
Abstract
The gut microbiota plays an important role in colorectal cancer (CRC), and the use of probiotics might be a promising intervention method. The aim of our study was to investigate the beneficial effect of Bifidobacterium bifidum CGMCC 15068 on an azoxymethane (AOM)/dextran sulphate sodium (DSS)-induced colitis-associated CRC (CAC) mouse model. CAC was induced by an intra-peritoneal injection of AOM (10 mg/kg) and three 7-day cycles of 2% DSS in drinking water with a 14-day recovery period between two consecutive DSS administrations. B. bifidum CGMCC 15068 (3 × 109 CFU/mL) was gavaged once daily during the recovery period. Then, the faecal microbial composition and metabolome were profiled using the 16S rRNA sequencing technology and gas chromatography-mass spectrometry (GC-MS), respectively. The administration of B. bifidum CGMCC 15068 attenuated tumourigenesis in the CAC mouse model. In addition, B. bifidum CGMCC 15068 pre-treatment increased the relative abundance of Akkermansia, Desulfovibrionaceae, Romboutsia, Turicibacter, Verrucomicrobiaceae, Ruminococcaceae_UCG_013, Lachnospiraceae_UCG_004, and Lactobacillus. Meanwhile, B. bifidum CGMCC 15068 altered metabolites involved in the citrate cycle (TCA cycle), glycolysis, butyrate metabolism, fatty acid biosynthesis, and galactose metabolism. Several significant correlations were identified between the differentially abundant microbes and metabolites. These findings supported the beneficial role of B. bifidum CGMCC 15068 in intestinal health by modulating dysbiosis and the gut metabolic profile. The manipulation of the gut microbial composition using probiotics might be a promising prevention strategy for CRC. Long-term and large-scale clinical trials are warranted for the potential clinical applications of this strategy in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and the second leading cause of cancer mortality worldwide (Bray et al. 2018). A majority of CRC incidences are associated with diet and lifestyle, particularly with the increased meat and decreased dietary fibre in the diet, which also affect the gut microbiota (Tan and Chen 2016). As the large intestine has the highest exposure to microorganisms, the involvement of gut microbial dysbiosis in colon tumourigenesis has been widely studied (Gao et al. 2015; Zou et al. 2018).
The application of probiotics might be a promising treatment and prevention strategy for CRC. Bifidobacterium bifidum CGMCC 15068 is a bacterial species belonging to the genus Bifidobacterium, which is one of the most widely used probiotic bacteria (O’Callaghan and van Sinderen 2016). Several researchers have investigated the potential role of bifidobacteria in the initiation and progression of CRC (Hibberd et al. 2017; Song et al. 2018; Mendes et al. 2018). However, the mechanism of its beneficial role in colon health is still not fully understood.
The metabolic activity is a crucial feature of the gut microbiota and is one of the potential mechanisms of the host–microbiota interaction (Louis et al. 2014; Zarrinpar et al. 2018). It also has been shown to have a strong influence on the development of CRC. These microbes contain a wide range of enzymes and thus can metabolise diverse substrates (Arthur and Jobin 2011). The gut microbial-generated metabolites include beneficial substances (Canani et al. 2011; Louis et al. 2014) as well as pro-inflammatory and pro-carcinogenic substances (Russell et al. 2011; Louis et al. 2014).
CRC has been strongly linked with inflammation, and previous epidemiology research has indicated the increased risk of developing CRC in patients with the inflammatory bowel disease (IBD), which is also known as colitis-associated cancer (CAC) (Jess et al. 2005). The injection of the pro-carcinogen azoxymethane (AOM) followed by the administration of the inflammatory agent dextran sulphate sodium (DSS) is a classic animal model for the study of inflammation-associated CRC (De Robertis et al. 2011). Thus, in the current study, we used the CAC mouse model to assess the influence of B. bifidum CGMCC 15068 on colon tumourigenesis, gut microbial composition, and gut metabolic profile.
Materials and methods
Bacterial strains
B. bifidum CGMCC 15068 was deposited at the China General Microbiological Culture Collection Center (CGMCC) and was cultured in Man–Rogosa–Sharpe broth (Thermo Fisher, Shanghai, China) anaerobically at 37 °C for 24 h. The cultures were centrifuged at 4 °C at 8000×g for 10 mins, washed twice with sterile phosphate buffer saline (PBS), and re-suspended to the centration of 3 × 109 colony-forming units (CFU)/mL for future use. The bacterial concentrations were determined by measuring the absorbance at 630 nm (O.D. range, 0.6–0.8). The experiments were conducted under strict anaerobic conditions.
Animals
Male C57BL/6 mice (aged 8 weeks) were purchased from Shanghai Laboratory Animal, Co., Ltd. (SLAC). The mice were acclimatised (1 week after their arrival) and raised in a specific pathogen-free (SPF) environment (23 °C, 12/12-h light/dark cycle, 50% humidity, and food and water available ad libitum). The experiment protocol was approved by the Animal Care Committee of Zhejiang University School of Medicine.
Experimental design and colitis-associated colon cancer mouse model
Azoxymethane (AOM) was purchased from Sigma-Aldrich (St. Louis, MO, USA), and dextran sodium sodium (DSS) was purchased from MP Biomedicals (molecular weight: 36–50 kDa; Santa Ana, CA, USA). The mice were randomly divided into three groups: the control group (PBS group; n = 10), AOM/DSS model group (AOM group; n = 12), and AOM/DSS model with B. bifidum CGMCC 15068 administration (AOM.BF group; n = 9). One of the main interests of the current study was to investigate the role of B. bifidum in the development of colitis-associated colon cancer. PBS group was set as the negative control group, and AOM/DSS model group was set as the positive control group, while AOM/DSS model with B. bifidum administration was set as the experimental group. The reason why the PBS group with B. bifidum administration was not added is based on previous research in which B. bifidum strain administration caused neither death nor colon-related illness (Grimm et al. 2015). Nevertheless, B. bifidum is one of the most important commensal bacteria in infant intestine and plays a role in maintaining host health status (Turroni et al. 2012). It has been extensively used as probiotics in both adult and paediatric populations (Gareau et al. 2010). The safety of B. bifidum strains has also been extensively assessed and confirmed in the previous studies (Kim et al. 2018; Abdelhamid et al. 2019). Each cage housed fewer than five mice. To induce colitis-associated colon cancer (Fig. 1a), 10 mg/kg of AOM was intra-peritoneally injected into all the mice in both the AOM group and the AOM.BF group. The mice in the PBS group were injected the same volume of 0.9% physiological saline. Three 7-day cycles of freshly prepared 2% DSS (wt/vol) were administered to the mice in both groups 7 days after the AOM injection. Between the DSS administrations, sterile drinking water was given to the mice as the recovery period. Meanwhile, the mice in the AOM.BF group were orally gavaged 0.2 mL of B. bifidum CGMCC 15068 (3 × 109 CFU/mL) once daily in the recovery period. In addition, the mice in the PBS group and the AOM group were orally administered the same amount of PBS.

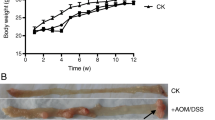
Bifidobacterium bifidum CGMCC 15068 attenuated intestinal tumourigenesis in AOM/DSS-induced colitis-related colon tumour in mice. a Mice experiment workflow. b Representative colon photo from the three groups. c Percentage of mice weight change from week 0 to week 10. d Number of macroscopic tumours (> 2 mm in diameter) in each group. Data are displayed as mean ± standard deviation. *p value < 0.05 **p value < 0.01
Sample collection
The weights of the mice were recorded in the experiment every 7 days. The mice were euthanised after the last cycle of DSS (Fig. 1a). The incidence of colon cancer was examined in each mouse. In addition, the faecal contents were collected at the time of sacrifice for the microbial composition and metabolome assessment. The samples were first frozen in liquid nitrogen and stored at − 80 °C until further processing.
DNA extraction and 16S rRNA sequencing
Total DNA was extracted using QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s handbook. After verifying the quality and the concentration of DNA with agarose gel and NanoDrop, the extracted DNA was diluted to the concentration of 1 ng/μL as the PCR template. The PCR amplification of the V3–V4 variable regions of the bacterial 16S rRNA genes was conducted using universal primers 343F and 798R and Takara Ex Taq (Takara Biomedicals, Kusatsu, Japan).
The amplicon was purified with AMPure XP beads (Agencourt, Beckman Coulter, Brea, CA, USA) and amplified with another round of PCR, and its quality was visualised with gel electrophoresis. The amplicon was then purified again before final quantification by using the Qubit dsDNA assay kit (Thermo Scientific, Waltham, Massachusetts, USA). The subsequent paired-end sequencing was performed on pooled purified amplicon at the Illumina MiSeq platform (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions.
Microbial composition analysis
The paired-end raw sequencing reads were trimmed using the Trimmomatic software (Bolger et al. 2014) with a quality score cutoff of 20. The trimmed reads were subsequently assembled by the FLASH software (version 1.2.8) (Magoč and Salzberg 2011). Then, the QIIME software (version 1.8.0) (Caporaso et al. 2010) was used for the sequence denoising as well as the chimaera detection and removal. After removing the primer sequence, clean reads with 97% similarity were clustered into the same operational taxonomic unit (OTU) with the VSEARCH software (Rognes et al. 2016). The representative read in each OTU was selected using the QIIME software (version 1.8.0) (Caporaso et al. 2010). The annotation of the representative reads in each OTU was based on the SILVA database (version 123) (Quast et al. 2013) by using the Ribosomal Database Project (RDP, database v.11.3) classifier (Cole et al. 2014). A principal coordinate analysis (PCoA) based on the Bray–Curtis distance was performed with the QIIME software (version 1.8.0) (Caporaso et al. 2010). The analysis of similarity (ANOSIM) was also conducted with 999 permutation tests to investigate the significance of the difference identified between groups. To further explore the microbiota diversity, a linear discriminant analysis effect size measurement (LEfSe) analysis was conducted on the Galaxy web platform (Segata et al. 2011). The effect size of the microbial taxa was calculated by the linear discriminant analysis (LDA), and the cutoff for the discriminative taxa was an LDA score of more than 2.
Untargeted metabolome profiling using GC–MS
For each accurately weighted faecal sample (50 mg), 600 μL of ice-cold methanol/water (4:1, v/v) (CNW Technologies GmbH, Düsseldorf, Germany) and 20 μL of internal standard (2-chloro-l-phenylalanine in methanol, 0.3 mg/mL) (Shanghai Hengchuang Bio Technology, Shanghai, China) were added and homogenised. After centrifugation (4 °C, 12000 rpm, 10 min), a quality control (QC) sample was prepared by mixing aliquots of all 31 faecal samples. The supernatant was vacuum-dried, and subsequently, 80 μL of methoxylamine hydrochloride (15 mg/mL, in pyridine) (CNW Technologies GmbH, Düsseldorf, Germany) was added. The mixture was vortexed for 2 min and then incubated at 37 °C for 90 min. Then, 80 μL of N,O-bistrifluoroacetamide (BSTFA) (with 1% trimethylsilyl chloride (TMCS)) (CNW Technologies GmbH, Düsseldorf, Germany) and 20 μL of n-hexane (CNW Technologies GmbH, Düsseldorf, Germany) were added into the mixture. The sample was vortexed for 2 mins and derivatised at 70 °C for 60 mins. The pre-processed samples, QC sample, and blanks were analysed on an Agilent 7890B gas chromatography system coupled to an Agilent 5977AMSD system (Agilent Technologies Inc., Santa Clara, CA, USA).
The raw data were pre-processed in the MS-DIAL software (Tsugawa et al. 2015). The metabolites were annotated by the in-house database (LUG database and untarget database of GC–MS from Lumingbio, Shanghai, China). Then, both principal component analysis (PCA) and orthogonal partial least squares-discriminant analysis (OPLS-DA) were performed to compare the difference in the metabolome profiles between the groups using SIMCA (version 14.1) (Sartorius Stedim Biotech, Umeå, Sweden). For OPLS-DA, we also carried out a sevenfold cross-validation and 200 response permutation tests to measure the model reliability and avoid model over-fitting. The variable importance in the projection (VIP) of each metabolite was obtained from the OPLS-DA model. In addition, two-tailed Student’s t tests were performed to compare the metabolite concentration between different groups using the R software (version 3.6.0) (R Core Team 2013). Metabolites with VIP > 1 and two-tailed Student’s t test p value < 0.05 were identified as differentially abundant metabolites among the groups. The differential metabolites were then uploaded to MetaboAnalyst 4.0 (www.metaboanalyst.ca/) for the metabolomics pathway analysis.
Microbiota and metabolome integration analysis
Both untargeted and targeted integration analyses were performed. First, the relationship between all the microbial taxa and metabolites within the three groups was evaluated using sparse partial least square-discriminant analysis (sPLS-DA). The analysis was conducted using mixOmics R package (Rohart et al. 2017). Subsequently, the Pearson correlations between the differentially abundant microbial taxa and metabolites between AOM.BF and AOM group were analysed and visualised using the R software (version 3.6.0) (R Core Team 2013).
Statistical analysis
The normality of the data was checked by the Kolmogorov–Smirnov test. The body weight change difference and the large tumour number difference were determined using one-way ANOVA and Tukey’s multiple pairwise comparisons. p value < 0.05 was considered significant. All of the data analyses were conducted using R (version 3.6.0) (R Core Team 2013).
Nucleotide sequence accession number
The 16S rRNA sequencing data of the mice faecal microbiota was deposited in the Sequence Read Archive (SRA) database and can be accessed by the accession number PRJNA594160.
Results
B. bifidum CGMCC 15068 attenuated intestinal tumourigenesis in mice
We replicated colitis-associated CRC using an intra-peritoneal injection of AOM followed by three cycles of water-administered DSS (Fig. 1b). The body weight loss was reported as an indicator of the tumourigenesis and was consistent with the inflammatory markers. The percentages of the weight change relative to the baseline weight were measured every 7 days during the experiment. As shown in Fig. 1c, there was a consistent decline in the mouse body weight after each round of DSS treatment. When the mice were sacrificed, the body weight gain was the highest in the PBS group (22.0% ± 7.5%) followed by that in the AOM.BF group and in the AOM group (22.0% ± 6.2%; 16.2% ± 3.3%; p value < 0.05). The number of macroscopic tumours (> 2 mm in diameter) was also compared among the groups (Fig. 1d). The mice in the AOM group developed almost twice more tumours per mouse than the CAC mouse model administered with B. bifidum CGMCC 15068 (1.6 ± 1.4 vs. 0.4 ± 0.7, p value < 0.05). The PBS group showed no tumour incidence. Overall, these results showed that B. bifidum CGMCC 15068 attenuated intestinal tumourigenesis in mice.
B. bifidum CGMCC 15068 induced shift in gut microbiota composition
In all, 874,912 reads (AOM, 357,204; AOM.BF, 274,967; PBS, 242,741) from 31 samples were obtained for the downstream analysis, and 1983 OTUs were identified with a 97% similarity cutoff. Chao1 and Shannon index (alpha diversity) are the predictors of taxonomic richness and evenness, respectively, and were not significantly different across the groups (p value > 0.05).
The inter-sample differences (beta diversity) were measured by the principal coordinate analysis (PCoA) on the basis of the Bray–Curtis distance (Fig. 2a). Together with the significance of the separation calculated by ANOSIM, the results revealed that the samples in the AOM group were separated from the PBS sample (p value = 0.001). In addition, the AOM.BF group was separated from the AOM group (p value = 0.009), and AOM.BF exhibited the tendency to cluster toward the PBS group, which demonstrated that the administration of B. bifidum CGMCC 15068 attenuated the AOM/DSS-induced gut microbiota dysbiosis.

Bifidobacterium bifidum CGMCC 15068 induced shift in gut microbiota composition. a Beta diversity comparison using PCoA analysis based on Bray–Curtis distance matrix. Each point represents one sample, and the colour indicates the mouse group (red, AOM; green, AOM.BF; blue, PBS). b–d Relative abundance distribution of the top 15 most abundant microbial taxa at the phylum (b), family (c), and genus (d) levels
Next, the relative microbial taxa abundances were compared between the three groups using ANOVA. The top 15 most abundant microbial taxa at the phylum, family, and genus levels are shown in Fig. 2b–d. At the phylum level, compared with the AOM group, the B. bifidum CGMCC 15068 administration increased the abundance of Proteobacteria and Verrucomicrobia and decreased the abundance of Bacteroidetes, Firmicutes, and Actinobacteria. At the family level, the abundance of Rikenellacea, Rhodospirillaceae, and Lachnospiraceae was more enriched in the PBS group than in the AOM group and AOM.BF group, whereas Coriobacteriaceae and Peptococcaceae were more abundant in the AOM group. At the genus level, the AOM group had higher abundance of Lachnoclostridium, Coriobacteriaceae_UCG_002, and Parasutterella.
To further investigate the significantly differentially abundant taxa with biological consistency, we then performed the LEfSe analysis. We found that the administration of B. bifidum CGMCC 15068 increased the abundance of Akkermansia, Desulfovibrionaceae, Romboutsia, Turicibacter, Verrucomicrobiaceae, Ruminococcaceae_UCG_013, Lachnospiraceae_UCG_004, and Lactobacillus (Fig. 3a–b, LDA score (−log10) > 2) and the AOM group had increased the abundance of Bacteroidetes, Parasutterella, Coriobacteriaceae, Actinobacteria, Peptococcaceae, Clostridiaceae, Ruminiclostridium_9, and Streptococcaceae (Fig. 3a–b, LDA score (−log10) > 2). The PBS control group showed higher abundance of Lachnospiraceae, Rikenellacea, Rhodospirillaceae, Deferribacteres, Butyricimonas, and Prevotellaceae (Fig. 3a–b, LDA score (−log10) > 2).

Identification of differentially abundant microbial taxa using linear discriminant analysis effect size (LEfSe) analysis. a LEfSe cladogram of the discriminative microbial taxa (LDA score > 2); the size of the circle shows the relative abundance of the taxa, and the colour shows the mouse group (red, AOM; green, AOM.BF; blue, PBS). b LEfSe score plot of the discriminative microbial taxa (LDA score > 2) that are more enriched in the AOM (red), AOM.BF (green), and PBS (blue) groups
To predict the metabolic functions of the microbiota, the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) analysis was conducted based on the Kyoto Encyclopaedia of Genes and Genomes (KEGG) metabolic pathways (Langille et al. 2013). Interestingly, in terms of the KEGG pathway analysis between the AOM group and the AOM.BF group, the colorectal cancer pathway and the p53 signalling pathway were more enriched in the AOM group (Online Resource Table S1).
B. bifidum CGMCC 15068 altered faecal metabolic composition
Overall, 342 metabolites were detected from all the faecal samples across the groups. The results of the unsupervised PCA analysis (Fig. 4a) indicated that the metabolome profile of the three groups could be separated from one another (R2X = 0.478). The score plot also showed that the AOM.BF group clustered between the AOM group and the PBS group, with a tendency toward the PBS group. The supervised OPLS-DA analysis was then performed between the groups (AOM vs. PBS; AOM.BF vs. AOM). As is depicted in Fig. 4b and c, there were significant separations in the metabolic profiles between these groups. The cross-validation permutation tests confirmed the validity of the supervised OPLS-DA models, which illustrated that the original model predicted the class label better than the other models with the permutated label (Online Resource Fig. S1; AOM vs. PBS: Q2inter = − 0.56; AOM.BF vs. AOM: Q2inter = − 0.569). Overall, the results showed that the CAC model group had a distinct metabolic composition from the PBS control group and the CAC model pre-treated with B. bifidum CGMCC 15068.

Bifidobacterium bifidum CGMCC 15068 altered faecal metabolic composition. a Score plot of PCA analysis comparing the metabolome profile among the groups (red, AOM; green, AOM.BF; blue, PBS). b Score plot of OPLS-DA analysis comparing the metabolome profile between AOM group (red) and the PBS group (blue). c Score plot of OPLS-DA analysis comparing the metabolome profile between the AOM.BF group (green) and the AOM group (red). d S-plot from OPLS-DA model between AOM.BF and AOM group. The marked metabolites located at the top-left and bottom-right side tend to play more important roles in discriminating the metabolic profile between the groups. e Heat map of the significantly differentially abundant metabolites between the AOM.BF group (red) and the AOM group (blue) (VIP > 1 and p value <0 .05). A row presents the data obtained from a metabolite, and a column represents a sample. The red and green colours correspond to the increased and the decreased levels of the metabolites, respectively
One of the main interests of the current study was to investigate the role of B. bifidum CGMCC 15068 in the development of CAC. Subsequently, we selected the metabolites that contributed to the change in the metabolic composition among the groups on the basis of the threshold of VIP score > 1 and p value < 0.05. As depicted in Fig. 4d–e, 109 differentially abundant metabolites were identified from the comparison between the AOM.BF group and the AOM group. The metabolic pathways these metabolites were involved with included citrate cycle (TCA cycle); galactose metabolism; glyoxylate and dicarboxylate metabolism; ascorbate and aldarate metabolism; glycolysis; alanine, aspartate, and glutamate metabolism; butyrate metabolism; and fatty acid biosynthesis. From the S-plot (Fig. 4d), the metabolites that were farthest from the origin were pointed out, including N-methylglutamic acid, L-lactic acid, cadaverine, and hexadecane. They might potentially contribute more to the separation of the metabolome profile between AOM.BF and AOM group.
Relationship between the microbiota and metabolome
First, Sparse partial least square-discriminant analysis (sPLS-DA) was performed to integrate the metabolome profile and the microbial composition from all the faecal samples in all the groups. The correlation circle plot (Fig. 5a) highlighted the close relationship between the microbial composition and faecal metabolite content. The pairwise correlations were further illustrated on a circus plot shown in Fig. 5b.

Sparse partial least square-discriminant analysis (sPLS-DA) of the microbiota and metabolome profile from all samples. a Correlation circle plot with the colours representing the nature of the data (red, microbiota; green, metabolome). The close cluster of the points indicated strong correlation between the microbial taxa and the metabolite level. b Circus plot displaying the correlations between various microbial taxa (red) and the metabolites (green) which are located at the side quadrants. Only correlation larger than 0.7 is shown; the colour of the lines indicates the direction of the correlation (orange, positive correlation; black, negative correlation). The abundance of the microbe and metabolites in all the groups is shown outside the circus (blue, AOM; orange, AOM.BF; grey, PBS)
We then continued to assess the targeted correlation between differentially abundant microbes and metabolites from the AOM.BF group and the AOM group. In particular, the most discriminative metabolites were further selected by fold change (FC) > 2. As shown in Fig. 6, several significant correlations (p value < 0.05) were identified. The relative abundance of Romboutsia was negatively correlated with the lactic acid level and positively correlated with the citric acid level. Furthermore, positive correlations with citric acid concentration were also found with the relative abundance of Turicibacter and Lachnospiraceae_UCG_004. The relative abundance of Ruminiclostridium_9 was, however, negatively correlated with the citric acid level. Additionally, Peptococcaceae and Ruminiclostridium_9 exhibited a negative relationship with cadaverine.

Pearson correlation analysis of the differentially abundant microbial taxa (LDA score > 2) and the differentially abundant metabolites (VIP > 1, p value < 0.05, fold change (FC) > 2). The significant correlations (p value < 0.05) are shown in the plot. The colour and the size of the circle represent the intensity level of the correlations, with the positive correlations shown in blue and the negative correlations shown in red
Discussion
In the present study, we investigated the microbial composition, metabolome profile, and the interaction between the microbiota and the metabolome to disentangle the complex role of B. bifidum CGMCC 15068 in the tumourigenesis of colorectal cancer. The significance of the current study was its investigation of the microbial metabolic function rather than merely focusing on the microbial taxonomy difference.
We first characterised the microbial composition differences across the three groups. On the one hand, we found microbial taxa that were more enriched in the AOM.BF group. In particular, Ruminococcaceae and Lachnospiraceae, which are two major families from the phylum Firmicutes, were enriched in the CAC mouse group administered with B. bifidum CGMCC 15068. Both microbial families are known as anti-inflammatory factors, and the microbes within the Lachnospiraceae family have been shown to be protective against CRC (Meehan and Beiko 2014). Additionally, Romboutsia is commonly related to the healthy status of the gut in the patients (Milani et al. 2016), and the increase in Romboutsia with the supplement of B. bifidum CGMCC 15068 indicated that B. bifidum CGMCC 15068 may help maintain the gut mucosa health and ameliorate tumourigenesis under the AOM/DSS condition. The group administered with B. bifidum CGMCC 15068 also increased the relative abundance of Lactobacillus, which is within the generally used probiotics and has been shown to prevent colon cancer in the mouse model (Foo et al. 2011; Zhu et al. 2014; Jacouton et al. 2017). Another interesting finding was the elevation of Akkermansia in the AOM.BF group. Akkermansia is the representative genus from the family Verrucomicrobiaceae and plays an important role in regulating the host metabolism (Derrien et al. 2004; Bian et al. 2019; Macchione et al. 2019). It has been extensively studied in metabolic diseases such as obesity (Everard et al. 2013) as well as gastrointestinal diseases such as ulcerative colitis (Bian et al. 2019). Additionally, the increase in Turicibacter was observed in the AOM.BF group, which has been reported in the previous studies as a possible marker for the gut health status (Collins et al. 2014). On the other hand, there were bacteria more enriched in the CAC mouse model. For instance, the relative abundance of Parasutterella and Coriobacteriaceae was increased. Previous research has suggested that Parasutterella may play a role in the chronic intestinal inflammation and gut pathophysiological changes (Chen et al. 2018). The relative abundance of Coriobacteriaceae has been reported to increase in CRC patients (Chen et al. 2012).
Accumulating evidence has indicated that the microbiota may influence the host’s health by its metabolites. As a functional readout of the intestinal microbiota (Zierer et al. 2018), the faecal metabolome was then profiled. We demonstrated significant differences in the metabolic profile across the AOM, AOM.BF, and PBS groups. Compared with the AOM group, the pre-treatment of B. bifidum CGMCC 15068 in the AOM.BF group regulated the level of 109 metabolites, including benzenoids, lipids, nucleotides, organic acids, and organic nitrogen/oxygen compounds. These metabolites were involved in several important cancer-related metabolic pathways including the TCA cycle, glycolysis, butyrate metabolism, fatty acid biosynthesis, and galactose metabolism. The TCA cycle was the most important central pathway for biosynthesis and energy metabolism. The role of the TCA cycle in cancer metabolism was demonstrated in the previous study (Anderson et al. 2018). Glycolysis is the feature of cancer cells to generate most of their energy even under aerobic conditions, which is also called the Warburg metabolism (Liberti and Locasale 2016). The regulation of the metabolites involved in these pathways by B. bifidum CGMCC 15068 demonstrated the gut microbiota as an important modulator of the host metabolism in health and disease (Kinross et al. 2011). In addition, the butyrate metabolism and fatty acid metabolism pathways were also regulated by the administration of B. bifidum CGMCC 15068. Butyrate is a short fatty chain acid and has been considered to mediate the repressive effect of dietary fibre in cancer (Scharlau et al. 2009). Furthermore, butyrate is an anti-cancer and anti-inflammatory microbial metabolite and is important for gut homeostasis (Rivière et al. 2016). Butyrate-generating bacteria have been reported to co-exist with bifidobacteria (Rivière et al. 2016). This is also shown in our finding that Akkermansia, which is butyrate-generating bacteria, was more abundant in the group that administered with B. bifidum CGMCC 15068. The significance of the fatty acid synthesis pathway in tumourigenesis has been linked to the excess need for cellular proliferation (Currie et al. 2013). For the modulation of the galactose level, bifidobacterial enzymes are involved in the galactose metabolism (Kitaoka 2012).
A number of microbes might be involved in the alteration of the faecal metabolome and thus affect colon health and CAC progression. Note that in the CAC mice treated with B. bifidum CGMCC 15068, the relative abundances of Turicibacter, Romboutsia, and Lachnospiraceae_UCG_004 were positively correlated with the citric acid level, and the abundance of Ruminiclostridium_9 was negatively correlated with the citric acid level. Citric acid is a crucial intermediate in the TCA cycle and has been shown to inhibit cancer growth in a range of tumour types (Wang et al. 2016; Ren et al. 2017) potentially by downregulating glycolysis (Lin et al. 2012). Furthermore, the relative abundance of the microbial taxa within Romboutsia family was negatively correlated with the level of lactic acid. It is a characteristic of tumour metabolism that excess lactic acid is generated because of the high consumption of glucose in the glycolysis in cancer cells (Liberti and Locasale 2016). The negative correlation between the microbes and the lactic acid suggests that these bacteria may play a role in lactic acid metabolism. There is a synergic metabolism between the lactic acid-generating bacteria such as B. bifidum and the lactic acid-utilising bacteria (Sato et al. 2008). The enrichment of B. bifidum CGMCC 15068 might favour the metabolic function of the lactic acid-utilising bacteria so as to reduce the amount of lactic acid. Another interesting finding was the negative correlation between Peptococcaceae and Ruminiclostridium_9 and the concentration of cadaverine (CAD). CAD is a microbial metabolite produced by the bacterial decarboxylation of lysine by the lysine decarboxylase (LDC) enzyme (de las Rivas et al. 2006). The influence of CAD on carcinogenesis was characterised in a previous study that treated the breast cancer cells with CAD (Kovács et al. 2019). The finding showed that CAD inhibited the tumour cell movement and invasion (Kovács et al. 2019). Furthermore, the abundance of DNA coding for the lysine decarboxylase (LDC) enzyme that generated CAD was found to be lower in the cancer faecal samples (Kovács et al. 2019). This indicated that the microbial taxa Peptococcaceae and Ruminiclostridium_9 might lack LDC enzyme or lead to the decrease of bacteria that contain such enzyme. Further functional studies are required to explore the relationships identified between the microbes and metabolites in the current study.
In summary, the consumption of probiotic bacteria B. bifidum CGMCC 15068 might modulate the intestinal microbial composition and the gut metabolic profile as part of the microbial function. These findings might offer mechanistic insights on the role of microbiota in colon tumourigenesis, and the manipulation of the gut microbial composition using probiotics might be a promising prevention strategy for CRC. Future long-term and large-scaled clinical trials are warranted for potential clinical applications.
References
Abdelhamid AG, El-Masry SS, El-Dougdoug NK (2019) Probiotic Lactobacillus and Bifidobacterium strains possess safety characteristics, antiviral activities and host adherence factors revealed by genome mining. EPMA J 10:337–350. https://doi.org/10.1007/s13167-019-00184-z
Anderson NM, Mucka P, Kern JG, Feng H (2018) The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9:216–237. https://doi.org/10.1007/s13238-017-0451-1
Arthur JC, Jobin C (2011) The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis 17:396–409. https://doi.org/10.1002/ibd.21354
Bian X, Wu W, Yang L, Lv L, Wang Q, Li Y, Ye J, Fang D, Wu J, Jiang X, Shi D, Li L (2019) Administration of Akkermansia muciniphila ameliorates dextran sulfate sodium-induced ulcerative colitis in mice. Front Microbiol 10: . https://doi.org/10.3389/fmicb.2019.02259
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A (2011) Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol 17:1519–1528. https://doi.org/10.3748/wjg.v17.i12.1519
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chen W, Liu F, Ling Z, Tong X, Xiang C (2012) Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One 7:e39743. https://doi.org/10.1371/journal.pone.0039743
Chen Y-J, Wu H, Wu S-D, Lu N, Wang Y-T, Liu H-N, Dong L, Liu T-T, Shen X-Z (2018) Parasutterella, in association with irritable bowel syndrome and intestinal chronic inflammation. J Gastroenterol Hepatol 33:1844–1852. https://doi.org/10.1111/jgh.14281
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM (2014) Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. https://doi.org/10.1093/nar/gkt1244
Collins JW, Chervaux C, Raymond B, Derrien M, Brazeilles R, Kosta A, Chambaud I, Crepin VF, Frankel G (2014) Fermented dairy products modulate Citrobacter rodentium-induced colonic hyperplasia. J Infect Dis 210:1029–1041. https://doi.org/10.1093/infdis/jiu205
Currie E, Schulze A, Zechner R, Walther TC, Farese RV (2013) Cellular fatty acid metabolism and Cancer. Cell Metab 18:153–161. https://doi.org/10.1016/j.cmet.2013.05.017
de las Rivas B, Marcobal A, Carrascosa AV, Muñoz R (2006) PCR detection of foodborne bacteria producing the biogenic amines histamine, tyramine, putrescine, and cadaverine. J Food Prot 69:2509–2514. https://doi.org/10.4315/0362-028x-69.10.2509
De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM (2011) The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog 10:9. https://doi.org/10.4103/1477-3163.78279
Derrien M, Vaughan EE, Plugge CM, De WV (2004) Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 54:1469–1476. https://doi.org/10.1099/ijs.0.02873-0
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. PNAS 110:9066–9071. https://doi.org/10.1073/pnas.1219451110
Foo N-P, Ou Yang H, Chiu H-H, Chan H-Y, Liao C-C, Yu C-K, Wang Y-J (2011) Probiotics prevent the development of 1,2-dimethylhydrazine (DMH)-induced colonic tumorigenesis through suppressed colonic mucosa cellular proliferation and increased stimulation of macrophages. J Agric Food Chem 59:13337–13345. https://doi.org/10.1021/jf203444d
Gao Z, Guo B, Gao R, Zhu Q, Qin H (2015) Microbiota disbiosis is associated with colorectal cancer. Front Microbiol 6: . https://doi.org/10.3389/fmicb.2015.00020
Gareau MG, Sherman PM, Walker WA (2010) Probiotics and the gut microbiota in intestinal health and disease. Nat Rev Gastroenterol Hepatol 7:503–514. https://doi.org/10.1038/nrgastro.2010.117
Grimm V, Radulovic K, Riedel CU (2015) Colonization of C57BL/6 mice by a potential probiotic Bifidobacterium bifidum strain under germ-free and specific pathogen-free conditions and during experimental colitis. PLoS One 10:e0139935. https://doi.org/10.1371/journal.pone.0139935
Hibberd AA, Lyra A, Ouwehand AC, Rolny P, Lindegren H, Cedgård L, Wettergren Y (2017) Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol 4:e000145. https://doi.org/10.1136/bmjgast-2017-000145
Jacouton E, Chain F, Sokol H, Langella P, Bermúdez-Humarán LG (2017) Probiotic strain Lactobacillus casei BL23 prevents colitis-associated colorectal cancer. Front Immunol 8:1553. https://doi.org/10.3389/fimmu.2017.01553
Jess T, Gamborg M, Matzen P, Munkholm P, Sørensen TIA (2005) Increased risk of intestinal cancer in Crohn’s disease: a meta-analysis of population-based cohort studies. Am J Gastroenterol 100:2724–2729. https://doi.org/10.1111/j.1572-0241.2005.00287.x
Kim MJ, Ku S, Kim SY, Lee HH, Jin H, Kang S, Li R, Johnston TV, Park MS, Ji GE (2018) Safety evaluations of Bifidobacterium bifidum BGN4 and Bifidobacterium longum BORI. Int J Mol Sci 19. https://doi.org/10.3390/ijms19051422
Kinross JM, Darzi AW, Nicholson JK (2011) Gut microbiome-host interactions in health and disease. Genome Med 3:14. https://doi.org/10.1186/gm228
Kitaoka M (2012) Bifidobacterial enzymes involved in the metabolism of human milk oligosaccharides123. Adv Nutr 3:422S–429S. https://doi.org/10.3945/an.111.001420
Kovács T, Mikó E, Vida A, Sebő É, Toth J, Csonka T, Boratkó A, Ujlaki G, Lente G, Kovács P, Tóth D, Árkosy P, Kiss B, Méhes G, Goedert JJ, Bai P (2019) Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci Rep 9:1–14. https://doi.org/10.1038/s41598-018-37664-7
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. https://doi.org/10.1038/nbt.2676
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41:211–218. https://doi.org/10.1016/j.tibs.2015.12.001
Lin C-C, Cheng T-L, Tsai W-H, Tsai H-J, Hu K-H, Chang H-C, Yeh C-W, Chen Y-C, Liao C-C, Chang W-T (2012) Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2:785. https://doi.org/10.1038/srep00785
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672. https://doi.org/10.1038/nrmicro3344
Macchione IG, Lopetuso LR, Ianiro G, Napoli M, Gibiino G, Rizzatti G, Petito V, Gasbarrini A, Scaldaferri F (2019) Akkermansia muciniphila: key player in metabolic and gastrointestinal disorders. Eur Rev Med Pharmacol Sci 23:8075–8083. https://doi.org/10.26355/eurrev_201909_19024
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Meehan CJ, Beiko RG (2014) A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol Evol 6:703–713. https://doi.org/10.1093/gbe/evu050
Mendes MCS, Paulino DS, Brambilla SR, Camargo JA, Persinoti GF, Carvalheira JBC (2018) Microbiota modification by probiotic supplementation reduces colitis associated colon cancer in mice. World J Gastroenterol 24:1995–2008. https://doi.org/10.3748/wjg.v24.i18.1995
Milani C, Ticinesi A, Gerritsen J, Nouvenne A, Lugli GA, Mancabelli L, Turroni F, Duranti S, Mangifesta M, Viappiani A, Ferrario C, Maggio M, Lauretani F, De Vos W, van Sinderen D, Meschi T, Ventura M (2016) Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep 6:1–12. https://doi.org/10.1038/srep25945
O’Callaghan A, van Sinderen D (2016) Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol 7. https://doi.org/10.3389/fmicb.2016.00925
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
R Core Team (2013) R: The R Project for Statistical Computing
Ren J-G, Seth P, Ye H, Guo K, Hanai J, Husain Z, Sukhatme VP (2017) Citrate suppresses tumor growth in multiple models through inhibition of glycolysis, the tricarboxylic acid cycle and the IGF-1R pathway. Sci Rep 7:1–13. https://doi.org/10.1038/s41598-017-04626-4
Rivière A, Selak M, Lantin D, Leroy F, De Vuyst L (2016) Bifidobacteria and butyrate-producing colon bacteria: importance and strategies for their stimulation in the human gut. Front Microbiol 7. https://doi.org/10.3389/fmicb.2016.00979
Rognes T, Flouri T, Nichols B, Quince C, Mahé F (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. https://doi.org/10.7717/peerj.2584
Rohart F, Gautier B, Singh A, Cao K-AL (2017) mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol 13:e1005752 . https://doi.org/10.1371/journal.pcbi.1005752
Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, Duncan G, Johnstone AM, Lobley GE, Wallace RJ, Duthie GG, Flint HJ (2011) High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr 93:1062–1072. https://doi.org/10.3945/ajcn.110.002188
Sato T, Matsumoto K, Okumura T, Yokoi W, Naito E, Yoshida Y, Nomoto K, Ito M, Sawada H (2008) Isolation of lactate-utilizing butyrate-producing bacteria from human feces and in vivo administration of Anaerostipes caccae strain L2 and galacto-oligosaccharides in a rat model. FEMS Microbiol Ecol 66:528–536. https://doi.org/10.1111/j.1574-6941.2008.00528.x
Scharlau D, Borowicki A, Habermann N, Hofmann T, Klenow S, Miene C, Munjal U, Stein K, Glei M (2009) Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat Res 682:39–53. https://doi.org/10.1016/j.mrrev.2009.04.001
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. https://doi.org/10.1186/gb-2011-12-6-r60
Song H, Wang W, Shen B, Jia H, Hou Z, Chen P, Sun Y (2018) Pretreatment with probiotic Bifico ameliorates colitis-associated cancer in mice: transcriptome and gut flora profiling. Cancer Sci 109:666–677. https://doi.org/10.1111/cas.13497
Tan J, Chen Y-X (2016) Dietary and lifestyle factors associated with colorectal cancer risk and interactions with microbiota: fiber, red or processed meat and alcoholic drinks. Gastrointest Tumors 3:17–24. https://doi.org/10.1159/000442831
Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, Arita M (2015) MS-DIAL: data independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods 12:523–526. https://doi.org/10.1038/nmeth.3393
Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuligni F, Gueimonde M, Margolles A, De Bellis G, O’Toole PW, van Sinderen D, Marchesi JR, Ventura M (2012) Diversity of bifidobacteria within the infant gut microbiota. PLoS One 7:e36957. https://doi.org/10.1371/journal.pone.0036957
Wang T-A, Zhang X-D, Guo X-Y, Xian S-L, Lu Y-F (2016) 3-bromopyruvate and sodium citrate target glycolysis, suppress survivin, and induce mitochondrial-mediated apoptosis in gastric cancer cells and inhibit gastric orthotopic transplantation tumor growth. Oncol Rep 35:1287–1296. https://doi.org/10.3892/or.2015.4511
Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, Knight R, Panda S (2018) Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat Commun 9:2872. https://doi.org/10.1038/s41467-018-05336-9
Zhu J, Zhu C, Ge S, Zhang M, Jiang L, Cui J, Ren F (2014) Lactobacillus salivarius Ren prevent the early colorectal carcinogenesis in 1, 2-dimethylhydrazine-induced rat model. J Appl Microbiol 117:208–216. https://doi.org/10.1111/jam.12499
Zierer J, Jackson MA, Kastenmüller G, Mangino M, Long T, Telenti A, Mohney RP, Small KS, Bell JT, Steves CJ, Valdes AM, Spector TD, Menni C (2018) The fecal metabolome as a functional readout of the gut microbiome. Nat Genet 50:790–795. https://doi.org/10.1038/s41588-018-0135-7
Zou S, Fang L, Lee M-H (2018) Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol Rep (Oxf) 6:1–12. https://doi.org/10.1093/gastro/gox031
Funding
This study was funded by the National Natural Science Foundation of China (grant nos. 81790631, 81570512), the National Key Research and Development Program of China (2018YFC2000500), and the Natural Science Foundation of Zhejiang Province, China (LQ19H030007).
Author information
Authors and Affiliations
Contributions
QW, KW, WW, and LL designed the experiment. XB, LY, QQW, YL, JY, and DF conducted the experiments. JJW, XJ, JX, and YML collected the sample. QW drafted the manuscript. LL reviewed the manuscript. All authors contributed to the project and reviewed the manuscripts.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Animal Care and Use Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (permit number: 2019-1072).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 93 kb)
Rights and permissions
About this article

Cite this article
Wang, Q., Wang, K., Wu, W. et al. Administration of Bifidobacterium bifidum CGMCC 15068 modulates gut microbiota and metabolome in azoxymethane (AOM)/dextran sulphate sodium (DSS)-induced colitis-associated colon cancer (CAC) in mice. Appl Microbiol Biotechnol 104, 5915–5928 (2020). https://doi.org/10.1007/s00253-020-10621-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10621-z