in condition #1 (Supplementary Table S1), confirming the high demand for ATP imposed by nitrate metabolism independent of oxygen to promote cell growth (de Barros Pita et al. 2013; Galafassi et al. 2013; Peña-Moreno et al. 2019).
Three proteins exclusively found in cells cultivated under condition #1 were related to cell morphogenesis: Bem2 (a Rho GTPase activating protein involved in cytoskeleton organisation), Syt1 (a guanine exchange factor for Arf proteins involved in vesicular transport and exocytosis) and Rvs161 (an amphiphysin-like lipid raft protein involved in cell polarity and endocytosis) (Supplementary Table S1). This profile indicates that cell progression was stimulated when nitrate was anaerobically assimilated.
Oxidative stress limits aerobic growth in nitrate
Proteins of D. bruxellensis cells that accumulated in condition #2 more than in condition #1 were used to explain the deficit of 40% in the growth rate when nitrate is assimilated aerobically (Fig. 1). These corresponded almost exclusively to proteins involved in oxidative stress responses (Fig. 3a), such as manganese-dependent superoxide dismutase (Sod2), thioredoxin peroxidase (Tsa2) and flavohaemoprotein (Yhp1) with nitric oxide oxidoreductase activity (Supplementary Table S2). In plants, nitric oxide oxidoreductase is solely produced when the cells are under nitric oxide stress (Corpas and Palma 2020). These results indicate that cells cultivated in condition #2 were submitted to oxidative stress, which might have limited the cell growth as reported above (Fig. 1). Five other proteins were exclusively identified in the proteome of these cells: Hsp10 (mitochondrial chaperonin), Mrp1 and Mrpl10 (mitochondrial ribosomal proteins), Apn1 (AP endonuclease of the base excision repair mechanism) and Rad17 (DNA damage checkpoint protein).
Energy demand is enhanced in nitrate-based medium
The comparison [B] represented the peptides grouped differentially when the D. bruxellensis cells were cultivated under condition #1 (nitrate) or condition #3 (ammonium) in the absence of oxygen, despite the fact that yeast cells grew at the same growth rate in both N sources (Fig. 1). Some proteins of the glycolytic pathway were overproduced, indicating an intense sugar metabolism in nitrate. This higher flux in the glycolysis could be responsible for high ATP production at the substrate level as well as enablement of the yeast cells to produce more ethanol (Peña-Moreno et al. 2019). Hence, more ATP can be produced at substrate level. It is worth noting that the central difference between both conditions was that the first phosphorylation reaction of glycolysis was represented exclusively by hexokinase (Hxk) (Supplementary Table S4) when ammonium was the N source, or it was represented exclusively by glucokinase (Glk) (Supplementary Table S3) when nitrate was the N source, which might represent differences in the regulation of cell metabolism.
In this regard, we observed an unexpected exclusive production of the glycerol kinase Gut1 in condition #1 (Supplementary Table S3) that contained sugar as C source. This enzyme is involved in the re-assimilation of the glycerol produced in anaerobiosis and normally subjected to glucose repression in S. cerevisiae. Exclusive enzymes involved in purine catabolism were observed in nitrate (Fig. 3b). GTP cyclohydrolase (Rib1) involved in the first reaction of the riboflavin and FAD biosynthetic pathway from GTP was exclusive to condition #1 (Supplementary Table S1), while the mitochondrial tetrahydrofolate synthetase (Mis1) was present in conditions #3 and #4 in different isoforms (Tables S3 and S4). Together with Gut1, these proteins indicated the differences in metabolic regulation where the oxidative metabolism was very stimulated to fulfil the high energy demand promoted by nitrate assimilation (Fig. 3b).
Ribosomal proteins S2 (Rps2) and S3 (Rps3) of the 40s ribosome subunit were found only in condition #1 (Supplementary Table S3; Fig. 3b), indicating intense protein synthesis in nitrate that resulted in a high growth rate in anaerobiosis regardless of the N source used in the medium (Fig. 1).
Relieved TORC1 regulation in nitrate-based growth
Differences in the metabolic regulation between ammonium and nitrate-growing cells were also observed in comparison [B] regarding proteins that respond to the complex 1 TOR signalling mechanism (TORC1). There was a greater production of S6-like serine/threonine kinase Sch9, involved in regulation of ribosome biogenesis and translation initiation, under condition #3 than condition #1 (Supplementary Tables S3 and S4), while the type 2A-related serine/threonine phosphatase activator Sit4 was exclusive to condition #3 (Supplementary Table S4). Sit4 is negatively regulated by TORC1 by the presence of ammonium in the canonical regulation in S. cerevisiae (Bertram et al. 2000). Its production in D. bruxellensis under condition #4 indicated differential regulation between these yeasts regarding nitrogen metabolism. Moreover, the absence of Sit4 in nitrate indicated an alternative TORC1-independent regulatory mechanism triggered by this N source.
Gene expression analysis corroborated the differential regulatory aspects of the yeast growth in nitrate and ammonium
The expression of genes encoding the DAPs identified in the proteomic profiles, together with genes previously used to compare growth in nitrate and ammonium (de Barros Pita et al. 2013; Peña-Moreno et al. 2019), was evaluated to help to understand the yeast metabolic differences. In comparison [A], upregulation of PFK1 (phosphofructokinase in the glycolytic pathway), GDH1 (NADPH-dependent glutamate dehydrogenase) and ATP1 (alpha subunit of the F1 sector of mitochondrial F1F0 ATP synthase) was detected (Fig. 4a). This corroborated the proteomic data that indicated a higher flux through the central metabolism and energy production to support the higher growth rate in nitrate in anaerobiosis over nitrate in aerobiosis (Fig. 1). In addition, the upregulation of FKS1 (the catalytic unit of beta glucan synthase in a cell wall) (Fig. 4a) agreed with the production of proteins involved in cell progression and morphogenesis. On the other hand, the expression of CTT1 (encoding cytosolic catalase) was downregulated in condition #1 (Fig. 4a), meaning that it was more expressed when the cells were cultivated in nitrate in the presence of oxygen. This gene responds to oxidative stress and its protein plays a paramount role against oxidative damage by hydrogen peroxide. Together with the overproduction of Tsa1, Sod1 and Yhb1 proteins (Supplementary Table S2), this result confirmed that aerobic growth in nitrate induces oxidative stress to D. bruxellensis.

Relative expression of genes of glycolysis, cell progression and synthesis of ATP and glutamate in D. bruxellensis GDB248 in the comparison [A] between condition #1 nitrate/anaerobiosis over condition #2 nitrate/aerobiosis (a) and in the comparison [B] between condition #1 nitrate/anaerobiosis over condition #3 ammonium/anaerobiosis (b)
Regarding comparison [B], data on gene expression showed that genes of the glucose metabolism, oxidative/energy metabolism and glutamate biosynthesis were downregulated in nitrate relative to ammonium (Fig. 4b), indicating that ammonium was still more inductive of the yeast central metabolism than nitrate in anaerobiosis. A slight upregulation of ADH1 was detected in nitrate (Fig. 4b), even though the Adh1 protein was more accumulated in ammonium (Supplemental Table S4). This indicated that the constant high metabolic flux towards fermentation in anaerobiosis might be maintained by two regulatory mechanisms that control either the expression of ADH1 gene in nitrate or the production of Adh1p in ammonium. As consequence, ethanol was produced at the same yield in both N sources (Peña-Moreno et al. 2019).
Discussion
Oxidative stress is the limiting factor for the aerobic growth of D. bruxellensis in nitrate
The capacity of the D. bruxellensis strain GDB 248 to assimilate nitrate as the sole N source has been described previously, although resulting in low growth rate and biomass production (de Barros Pita et al. 2011; 2013; Cajueiro et al. 2017; Parente et al. 2017). This led to the assumption that nitrate is a non-preferential N source. However, this scenario contrasts with the high growth rate of 0.21 h−1 calculated in nitrate when D. bruxellensis cells were cultivated in anaerobiosis that was similar to growth on ammonium (Peña-Moreno et al. 2019; Fig. 1). Therefore, nitrate can be considered the preferred N source like ammonium when the cells are growing in anaerobiosis. This was the first question addressed in this work: what makes this yeast grow slowly in nitrate in aerobiosis?
Nitrate assimilation requires 4 mol of NAD(P)H for each mole of nitrate in two consecutive redox reactions of nitrate reductase (Ynr1) and nitrite reductase (Yni1), plus 1 mol of NADPH to convert ammonium to glutamate by glutamate dehydrogenase (Gdh1) (Siverio 2002). The first aspect to be considered is the fact that the nitrate assimilatory genes YNR1, YNI1 and YNT1 are highly expressed in D. bruxellensis both in aerobiosis (de Barros Pita et al. 2011; de Barros Pita et al. 2013; Cajueiro et al. 2017) and in anaerobiosis (Peña-Moreno et al. 2019). The second aspect relies on the fact that, in this yeast species, genes of the pentose phosphate pathway and the PDH-bypass, two metabolic pathways that provide NADPH, are induced by nitrate both in aerobiosis (de Barros Pita et al. 2013) and in anaerobiosis (Peña-Moreno et al. 2019). These observations led to the conclusion that the lower yeast growth rate in aerobiosis resulted neither from the inability of the nitrate assimilatory pathway nor the shortage of reduced cofactors. Therefore, it could only be explained by the action of some indirect metabolic burden that impeded the cell cycle progression in nitrate when oxygen is present.
The results showed high accumulation levels of Tsa1p and Sod2p and other oxidative stress-related proteins in aerobiosis, but not in anaerobiosis (Fig. 3). This is indicative that the D. bruxellensis cells might be under oxidative stress when nitrate is assimilated in the presence of oxygen. This assumption was confirmed by the fact that the CTT1 gene encoding cytosolic catalase is more highly expressed when nitrate is assimilated aerobically than anaerobically (Fig. 4). Besides causing direct damages to cell molecules, oxidative stress increases the demand for NADPH to maintain a high level of anti-oxidant defence molecules (mainly containing sulfhydryl groups) to detoxify the reactive oxygen species (ROS) (Steensels et al. 2015; Vigentini et al. 2013). This should promote a competition between the nitrate assimilation pathway and the oxidative stress response for NADPH pool, ultimately compromising cell growth. As D. bruxellensis seems to be more sensitive to hydrogen peroxide than S. cerevisiae, we hypothesised that the mechanisms for tolerance to oxidative stress are less efficient in the first yeast (Leite et al. 2013).
Yhb1p was another relevant DAP exclusive to aerobiosis (Fig. 5). In S. cerevisiae, the expression of the YHB1 gene is enhanced when the respiration chain is compromised, when the cells are exposed to oxidative stress or when the oxidative stress response is deficient (Zhao et al. 1996). Yhb1p is a flavohaemoglobin-like protein that provides protection against nitric oxide (NO) through its C-terminal domain using NADPH as an electron donor for detoxification reactions (Mukai et al. 2001; Gardner 2012). In plants, NO may be produced as a partially reduced intermediate of nitrate conversion to ammonium by the action of nitrate/nitrite reductases (Corpas and Palma 2020). Our result indicated that NO might be produced in the course of aerobic assimilation of nitrate by D. bruxellensis, damaging the cells and reducing the growth rate. The aerobic interactome showed a subnet composed by Yhb1, Tsa1 and Tpi1 linked to a cluster of mitochondrial stress proteins (Hsp10 mitochondrial chaperonin and Mrp1/Mrpl10 mitochondrial ribosomal proteins) and to a cluster of DNA repair proteins (Apn1 AP endonuclease and Rad17 DNA damage checkpoint protein). The expression of DNA repair proteins is indicative of oxidative damages produced during nitrate metabolization. This might lead to oxidation of nitrogen bases in the DNA, producing, for example, 8-oxoguanosine, that is subjected to base excision repair by AP endonucleases (Lu et al. 2001). Therefore, oxidative stress is produced when nitrate is aerobically assimilated and causes damages to different cell structures and reduced cell cycle progression and cell growth. This might be the major cause of the low aerobic yeast growth in nitrate.

Interactome of Dekkera bruxellensis GDB248 based on the profile of differentially accumulated proteins (DAPs). a DAPs identified in the proteome of cells growing in nitrate in anaerobiosis (red circles), in aerobiosis (blue circles) or in both conditions as different isoforms (yellow circles). b DAPs identified in the proteome of cells growing in anaerobiosis in nitrate (red circles), in ammonium (blue circles) or under both conditions as different isoforms (yellow circles). The thickness of nodes represents the relative quantity of DAPs, and the width of connecting lines represents the strength of connection
Anaerobic growth of D. bruxellensis in nitrate is boosted by an overflow in both carbon and nitrogen metabolisms
After presenting an explanation for the poor growth in nitrate in aerobiosis, the next question is, what are the metabolic factors that enabled an accelerated growth of D. bruxellensis in nitrate in the absence of oxygen. In this environment, yeast cells grew at the same rate both in ammonium and in nitrate (Fig. 1), indicating that nitrate is very efficiently assimilated as an N source. Proteins involved in the glycolytic pathway were accumulated in both N sources (Figs. 3 and 5). However, one key difference was observed in the first step of glycolysis regarding the type of kinase that converts glucose to glucose-6-phosphate (Fig. 6). In ammonium, this reaction is ruled by the hexokinase (Hxk) (Fig. 5b), which resembles a common feature of S. cerevisiae cells cultivated in media containing sugars as C source (Rodríguez et al. 2001). On the other hand, in nitrate this reaction seems to be exclusively ruled by the glucokinase (Glk) (Fig. 5b). In S. cerevisiae, the GLK1 gene is subjected to glucose catabolite repression (GCR), meaning that it is not expressed when glucose is present in the medium (Rodríguez et al. 2001). This means that S. cerevisiae and D. bruxellensis differ in the mechanism that regulates the functioning of the carbon central metabolism, with D. bruxellensis being alleviated in this mechanism as proposed (Leite et al. 2016).

A general scheme of the central metabolism of Dekkera bruxellensis connecting glycolysis, TCA cycle, nitrogen assimilation and mitochondrial tetrahydrofolate (THF) pathways, as putatively depicted from differential proteome analyses. The red boxes represent proteins found in nitrate anaerobiosis; green boxes show proteins found in nitrate aerobiosis; blue boxes represent proteins found in ammonia anaerobiosis; and grey boxes represent proteins whose genes were evaluated for relative expression in the present work
This increase in the glycolytic flux in anaerobiosis leads to a high production of ethanol at the level of S. cerevisiae (Peña-Moreno et al. 2019). This is two or four times higher than that observed in ammonium or nitrate in aerobiosis, respectively (Pereira et al. 2012, 2014; de Barros Pita et al. 2013; Leite et al. 2013). This profile is consequence of the stimulated expression of genes (Fig. 4; Peña-Moreno et al. 2019) and production of respective proteins (Fig. 5) of the fermentative metabolism (PDC and ADH). It explains why distilleries with a high count of D. bruxellensis cells in the fermentation tanks report normal levels of ethanol production (Passoth et al. 2007; De Souza et al. 2012). This shows the industrial relevance of these results, confirming that D. bruxellensis can be considered as a fermenting yeast for ethanol production and other biotechnological processes, as has been postulated (De Barros Pita et al. 2019).
In addition to increased fermentative activity, part of the pyruvate produced at the end of glycolysis is also metabolised in the TCA cycle as indicated by the overproduction of proteins of the Pdb complex, citrate synthase (Cit) and isocitrate dehydrogenase (Idh) in nitrate/anaerobiosis (Fig. 6). Together with the upregulation of TCA genes (Peña-Moreno et al. 2019), this indicates an intense metabolic activity inside the mitochondria. The combined activity of these three enzymes produces 2-oxoglutatare, which is essential in the central nitrogen metabolism (Fig. 6). This molecule is the meeting point between the central metabolism of carbon and nitrogen (Fig. 6).
Assimilation of nitrate into the central nitrogen metabolism of D. bruxellensis has been reported based on observed intensive expression of nitrate assimilating genes (de Barros Pita et al. 2011; Galafassi et al. 2013; Cajueiro et al. 2017; Peña-Moreno et al. 2019). The considerable reducing power required (Siverio 2002) might be provided by the coordinated action of the pentose phosphate (Peña-Moreno et al. 2019), PDH-bypass (Teles et al. 2018) and the tetrahydrofolate (THF) pathways (Fig. 6). The induction of THF pathway was indicated by the overproduction of GTP cyclohydrolase II Rib1 and C1-tetrahydrofolate synthase Mis1 that involves extra supply of mitochondrial ATP and NADPH (Fig. 6). This pathway provides the ethyl radicals that could be used in various anabolic methylations such as synthesis of amino acids, purines and deoxi-thymidine monophosphate (Denis and Daignan-Fornier 1998; Grawert et al. 2013; Bradshaw 2019). It shows the importance of the THF pathway towards sustaining high growth rates in anaerobiosis. The coordinated action of enzymes in the nitrate metabolism together with the enzymes that provide NAD(P)H and ATP resulted in the conversion of nitrate to ammonium (Fig. 6). The ammonium produced was further assimilated by the central metabolism of nitrogen to produce amino acids. For this purpose, 2-oxoglutarate from TCA is aminated to glutamate by Gdh1 (glutamate dehydrogenase) (Figs. 5 and 6). Glutamate is further aminated to glutamine by Gln1 (glutamine synthase) (Figs. 5 and 6). Glutamate and glutamine serve as the major amino donors for the production of other amino acids by transamination reactions. The glutamine ultimately functions as internal sensor of nitrogen availability to stimulate D. bruxellensis growth (Cajueiro et al. 2017; Parente et al. 2017). Therefore, the efficient nitrate assimilation to glutamine signals the availability of N in the medium and stimulates growth at high rate. Therefore, it seems not by chance that Gdh1, Gln1 and Idh1 occupy the central position of the protein interactome of D. bruxellensis cultivated in nitrate/anaerobiosis (Fig. 5a).
The mode of regulation is paramount for the correct functioning of metabolic routes. In S. cerevisiae, the expression of GDH1 and GLN1 genes is subjected to regulation by the Gln3 transcriptional activator that responds to the target of the rapamycin complex C1 (TORC1) signalling pathway and also subjected to the availability of 2-oxoglutarate from the TCA cycle (Bertram et al. 2000; Conrad et al. 2013). Contrary to its orthologous in S. cerevisiae, the GDH1 gene in D. bruxellensis does not seem to respond to nitrogen catabolite repression (NCR) under the aerobic condition either ammonium or nitrate (Cajueiro et al. 2017). However, Parente et al. (2017) showed upregulation GDH1 gene in the presence of all 20 amino acids in aerobiosis and anaerobiosis, or as glutamate in anaerobiosis. This indicates that the gene might be subjected to an alternative regulatory circuit, which is less dependent on the N source and more dependent on the oxygen in the medium. The pathways commanded by TORC1 sense the availability of nitrogen and control cell growth (Conrad et al. 2013). Indeed, we identified several ribosomal proteins (RP) involved in ribosome biogenesis (RiBi) and protein synthesis (Fig. 5) that are controlled by TORC1 (Conrad et al. 2013; Haugen et al. 2004). Thus, we were able to build a proposed model of regulation based on the regulatory circuit in S. cerevisiae (Fig. 7a). In brief, ammonium (as well as other sources) activates TORC1 which induces the expression of RP and RiBi genes via Sch9 kinase while blocking the expression of NCR-controlled genes via Tap42 activation. In non-preferential N sources, the strength of the signal that activates TORC1 is reduced, which releases the repression over the NCR-controlled genes (Bertram et al. 2000; Butow and Avadhani 2004; Conrad et al. 2013). The present work showed that the TORC1-activated Sch9 kinase was detected in the proteome of D. bruxellensis in both ammonium and in nitrate (Fig. 5). In this case, the activity of Sch9p might phosphorylate and activate transcription factors responsible for the expression of gene encoding for ribosomal proteins and translation factors (Fig. 7b, c). As a consequence, protein synthesis might occur at a level that allows the cells to grow faster in both N sources. However, the 2A-related phosphatase Sit4p was exclusively produced in ammonium (Fig. 5). This suggests alleviation of NCR even in ammonium, activation of Gln3 and expression of NCR-controlled genes (Fig. 7b). In fact, the promoters of nitrate genes contain binding sites for Gln3 and Gat1 (Cajueiro et al. 2017), indicating their control via the Sit4 pathway, enabling potential co-assimilation of nitrate with ammonium (de Barros Pita et al. 2011; Peña-Moreno et al. 2019), which is very relevant from an industrial point of view. The absence of Sit4 in the proteome of nitrate-growing cells indicates the action of alternative regulatory circuits, as has been proposed (Parente et al. 2017; Cajueiro et al. 2017). In this case, we suggest that Sit4 is replaced by the PP4 type-2A phosphatase complex (Ronne et al. 1991; Bertram et al. 2000) that will be in charge of releasing the expression of NCR genes when the cells are cultivated in nitrate (Fig. 7c). This opens the possibility for the nitrate-growing cells to scavenge different N sources in the environment. Another relevant aspect is that S. cerevisiae Sit4 triggers the repressor activity of Hxk2 and participates in GCR (Rodríguez et al. 2001). Interestingly, neither Hxk nor Sit4 D. bruxellensis orthologous proteins accumulated in nitrate (Fig. 5b). In this case, the species lacks an important GCR signal via Hxk and releases the expression of GCR-controlled genes so that that the cells can scavenge different C sources in the environment.

Regulatory network commanded by target of rapamycin complex 1 (TORC1) in response to the availability of nitrogen in the environment. The reference regulatory circuit in Saccharomyces cerevisiae growing in ammonium (a) was used to build up a proposed regulatory circuit for Dekkera bruxellensis growing in ammonium (b) or in nitrate (c). Regulatory proteins are represented in the black boxes. P in black circles represent phosphate residues and TFs mean transcription factor. Genes of retrograde (RTG) response, nitrogen catabolite repression (NCR), ribosomal proteins (RP) and ribosomal biogenesis (RiBi) are the target of the regulatory networks. Straight arrows represent activation, a T-shaped line represents inhibition, bent arrows represent dephosphorylation and L-shaped arrows represent gene transcription
TORC1 also regulates by the retrograde (RTG) mechanism responsible for the mitochondria-to-nucleus communication that controls metabolic activities, cytoskeleton organisation and cell cycle progression (Aronova et al. 2017). RTG is also involved in the anaerobic expression of TCA genes involved in the anaplerotic production of oxaloacetate used for the synthesis of citrate and production of 2-oxoglutarate for the synthesis of glutamate (Conrad et al. 2013; Butow and Avadhani 2004). The results in the present work showed the overproduction of proteins controlled by the RGT mechanism in both N sources, such as those related to cytoskeleton organisation (Rvs161 and Bem2), vacuolar transport (Rvs167, Vps35 and Vps3) and TCA (Idh1) (Fig. 5 and Supplementary Tables S1–S4). Hence, in D. bruxellensis, the alleviation of TORC1 in ammonium (Fig. 7b) and its inactivation in nitrate (Fig. 7c) should relieve the negative regulation of TORC1 over the RTG mechanism, allowing faster cell growth rate.
Therefore, as the response the second question addressed above, we concluded that the success of anaerobic cell growth in nitrate derives, on the one hand, from an increased flow through the glycolytic pathway, producing energy (ATP and reducing equivalents) and TCA intermediates; on the other hand, it is a result of deregulation of the central nitrogen metabolism that allows the synthesis of ammonium and amino acids for protein synthesis and ultimately promotes cell expansion and division. The scenario revealed by the proteomic analyses presented herein and in previous work (Neto et al. 2014) when allied to previous genetic and physiological results (de Barros Pita et al. 2011, 2013; Cajueiro et al. 2017; Parente et al. 2017; Peña-Moreno et al. 2019) showed that nitrate is a readily assimilable source of nitrogen by D. bruxellensis cells. Its metabolization triggers a reorientation in the yeast metabolism, allowing the cells to widen their capacity to assimilate different sources of carbon and nitrogen present in the environment and/or substrate. These results represent a milestone regarding knowledge about the metabolism of nitrate that may be explored in the future for the industrial use of D. bruxellensis as an attractive yeast.
Data availability statement
All data generated or analysed during this study are included in this published article (and its supplementary information files) and further information are available from the corresponding author on reasonable request.
References
Aronova S, Wedaman K, Anderson S, Yates Y, Powers T (2017) Probing the membrane environment of the TOR kinases reveals functional interactions between TORC1, actin, and membrane trafficking in Saccharomyces cerevisiae. Mol Biol Cell 18:2779–2794. https://doi.org/10.1091/mbc.e07-03-0274
Bertram PG, Choi JH, Carvalho J, Ai W, Zeng C, Chan TF, Zheng XF (2000) Tripartite regulation of Gln3p by TOR, Ure2p, and phosphatases. J Biol Chem 46:5727–5733. https://doi.org/10.1074/jbc.M004235200
Blomqvist J, Eberhard T, Schnürer J, Passoth V (2010) Fermentation characteristics of Dekkera bruxellensis strains. Appl Microbiol Biotechnol 87:1487–1497. https://doi.org/10.1007/s00253-010-2619-y
Blomqvist J, Nogue VS, Gorwa-Grauslund M, Passoth V (2012) Physiological requirements for growth and competitiveness of Dekkera bruxellensis under oxygen-limited or anaerobic conditions. Yeast 29:265–274. https://doi.org/10.1002/yea.2904
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Bradshaw PC (2019) Cytoplasmic and mitochondrial NADPH-coupled redox systems in the regulation of aging. Nutrients 3:504. https://doi.org/10.3390/nu11030504
Butow RA, Avadhani NG (2004) Mitochondrial signaling: the retrograde response. Mol Cell 14:1–15. https://doi.org/10.1016/S1097-2765(04)00179-0
Cajueiro DBB, Parente DC, Leite FCB, de Morais Jr MA, de Barros PW (2017) Glutamine: a major player in nitrogen catabolite repression in the yeast Dekkera bruxellensis. Antonie Van Leeuwenhoek 110:1157–1168. https://doi.org/10.1007/s10482-017-0888-5
Carmona L, Varela J, Godoy L, Ganga MA (2016) Comparative proteome analysis of Brettanomyces bruxellensis under hydroxycinnamic acid growth. Electron J Biotechnol 23:37–43. https://doi.org/10.1016/j.ejbt.2016.07.005
Conrad M, Schothorst J, Kankipati HN, Zeebroeck GV, Rubio-Texeira M, Thevelein JM (2013) Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38:254–299. https://doi.org/10.1111/1574-6976.12065
Corpas FJ, Palma JM (2020) Assessing nitric oxide (NO) in higher plants: an outline. Nitrogen 1:12–20. https://doi.org/10.3390/nitrogen1010003
da Silva JM, Silva GHTG, Parente DC, Leite FCB, Silva CS, Valente P, Ganga AM, Simões DA, de Morais MA Jr (2019) Biological diversity of carbon assimilation among isolates of the yeast Dekkera bruxellensis from wine and fuel-ethanol industrial processes. FEMS Yeast Res 19:foz022. https://doi.org/10.1093/femsyr/foz022
de Barros Pita W, Leite FCB, de Souza Liberal AT, Simões DA, de Morais MA Jr (2011) The ability to use nitrate confers advantage to Dekkera bruxellensis over S. cerevisiae and can explain its adaptation to industrial fermentation processes. Antonie van Leeuwenhoek 1:1–9. https://doi.org/10.1007/s10482-011-9568-z
de Barros Pita W, Leite FC, de Souza Liberal AT, Pereira LF, Carazzolle MF, Pereira GA, de Morais MA Jr (2012) A new set of reference genes for RT-qPCR assays in the yeast Dekkera bruxellensis. Can J Microbiol 12:1362–1367. https://doi.org/10.1139/cjm-2012-0457
de Barros Pita W, Castro-Silva D, Simões-Ardaillon D, Volkmar P, de Morais MA Jr (2013) Physiology and gene expression profiles of Dekkera bruxellensis in response to carbon and nitrogen availability. Antonie van Leeuwenhoek 5:855–868. https://doi.org/10.1007/s10482-013-9998-x
de Barros Pita W, Teles GH, Peña-Moreno IC, da Silva JM, Ribeiro KC, de Morais Junior MA (2019) The biotechnological potential of the yeast Dekkera bruxellensis. World J Microbiol Biotechnol 35: 103. https://doi.org/10.1007/s11274-019-2678-x
de Groot MLJ, Daran-Lapujade P, van Breukelen B, Knijnenburg TA, de Hulster EAF, Reinders MJT, Pronk JT, Heck AJR, Slijper M (2007) Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals posttranscriptional regulation of key cellular processes. Microbiol 153:3864–3878. https://doi.org/10.1099/mic.0.2007/009969-0
de Souza RB, dos Santos BM, de Fátima Rodrigues de Souza R, da Silva PF, Lucena BT, de Morais MA Jr (2012) The consequences of Lactobacillus vini and Dekkera bruxellensis as contaminants of the sugarcane-based ethanol fermentation. Ind Microbiol Biotechnol 11:1645–1650. https://doi.org/10.1007/s10295-012-1167-0
Denis V, Daignan-Fornier B (1998) Synthesis of glutamine, glycine and 10-formyl tetrahydrofolate is coregulated with purine biosynthesis in Saccharomyces cerevisiae. Mol Gen Genet 259:246–255. https://doi.org/10.1007/s004380050810
Galafassi S, Capusoni S, Moktaduzzaman M, Compagno C (2013) Utilization of nitrate abolishes the “Custers effect” in Dekkera bruxellensis and determines a different pattern of fermentation products. J Ind Microbiol Biotechnol 34:297–303. https://doi.org/10.1007/s10295-012-1229-3
Gardner PR (2012) Hemoglobin: a nitric-oxide dioxygenase. Scientifica 683729:1–34. https://doi.org/10.6064/2012/683729
Grawert T, Fischer M, Bacher A (2013) Structures and reaction mechanisms of GTP cyclohydrolases. IUBMB Life 4:310–322. https://doi.org/10.1002/iub.1153
Haugen AC, Kelley R, Collins JB, Tucker CJ, Deng C, Afshari CA, Brown JM, Ideker T, Van Houten B (2004) Integrating phenotypic and expression profiles to map arsenic-response networks. Genome Biol 5:R95. https://doi.org/10.1186/gb-2004-5-12-r95
Jouhten P, Rintala E, Huuskonen A, Tamminen A, Toivari M, Wiebe M, Ruohonen L, Penttila M, Maaheimo H (2008) Oxygen dependence of metabolic fluxes and energy generation of Saccharomyces cerevisiae. BMC Syst Biol 2:60. https://doi.org/10.1186/1752-0509-2-60
Leite FCB, Basso TO, de Pita BW, Gombert AK, Simoes DA, de Morais MA Jr (2013) Quantitative aerobic physiology of the yeast Dekkera bruxellensis, a major contaminant in bioethanol production plants. FEMS Yeast Res 1:34–43.
Leite FCB, Leite DVR, Pereira LF, de Barros PW, De Morais Jr MA (2016) High intracellular trehalase activity prevents the storage of trehalose in the yeast Dekkera bruxellensis. Lett Appl Microbiol 63:210–214. https://doi.org/10.1111/lam.12609
Lisacek F (2006) Web-based MS/MS data analysis. Proteomics 6(Suppl 2):22–32. https://doi.org/10.1002/pmic.200600524
Lu AL, Li X, Gu Y, Wright PM, Chang DY (2001) Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys 35:141–170. https://doi.org/10.1385/CBB:35:2:141
Mukai M, Mills SE, Poole RK, Yeh SR (2001) Flavohemoglobin, a globin with a peroxidase-like catalytic site. J Biol Chem 10:7272–7277. https://doi.org/10.1074/jbc.M009280200
Neto AG, Pestana-Calsa MC, de Morais Jr MA, Calsa T (2014) Proteome responses to nitrate in bioethanol production contaminant Dekkera bruxellensis. J Proteomics 104:104–111. https://doi.org/10.1016/j.jprot.2014.03.014
Pacheco CM, Pestana-Calsa MC, Gozzo FC, Mansur Custodio Nogueira RJ, Menossi M, Calsa T Jr (2013) Differentially delayed root proteome responses to salt stress in sugar cane varieties. J Proteome Res 12:5681–5695. https://doi.org/10.1021/pr400654a
Parente DC, Vidal EE, Leite FC, de Barros PW, de Morais MA Jr (2015) Production of sensory compounds by means of the yeast Dekkera bruxellensis in different nitrogen sources with the prospect of producing cachaca. Yeast 32:77–87. https://doi.org/10.1002/yea.3051
Parente DC, Cajueiro DBB, Moreno ICP, Leite FCB, de Barros PW, de Morais Jr MA (2017) On the catabolism of amino acids in the yeast Dekkera bruxellensis and the implications for industrial fermentation processes. Yeast 3:299–309. https://doi.org/10.1002/yea.3290
Passoth V, Blomqvist J, Schnürer J (2007) Dekkera bruxellensis and Lactobacillus vini form a stable ethanol-producing consortium in a commercial alcohol production process. Appl Environ Microbiol 73:4354–4356. https://doi.org/10.1128/AEM.00437-07
Peña-Moreno IC, Castro Parente D, da Silva JM, Andrade Mendonça A, Rojas LAV, de Morais Jr MA, de Barros PW (2019) Nitrate boosts anaerobic ethanol production in an acetate-dependent manner in the yeast Dekkera bruxellensis. J Ind Microbiol Biotechnol 46:209–220. https://doi.org/10.1007/s10295-018-2118-1
Pereira LF, Bassi AFG, Avansini SH, Neto AGB, Brasileiro BTRV, Ceccato-Antonini SR, de Morais Jr MA (2012) The physiological characteristics of the yeast Dekkera bruxellensis in fully fermentative conditions with cell recycling in mixed cultures with Saccharomyces cerevisiae. Antonie Van Leeuwenhoek 101:529–539. https://doi.org/10.1007/s10482-011-9662-2
Pereira LF, Lucatti E, Basso LC, de Morais Jr MA (2014) The fermentation of sugarcane molasses by Dekkera bruxellensis and the mobilization of reserve carbohydrates. Antonie Van Leeuwenhoek 105:481–489. https://doi.org/10.1007/s10482-013-0100-5
Reis ALS, Damilano LD, Menezes RSC, de Morais Jr MA (2016) Second-generation ethanol from sugarcane and sweet sorghum bagasses using the yeast Dekkera bruxellensis. Ind Crops Prod 92:255–262. https://doi.org/10.1016/j.indcrop.2016.08.007
Rodríguez A, De La Cera T, Herrero P, Moreno F (2001) The hexokinase 2 protein regulates the expression of the GLK1, HXK1 and HXK2 genes of Saccharomyces cerevisiae. Biochem J 355:625–631. https://doi.org/10.1042/bj3550625
Ronne H, Carlberg M, Hu GZ, Nehlin JO (1991) Protein phosphatase 2A in Saccharomyces cerevisiae: effects on cell growth and bud morphogenesis. Mol Cel Biol 10:4876–4884. https://doi.org/10.1128/mcb.11.10.4876
Siverio JM (2002) Assimilation of nitrate by yeasts. FEMS Microbiol Rev 3:277–284. https://doi.org/10.1111/j.1574-6976.2002.tb00615.x
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45:D362–D368. https://doi.org/10.1093/nar/gkw937
Steensels J, Daenen L, Malcorps P, Derdelinckx G, Verachtert H, Verstrepen K (2015) Brettanomyces yeasts—from spoilage organisms to valuable contributors to industrial fermentations. Int J Food Microbiol 206:24–38. https://doi.org/10.1016/j.ijfoodmicro.2015.04.005
Teles GH, Da Silva JM, Mendonça AA, de Morais Jr MA, de Barros PW (2018) First aspects on acetate metabolism in the yeast Dekkera bruxellensis: a 2 few keys for improving ethanol fermentation. Yeast 10:577–584. https://doi.org/10.1002/yea.3348
Tiukova IA, Petterson ME, Tellgren-Roth C, Bunikis I, Eberhard T, Pettersson OV, Passoth V (2013) Transcriptome of the alternative ethanol production strain Dekkera bruxellensis CBS 11270 in sugar limited, low oxygen cultivation. PloS One 3:e58455. https://doi.org/10.1371/journal.pone.0058455
van Dijken JP, Scheffers WA (1986) Redox balances in the metabolism of sugars by yeasts. FEMS Microbiol Letters 32:199–224. https://doi.org/10.1016/0378-1097(86)90291-0
Van Rossum HM, Kozak BU, Pronk JT, van Marris AJA (2016) Engineering cytosolic acetyl-coenzyme A supply in Saccharomyces cerevisiae: pathway stoichiometry, free-energy conservation and redox-cofactor balancing. Metabol Eng 36:99–115. https://doi.org/10.1016/j.ymben.2016.03.006
Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L (2011) SearchGUI: an open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 11:996–999. https://doi.org/10.1002/pmic.201000595
Vigentini I, Lucy Joseph CM, Picozzi C, Foschino R, Bisson LF (2013) Assessment of the Brettanomyces bruxellensis metabolome during sulphur dioxide exposure. FEMS Yeast Res 13:597–608. https://doi.org/10.1111/1567-1364.12060
Zhao XJ, Raitt D, Burke VP, Clewell AS, Kwast KE, Poyton RO (1996) Function and expression of flavohemoglobin in Saccharomyces cerevisiae. Evidence for a role in the oxidative stress response. J Biol Chem 41:25131–25138. https://doi.org/10.1074/jbc.271.41.25131
Acknowledgements
The authors gratefully thank the technical staff of the Northern Centre for Technological Strategies CETENE (Recife, Pernambuco, Brazil at https://www.cetene.gov.br/) for their kind help with MALDI-ToF and MS analyses and Dr. Jimmy Eng of the Centre for Advanced Proteomics of the University of Washington (Seattle, WA, USA at http://proteomicsresource.washington.edu/) for the use of MASCOT with the specific D. bruxellensis database. The English text of this paper has been revised by Sidney Pratt, Canadian, MAT (The Johns Hopkins University), RSAdip - TESL (Cambridge University).
Funding
This work was sponsored by grants of the National Council of Science and Technology (CNPq/process 409767/2018-2 and CNPq/process 303551/2017-8) and by the Bioethanol Research Network of the State of Pernambuco (CNPq-FACEPE/PRONEM APQ-1452-2.01/10).
Author information
Authors and Affiliations
Contributions
MAMJ, WBP and TCJ conceived and designed the research. ICPM, DCP, KMS and EPNP conducted the experiments. ICPM, DCP, EPNP and FACS performed the proteome annotation and GO analyses. ICPM, WBP, TCJ and MAMJ prepared the manuscript. All authors read and approved the submitted manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 408 kb)
Rights and permissions
About this article

Cite this article
Peña-Moreno, I.C., Parente, D.C., da Silva, K.M. et al. Comparative proteomic analyses reveal the metabolic aspects and biotechnological potential of nitrate assimilation in the yeast Dekkera bruxellensis. Appl Microbiol Biotechnol 105, 1585–1600 (2021). https://doi.org/10.1007/s00253-021-11117-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11117-0