
Abstract
Foot-and-mouth disease virus (FMDV) has led to serious losses in the farming industry worldwide, particularly in cattle and swine. In developing countries, the control and eradication of FMD rely upon vaccination, in which the inactivated vaccine is predominant. In the preparation of inactivated vaccine, a series of purification methods were used to remove non-structural proteins (NSPs). It is necessary to develop a quantitative detection method of residual NSP and confirm a threshold value for the evaluation of the vaccine. Meanwhile, it is also important to develop a sensitive and rapid diagnostic method to distinguish infected animals from vaccinated animals (DIVA). In this study, three monoclonal antibodies (MAbs) against NSP 3ABC, designated 2G5, 9E2, and 1E10, were used. Subsequently, a series of overlapping peptides were expressed using a prokaryotic expression system to determine the minimal epitopes identified by the MAbs. Three linear B cell epitopes (BCEs), “92EYIEKA97” “23EGPYAGPLE31” and “209EPHH212”, were identified by MAbs 2G5, 9E2, and 1E10, respectively. Alanine-scanning mutagenesis analysis confirmed the critical amino acid in these epitopes. The epitope “92EYIEKA97” is located in 3A, which is deleted in some natural deletion mutants that result in a change in virus tropism. MAb 9E2 that identified the epitope “23EGPYAGPLE31” reacted with 3B1 and 3B2, but did not react with 3B3. In combination with sequence alignment analysis, the epitope “23EGPYAGPLE31” is highly conserved among different FMDV isolates. Preliminary screening using the known positive and negative sera indicated the MAb 9E2 has the potential for the development of a diagnostic method for DIVA. The residual NSP in inactivated vaccines can be detected using 9E2-HRP, which indicated the MAb 9E2 is able to evaluate inactivated vaccines. The four–amino acid epitope is the first reported to date that is recognized by 1E10. These results provide valuable insight into the diagnosis of DIVA and the NSP residual evaluation in inactivated vaccines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Foot-and-mouth disease (FMD) is a highly contagious and economically devastating viral disease of ruminants and swine, which seriously hinders the trade of animal and animal products. The causative agent, foot-and-mouth disease virus (FMDV), is a positive-sense and single-stranded RNA virus belonging to the Aphthovirus genus in the Picornaviridae family. Its genome contains 8400 nucleotides that code for four structural proteins (SPs: VP4, VP2, VP3, and VP1) and eight non-structural proteins (NSPs: L, 2A, 2B, 2C, 3A, 3B, 3C, and 3D). Some of these (e.g., 3ABC) exist as uncleaved precursors (Inoue et al. 2006). The SPs make up the shell of the virion, and the NSPs play a part in viral replication as well as interaction with the host cell (Jaworski et al. 2011; Liu et al. 2017b).
FMD control depends on vaccination with inactivated vaccines in enzootic countries and slaughtering the infected and susceptible animals in FMD-free countries (Fu et al. 2017). Generally, most NSPs are removed from the inactivated vaccine by a series of purification methods during production. Therefore, differentiation of FMDV-infected from inactivated vaccinated animals (DIVA) relies on the detection of antibodies against NSPs that exist only in infected animals. Thus far, many NSPs have been used as antigens in the development of the DIVA test. The 3D polymerase, with high immunogenicity and antigenicity across all serotypes of FMDV, has been investigated for DIVA. The results indicated that animals repeatedly vaccinated can produce 3D-specific antibodies (Mahajan et al. 2015; Sørensen et al. 1998). Therefore, 3D I-ELISA possesses a better diagnostic sensitivity but lower specificity in vaccinated animals so that it just can be adopted for seroepidemiological investigations in non-vaccinated regions (Mahajan et al. 2015). In addition, other NSPs have been used as antigens for DIVA, such as recombinant fusion proteins of 2B (Biswal et al. 2014), 2C (Meyer et al. 1997), 3A (Kumar et al. 2007), 3B (Mohapatra et al. 2014), 3AB (Mohapatra et al. 2011), 2C3AB (Lu et al. 2010), or 3ABC (Hosamani et al. 2015; Liu et al. 2017b). Due to the differences among species and individuals, not all NSP antibodies can be detected in all infected animals. Therefore, it has been proposed that a diagnostic method based on more than one NSP might improve the prospect of detecting and confirming infected animals (Brocchi et al. 2006; Mohapatra et al. 2014).
Antibodies against NSPs were detected in some repeatedly vaccinated cattle. This is induced by NSP contamination in the inactivated vaccine. Another possible explanation may be cross-reactivity with antibodies to other picornaviruses (Fan et al. 2006; Gao et al. 2012). In order to decrease cross-reactivity with other picornaviruses, some researchers have employed several peptides derived from the NSPs 2B, 2C, 3A, and 3B for DIVA. Table 1 shows that the peptides in previous research have good diagnostic performance.
In addition to being used for DIVA, the MAbs also can be used to detect NSP residual in inactivated vaccines in combination with chemiluminescence and nanotechnology. Therefore, it is necessary to identify and confirm the epitopes of 3ABC recognized by the antibodies. Generally, phage-displayed random peptide libraries, overlapping peptide fragments that were synthesized chemically or expressed as recombinant proteins in combination with ELISA or Western blot, were used to identify the minimal epitopes (Bi et al. 2019; Fu et al. 2017; Li et al. 2016; Liu et al. 2017a).
In the present study, three FMDV serotype-independent MAbs, named 2G5, 9E2, and 1E10, were generated against NSP 3ABC. The epitopes “92EYIEKA97” “23EGPYAGPLE31” and “209EPHH212” were identified using the GST-fusion protein and synthetic peptides. Results showed that the “92EYIEKA97” epitope was recognized by 2G5 and was located in 3A, which was deleted in some natural deletion mutants. The “23EGPYAGPLE31” epitope was recognized by 9E2 and was located in the 3B1 (the first Glu was located in 3A) and 3B2 (the first Glu was located in 3B1) regions, which was highly conserved among different FMDV isolates. The studies provide a guide for the diagnosis of DIVA and NSP residual evaluation in inactivated vaccines.
Materials and methods
Viruses, cells, and sera
FMDV O/NC/CHA/2010, O/BY/CHA/2010 (GeneBank associate JN998085), and A/GDMM/CHA/2013 used for indirect immunofluorescence assays (IFAs) were preserved at the Lanzhou Veterinary Research Institute (LVRI), Chinese Academy of Agricultural Sciences. Viruses were propagated in the baby hamster kidney cell line BHK-21 and harvested when the cytopathic effect was up to 80%. BHK-21 cells were cultured in Dulbecco’s modified eagle medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, BI) and antibiotics (100 U/mL of penicillin-streptomycin) at 37 °C under 5% CO2 atmosphere.
Thirty known positive and negative sera were collected from pig, sheep, and cattle and stored at − 20 °C. Among these sera, ten were collected from pigs containing five from pigs experimentally challenged with the O/Mya-98 FMDV strain, two from clinically healthy pigs, and three from pigs inoculated three times with the univalent inactivated vaccine FMDV O, A, and Asia I. Ten sera were collected from cattle containing three from cattle experimentally challenged with the O/Mya-98 FMDV strain, one from the A/GDMM/2013 strain, one from the Asia I/Js05 strain, three from clinically healthy cattle, and two from cattle inoculated with a univalent vaccine of FMDV O. Ten sera were collected from sheep containing five from clinically infected sheep, two from sheep inoculated three times with the univalent inactivated vaccine FMDV O, one from the univalent inactivated vaccine of FMDV A, and two from the univalent inactivated vaccine FMDV Asia I. The sera from experimentally challenged animals were collected in the Animal Biological Safety Level 3 (ABSL-3) Laboratory at LVRI. The sera from animals inoculated three times with the univalent inactivated vaccine FMDV O, A, and Asia I were prepared and stored in the laboratory. All procedures involving animals were approved by the Animal Ethics Committee of LVRI, Chinese Academy of Agricultural Sciences.
MAb production
A recombinant prokaryote expressing the NSP 3ABC was constructed in our laboratory. The expression and purification of the recombinant proteins were performed using the method previously described by Liu et al. (2017b). Inclusion bodies, the expression form of 3ABC recombinant proteins, were solubilized with IB solubilization buffer (20 mM NaH2PO4, 500 mM NaCl, 8 M urea, pH 8.0) and were purified by Ni nitrilotriacetic acid (Ni-NTA). Then, the purified protein was renatured using dialysis against a solution where the concentration of urea and imidazole was gradually reduced. In brief, the purified protein was loaded in the dialysis bag (Spectrum labs), and the dialysis bag was placed in renaturation buffer 1 (4 M urea, 200 mM imidazole, 0.1% Triton-100 in PBS buffer, pH 8.0) and refolded at 4 °C for 12 h. Then, the dialysis bag was transferred to renaturation buffer 2 (2 M urea, 100 mM imidazole, 0.1% Triton-100 in PBS buffer, pH 8.0) for 12 h and then buffer 3 (1 M urea, 50 mM imidazole in PBS buffer, pH 8.0) and finally into PBS (pH 8.0).
MAbs against the 3ABC renatured protein were produced by Wuhan GeneCreate Biological Engineering Company Limited (Wuhan, China). Positive hybridomas were sub-cultured three times by limiting dilution of cells. The selected clones were injected into the peritoneal cavities of Balb/c mice to obtain ascites. In brief, 3 days after the female Balb/c mice were treated with 0.5 mL/mouse Freund’s incomplete adjuvant, ~ 7.0 × 105 hybridoma cells were intraperitoneally injected into mice. After 9–10 days, ascites fluid was obtained from the peritoneal cavity of mice and purified by GenScript Biotech Corporation (Nanjing, China). The subclass specificity of mAbs was identified by the SBA Clonotyping System-HRP (SouthernBiotech).
Indirect immunofluorescence assay
BHK-21 cells were seeded into 6-well plates to produce monolayers. Cells were infected with FMDV O/NC/CHA/2010, O/BY/CHA/2010, or A/GDMM/CHA/2013 at a multiplicity of infection (MOI) of 0.1 for 8 h, respectively. Then, cells were rinsed with PBS and fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for 10 min. After blocking with 2% BSA for 1 h, the cells were incubated with 1 mL of hybridoma supernatant diluted (1:200) for 1 h. After washing three times with PBS, 1 mL of fluorescein isothiocyanate (FITC)–conjugated goat anti-mouse IgG (Abcam) at a dilution of 1:3000 was added to each well for 1 h at room temperature. Followed by washing three times with PBS, cells were then observed with a fluorescence microscope (Nikon).
Expression of the fusion protein
To accurately map the epitope position to the MAbs 2G5, 9E2, and 1E10, the non-structural proteins 3A, 3B, and 3C and a series of overlapping peptides were expressed in E. coli BL21 (DE3) using the expression vector pGEX-4T-1. The complete 3A, 3B, and 3C coding regions of FMDV A/GDMM/CHA/2013 were generated from the constructed plasmid of pProEXHTb-3ABC (full-length mutated) using the forward and reverse primers 3A F and R, 3B F and R, and 3C F and R and cloned into the pGEX-4T-1 by using BamH I and Xho I restriction sites. After being validated by DNA sequencing analysis, the constructed plasmid and pGEX-4T-1 null vector were transformed into BL21 (DE3) and were induced with 1 mM isopropyl-β-d-thiogalactoside (IPTG) for 6 h in LB medium. Pellets of bacterial cells were harvested by centrifugation at 6000×g for 5 min and then resuspended with PBS buffer and mixed with 4× loading buffer. The null vector was the control. Whole-cell lysates were analyzed by SDS-PAGE and western blot (WB) analysis.
Complementary primer pairs containing BamH I and Xho I restriction sites were synthesized. Each pair of primers was diluted to the working concentration and was equally mixed. The mixtures were annealed at 58 °C for 1 min and cloned into the pGEX-4T-1 previously digested with the same restriction endonucleases. After the products were validated by DNA sequencing analysis, protein expression was induced as above. All primers used are listed in Table S1–S3.
SDS-PAGE and Western blot
Approximately equal amounts of each GST-fusion protein were mixed with 4× loading buffer, boiled for 10 min, and then loaded onto 12% SDS-PAGE. The gel was stained with Coomassie blue staining solution or electrophoretically transferred to polyvinylidene difluoride (PVDF, Bio-Rad) membranes. After the membrane was blocked with 5% skimmed milk powder in PBST at ambient temperature for 2 h, the supernatant of hybridoma cell 2G5, 9E2, and 1E10 was diluted (1:40) and added to the above solution for overnight incubation at 4 °C. Membranes were washed five times with PBST and probed with a 1:5000 dilution of HRP-conjugated goat anti-mouse IgG (Sigma) for 1 h at ambient temperature. After washing five times, membranes were developed using chemiluminescence (ECL) reagents.
Critical residues in three epitopes responsible for MAb binding
To determine which residues are crucial in the epitopes recognized by the three MAbs, a panel of GST-fusion proteins were generated that each amino acid of three epitopes was substituted in turn by using alanine. These proteins were expressed and analyzed by WB to identify the crucial residues in the epitopes. The primers used are listed in Table S4.
Sequence comparisons
To demonstrate the conservation of the three epitopes among different serotypes of FMDV, 3ABC sequences were aligned using the ClustalW method. Twenty representative sequences from different serotypes were compared to show if the mutation occurred in these epitopes in order to determine which amino acids were mutated.
Selection of three MAbs binding to native epitopes
To determine the ability of the three MAbs binding to native epitopes, a competitive method was performed using checkerboard titration to determine the optimum coating concentrations of antigens, the optimum MAbs dilution, and the optimum serum dilution. Briefly, 96-well white plates (Corning) were coated with recombinant NSP 3ABC diluted in carbonate buffer (pH 9.6) and incubated at 4 °C overnight. After three washes with PBST, test sera from FMDV-infected (positive control) or non-infected (negative control) and MAbs were added to the plate in a volume of 100 μL per well (with 50 μL of sera and MAbs, respectively) and incubated at 37 °C for 30 min. After washing, a 1:100,000 dilution of HRP-conjugated goat anti-mouse IgG (Sigma) was added and incubated for 30 min. Finally, after five washes, 100 μL of chemiluminescence (CL) substrate was added. After the 5-min reaction, CL signals were measured using Varioskan lux (Thermo Scientific, USA). In the condition of optimum coating concentrations of antigens and the optimum serum dilution, the ratios of CL values of negative serum to positive serum (N/P) were calculated. A greater N/P ratio indicated that FMDV-infected sera have a greater ability to block the binding of the MAbs to NSP 3ABC. To verify the feasibility of the three MAbs for DIVA, the thirty sera from pig, sheep, and cattle known positive and negative sera were measured under the optimized conditions.
MAb 9E2 detection of NSP in the inactivated vaccine
To determine the ability of the MAb 9E2 to detect NSP of in the inactivated vaccine, a double-antibody sandwich method was performed using checkerboard titration. Briefly, 96-well white plates were coated with 0.5 μg/mL purified polyclonal antibody of NSP 3ABC diluted in carbonate buffer and incubated at 4 °C overnight. Then, the plates were sealed with blocking buffer (PBS containing 1% gelatin and 0.05% Tween-20) at 37 °C for 2 h. After three washes with PBST, different concentrations of NSP 3ABC were added to the plate in a volume of 100 μL per well and incubated at 37 °C for 30 min. After washing, a 1:10,000 dilution of 9E2-HRP was added and incubated for 30 min. CL signals were recorded as above, and a calibration curve was plotted.
To verify the feasibility of the method detecting NSP in the inactivated vaccine, three inactivated vaccines with different rounds of purification including one unpurified, one purified once, and one purified twice were measured using the above method. All data are the mean of the three measurements.
Results
Identification and characterization of MAbs
After purified and renatured, the 3ABC recombinant protein was delivered to the Wuhan GeneCreate Biological Engineering Company Limited to generate hybridoma cell secreting antibodies against NSP 3ABC of FMDV. Three hybridoma cells named 2G5, 9E2, and 1E10 were obtained. Antibody isotype analysis indicated that 2G5, 9E2, and 1E10 were identified as subclass IgG1, IgG1, and IgG2a, and three MAbs possessed κ-type light chains.
The specificity of three MAbs
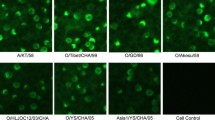
The specificity of three MAbs was verified using BHK-21 cells infected with FMDV O/NC/CHA/2010, O/BY/CHA/2010, or A/GDMM/CHA/2013 by IFA. IFA showed that MAbs 9E2 and 1E10 reacted with FMDV types A and O (Fig. 1), suggesting that the reactivity of MAbs 9E2 and 1E10 against FMDV was serotype-independent. MAb 2G5 did not react with FMDV types A and O in IFA.

Identification of MAbs 2G5, 9E2, and 1E10 by indirect immunofluorescence assay (IFA). The reaction of the three MAbs with BHK-21 cells infected with FMDV O and A isolates at a multiplicity of infection (MOI) of 0.1 for 8 h was submitted. The isolates of FMDV O/NC/CHA/2010, O/BY/CHA/2010, and A/GDMM/CHA/2013 were used in IFA. Non-infected BHK-21 were used as a negative control
To further confirm the specificity and the recognition sites, the GST-fused 3A, 3B, and 3C proteins were expressed and recognized using WB, indicating that the MAbs 2G5, 9E2, and 1E10 identified FMDV NSP 3A, 3B, and 3C, respectively, and demonstrated that these epitopes were linear (Fig. 2).

The specificity and the recognition sites of MAbs 2G5, 9E2, and 1E10. Approximately equal amounts of GST-fusion proteins were electrophoresed on 12% SDS-PAGE gels and then transferred to a nitrocellulose membrane and probed with the three MAbs as primary antibodies. GST control is shown in the first lane of the membrane
Mapping of the epitopes recognized by MAbs 2G5, 9E2, and 1E10
To identify the epitopes recognized by 2G5, 9E2, and 1E10, a series of overlapping peptide fragments (four on 3A, five on 3B, and four on 3C) spanning the 3A, 3B, and 3C were expressed and analyzed by WB. The results showed that the fusion peptides 3A-c, 3B-b, and 3C-d reacted with 2G5, 9E2 (all of the 3B-a, 3B-b, and 3B-c were recognized by 9E2, but the reactivity of 3B-b was strongest), and 1E10, respectively (Fig. 3a). Subsequently, the 3A-c fragment was further divided into four shorter fragments to analyze the reaction with 2G5. In addition, five shorter fragments on 3B-b and four shorter fragments on 3C-d were expressed and analyzed by WB. The results revealed that the epitopes recognized by 2G5, 9E2, and 1E10 were located in 3A-c-3, 3B-b-3, and 3C-d-5 (Fig. 3b).

Mapping of MAbs 2G5, 9E2, and 1E10 recognized epitopes. a, b A series of GST-fused overlapping peptides spanning the 3A, 3B, and 3C were expressed. Approximately equal amounts of lysates were electrophoresed and then transferred to nitrocellulose membrane and tested with MAbs in western blot (WB). c Approximately equal amount of GST-fused truncation peptides sequentially subtracted two or three amino acids from the N-terminus or the C-terminus to react with these MAbs on WB. d A series of GST-fused peptides containing terminator were expressed and analyzed using WB to confirm the minimal epitope. GST control is shown in the first lane of the membrane. A bullet denotes the terminator
To precisely map the epitopes, the series of 3A-c-3, 3B-b-3, and 3C-d-5 peptides were sequentially truncated by two or three amino acids from the N-terminus or the C-terminus. As shown in Fig. 3c, all fragments from 3A-c-3-a to 3A-c-3-d and 3A-c-3-f could react with MAb 2G5. In addition, a series of 3B-b-3 truncated peptides (3B-b-3-a, -b, -e) and 3C-d-5 truncated peptides (3C-d-5-f to 3C-d-5-j) were recognized by MAbs 9E2 and 1E10, respectively. Because the termination codon (TAA) was not included in the terminal of interesting peptides, the C-terminal of interesting peptides was connected with the carrier protein “LERPHRD.” Thus, two or three amino acids were added in the C-terminal region of interesting peptides and then one by one the amino acids for confirming the minimal core unit of epitope were deleted. The results indicated that the “92EYIEKA97” epitope was the minimal core unit recognized by MAb 2G5. The residue Ala97 of the 3A epitope can be substituted, but not deleted as shown in Fig. 3c and d. The “23EGPYAGPLE31” and “209EPHH212” epitopes were the minimal epitopes that were recognized by MAbs 9E2 and 1E10, respectively (Fig. 3d).
Crucial residues responsible for MAb binding
To identify which residues in these epitopes that contribute to antibody binding, a panel of alanine mutagenesis recombinant proteins were expressed and analyzed by WB. The results indicated that residues Glu92, Glu95, and Lys96 of the 3A epitope recognized by 2G5 were critical amino acids. Residues Tyr26 and Gly28 of the 3B epitope that recognized by 9E2 were critical amino acids, and residues Glu209, Pro210, His211, and His212 of the 3C epitope that recognized by 1E10 were critical amino acids. If the abovementioned mutations occurred, these epitopes will not react with their corresponding antibodies. When the Try93 residue of the 3A epitope and the Glu23 and Pro24 residues of the 3B epitope recognized by 2G5 and 9E2 respectively were replaced by Ala, the reactions between these epitopes and antibodies were significantly decreased (Fig. 4).

Alanine mutagenesis to determine the critical amino acids in the epitopes “92EYIEKA97” of 3A, “23EGPYAGPLE31” of 3B, and “209EPHH212” of 3C. GST-fused peptides with a single alanine substitution in the epitopes of 3A, 3B, and 3C were expressed, and approximately equal amounts of proteins were analyzed by WB. GST control is shown in the first lane of the membrane. A bullet denotes the terminator
Serological tests for FMDV NSP 3ABC and the identified epitopes
This study was designed to verify the immunogenic reactivity of FMDV NSP 3ABC and epitopes after FMDV infection and to further confirm minimal epitopes. We used the 3ABC recombinant protein or synthetic peptides EYIEKAC-OVA, EGPYAGPLEC-OVA, and EPHHC-OVA (OVA coupled in the C-terminal of the minimal epitopes) as coating antigens for the indirect ELISA. As shown in Fig. 5, in the 3ABC recombinant protein-coated and the peptide-coated ELISA assays, the OD value of FMDV-positive serum was significantly higher than that of FMDV-negative serum. The OD value of the MAbs was higher than that of the negative control. These results further indicated that the minimal epitopes are “92EYIEKA97” “23EGPYAGPLE31” and “209EPHH212”, and anti-FMDV serum recognized the three epitopes.

Recognition of minimal epitopes and serological detection. ELISA was performed using either 3ABC recombinant protein (a) or synthetic peptide coupled OVA EYIEKAC-OVA (b), EGPYAGPLEC-OVA (c), and EPHHC-OVA (d) to verify the reactivity of FMDV NSP 3ABC and novel epitopes using FMDV-infected serum. Inset shows the reactivity of 1E10 with three synthetic peptides 3AA, 3AC1, and 3AC2
The four–amino acid epitope that is recognized by MAb has not been reported thus far. To further confirm the epitope “209EPHH212”, we used synthetic peptides 3AA (DDAVNEYIEKANITTD), 3AC1 (DDAVNEPHHEANITTD), and 3AC2 (DDAVNEPHHANITTD), which were based on the NSP 3A region as frame structure recognized by 2G5, as coating antigens for the abovementioned indirect ELISA. The result showed that the MAb 1E10 reacted with 3AC1 (inset of the epitope “209EPHHE213” in the 3A region) and 3AC2 (inset of the epitope “209EPHH212” in the 3A region), but did not react with 3AA nor with the negative control (inset in Fig. 5). The wells of the negative control were coated with 3AC2 and then incubated with PBST without 1E10. After washing, HRP-conjugated goat anti-mouse IgG was added for detection.
The conservation of three epitopes among different isolates
Sequence alignment of three epitopes among different FMDV isolates was performed to demonstrate the conservation. As shown in Fig. 6, the linear epitope “92EYIEKA97” of 3A identified by 2G5 was relatively conserved among most isolates, but the epitope was absent in GD/CHA/JH12/2013. The critical amino acid Glu95 was replaced by Asp95 among some isolates of Asia I, which resulted in non-reactivity with 2G5. The linear epitope “23EGPYAGPLE31” that was present in 3B1, 3B2, and 3B3 was quite conserved in 3B1 and 3B2 among all isolates. The mutations of Ala4 and Leu7 in 3B1 (Glu0 was located in 3A) and Leu30 in 3B2 (Glu23 was located in 3B1) often happen among some isolates, but the mutations are not critical and do not affect the reaction. However, some amino acids (location 51, 54, and 55) in 3B3 were different from 3B1 and 3B2. The changes of all three amino acids resulted in non-reaction with 9E2, as shown in Fig. 3a (3B-d and 3B-e). The linear epitope “209EPHH212” was conserved in FMDV type A, O, and Asia I.

The conservation of three identified epitopes. Amino acid sequence of 3A, 3B, and 3C proteins covering the 2G5, 9E2, and1E10 recognized epitopes among different isolates were aligned
The N/P values of the three MAbs
A checkerboard titration was performed to identify whether or not the three MAbs could be applied to FMDV NSP diagnosis for DIVA. The results of the checkerboard titration indicated that the optimum concentrations of the coating antigens of 2G5, 9E2, and 1E10 were 0.25 μg/mL, 0.25 μg/mL, and 1 μg/mL, respectively. And the optimum serum dilution was 1:5. As shown in Fig. 7, when the concentration of 2G5, 9E2, and 1E10 was 0.25 μg/mL, 0.25 μg/mL, and 1 μg/mL, respectively, in the condition of optimum coating concentration and optimum serum dilution, the N/P values reached the highest levels. The N/P value of 1E10 was lower, compared with 2G5 and 9E2. To verify the feasibility of the competitive method, 30 samples from pig, sheep, and cattle known positive and negative sera were measured using the above-optimized methods. The preliminary results indicated that the MAbs 2G5 and 9E2 were able to differentiate between the positive and negative sera. However, a false negative was present using MAb 1E10.

The N/P values of the three MAbs at different dilutions in the condition of optimum coating concentrations of NSP 3ABC antigens (0.25 μg/mL of 2G5 and 9E2, 1 μg/mL of 1E10) and the optimum serum dilution (final concentration 1:5)
The detection of NSP in the inactivated vaccine using 9E2-HRP
Under the optimized conditions, the calibration curve was plotted against the NSP 3ABC concentration. As shown in Fig. 8, the chemiluminescence intensity (I) was linear in the 3ABC concentration range from 0.1 to 10 ng/mL. The detection limit (DL) at a signal/noise ratio of 3 (S/N = 3) was calculated according to the Eq. (1) (Shourian et al. 2015).
where SD is the standard deviation of the response and S is the slope of the calibration curve. The regression equation for 3ABC calibration curve was: I = 360.4 [3ABC] − 8.415 (R2 = 0.993, n = 3). The detection limit was calculated to be as low as 30 pg/mL.

The calibration curve for determination of 3ABC. The figure shows the linear range. The chemiluminescence intensity was recorded in different concentrations of 3ABC using the double-antibody sandwich
To verify the feasibility of the double-antibody sandwich method, three inactivated vaccines with different rounds of purification were measured using the method. The results showed that the NSP residual in the unpurified vaccine was 3.63 ng/mL, the NSP residual in the vaccine purified once was 0.42 ng/mL, and in the vaccine purified twice was 0.11 ng/mL. These data indicated that the NSP residual in inactivated vaccine purified twice was significantly reduced. Therefore, the method was able to detect the residual NSP in the inactivated vaccine.
Discussion
FMD is one of the most important economic diseases of livestock due to the restrictions on international trade in animal products and reduced animal productivity in FMD-infected countries (Jamal and Belsham 2013; Li et al. 2016). The DIVA test is of paramount importance for serological surveys, by providing evidence of FMD infection or lack of infection from FMD in vaccinated herds (Dreumel et al. 2015). The consensus is that 3ABC is considered the most reliable indicator of infection because it has high immunogenicity and can be induced to produce abundant antibodies that can persist longer in infected animals (Liu et al. 2017b; Robiolo et al. 2006). Therefore, a diagnostic method using MAbs against the NSP 3ABC for DIVA may show potential. In addition, MAbs can also be used to detect residual NSP in inactivated vaccines to confirm a threshold value of NSP. Therefore, epitope identification of NSP 3ABC can improve our understanding of these MAb characteristics and provide a basis for the development of MAb-based or epitopes-based diagnostic assays.
In the present research, we identified three linear BCEs on 3A, 3B, and 3C protein that were recognized by 2G5, 9E2, and 1E10, respectively. Minimal epitopes of the three MAbs (“92EYIEKA97” on 3A recognized by 2G5, “23EGPYAGPLE31” on 3B recognized by 9E2, and “209EPHH212” on 3C recognized by 1E10) were identified using the truncated peptides expressed as GST-fusion proteins (Fig. 3). In this case, GST was coupled at the N-terminal region of the minimal epitopes. The minimal epitopes were further confirmed by using synthetic peptides where the C-terminal was coupled with OVA (Fig. 5). The epitopes “109AEKNPLE115” that was recognized by MAb 3A24 (Fu et al. 2017) and “126ERTLP130” that reacted with MAb 3A10 (Wang et al. 2019) on NSP 3A reported are different from the epitope “92EYIEKA97” recognized by 2G5 in this study. The epitope “92EYIEKA97” in the study may be the subset of the reported peptide “NEYIEKANITTDDK” that reacted with sera of cattle infected with FMDV (Höhlich et al. 2003a, b). “92EYIEKA97” is relatively conserved among most isolates, except for a few isolates such as the GD/CHA/JH12/2013 isolate where the epitope was absent and a few Asia I isolates such as the Asia I/Jiangsu/2005 isolate and the FMDV HN/CHA/06 isolate in which the critical amino acid Glu95 was mutated (Fig. 6). Previous studies demonstrated that 3A plays an important role in host range and virulence (Pacheco et al. 2013; Stenfeldt et al. 2018; Yang et al. 1999). Some deletions such as the 93–102 natural deletion mutant and egg-adapted attenuated strains are associated with the tropism of FMDV. The deletion mutants severely affected the swine, but did not spread to the cattle (Yang et al. 1999). This is a consequence of reduced viral replication efficiency in bovine cells, rather than a difference in the host response (Pacheco et al. 2013; Stenfeldt et al. 2018). Due to the deletion and mutation of the epitope identified by MAb 2G5, the MAb is unsuitable to use alone for developing the diagnostic method for DIVA, though using the MAb can differentiate the known positive and negative sera in this study.
The three 3B proteins (3B1, 3B2, 3B3), which are 23 or 24 amino acids long, are similar but non-identical (Gao et al. 2016; Höhlich et al. 2003a). Three epitopes that were located in 3B were identified in previous studies, i.e., “QKPLK” that was the core motif of several B cell epitope (Höhlich et al. 2003a), “PxxGP” that was recognized by MAb 5D12 (Li et al. 2016), and “GPLERQ” that was reported by Chung (Chung et al. 2018). However, these are different from the epitope “23EGPYAGPLE31” recognized by 9E2 in this research. “23EGPYAGPLE31” is similar to the epitope “GPYAGPMER” that was previously reported by Fu et al. (2014). Although the minimal epitope was not identified in Fu’ research, the MAb was reported to react with 3B1 and 3B2, without 3B3, which is similar to the results presented here (Fig. 3a). Furthermore, the epitope “23EGPYAGPLE31” in the study may be the subset of peptides “GPYAGPLETQKPLK” and “GPYAGPMERQKPLK” that reacted with sera of cattle infected with FMDV (Höhlich et al. 2003a, b). “23EGPYAGPLE31” in the study that is located in 3B1 and 3B2, particularly in 3B2, is well-conserved among different serotypes of FMDV by using ClustalW for alignment (Fig. 6). The high N/P value, as well as the preliminary verification using the known positive and negative sera, indicated MAb 9E2 has the potential for developing the diagnostic method for DIVA. The double-antibody sandwich method using polyclonal antibody of NSP 3ABC as the capture antibody and 9E2-HRP as the detection antibody was developed. The method was verified using three inactivated vaccines with different rounds of purification. The results indicated that the residual NSP was detected in inactivated vaccines. However, the threshold value needs to be determined in further studies by establishing the correlation between residual NSP in the inactivated vaccines with different rounds of purification and the NSP antibody in animals that were inoculated different times with the corresponding inactivated vaccine for evaluation of the vaccine.
The epitope “209EPHH212” recognized by MAb 1E10 is located in the C-terminal of 3C (Fig. 6). In addition, this represents the first report of a four–amino acid epitope identified by the MAb. However, the reaction with antigen and N/P value was lower (Fig. 7).
In summary, three MAbs 2G5, 9E2, and 1E10 were produced against FMDV NSP 3ABC, and their corresponding linear BCEs were identified. Among the three identified epitopes, “92EYIEKA97”, recognized by 2G5 located in the NSP 3A, was deleted in some natural deletion mutants and egg-adapted attenuated strains. Hence, the MAb was unsuitable to be applied alone for DIVA. The epitope “23EGPYAGPLE31” which was recognized by 9E2 and located in NSP 3B was well-conserved in 3B2. This shows potential application value of MAb 9E2 for developing a diagnostic method for DIVA and detecting the residual NSP in the inactivated vaccine. This was initially verified using known positive and negative sera and inactivated vaccines with different rounds of purification. The four–amino acid epitope is the first reported that is identified by MAb 1E10. Such studies provide a foundation for diagnosis of DIVA and evaluation of inactivated vaccines.
References
Bi C, Shao Z, Li J, Weng C (2019) Identification of novel epitopes targeting non-structural protein 2 of PRRSV using monoclonal antibodies. Appl Microbiol Biotechnol 103(6):2689–2699
Biswal JK, Jena S, Mohapatra JK, Bisht P, Pattnaik B (2014) Detection of antibodies specific for foot-and-mouth disease virus infection using indirect ELISA based on recombinant nonstructural protein 2B. Arch Virol 159(7):1641–1650
Brocchi E, Bergmann IE, Dekker A, Paton DJ, Sammin DJ, Greiner M, Grazioli S, Simone FD, Yadin H, Haas B (2006) Comparative evaluation of six ELISAs for the detection of antibodies to the non-structural proteins of foot-and-mouth disease virus. Vaccine 24(47):6966–6979
Chung CJ, Clavijo A, Bounpheng MA, Uddowla S, Sayed A, Dancho B, Olesen IC, Pacheco J, Kamicker BJ, Brake DA, Bandaranayaka-Mudiyanselage CL, Lee SS, Rai DK, Rieder E (2018) An improved, rapid competitive ELISA using a novel conserved 3B epitope for the detection of serum antibodies to foot-and-mouth disease virus. J Vet Diagn Invest 30(5):699–707
Dreumel AKV, Michalski WP, Mcnabb LM, Shiell BJ, Singanallur NB, Peck GR (2015) Pan-serotype diagnostic for foot-and-mouth disease using the consensus antigen of nonstructural protein 3B. J Clin Microbiol 53(6):1797–1805
Fan L, Jong MH, Yang DW (2006) Presence of antibodies to non-structural proteins of foot-and-mouth disease virus in repeatedly vaccinated cattle. Vet Microbiol 115(1):14–20
Fu Y, Lu Z, Li P, Cao Y, Sun P, Tian M, Wang N, Bao H, Bai X, Li D (2014) Development of a blocking ELISA based on a monoclonal antibody against a predominant epitope in non-structural protein 3B2 of foot-and-mouth disease virus for differentiating infected from vaccinated animals. PLoS One 9(11):e111737
Fu Y, Li P, Cao Y, Wang N, Sun P, Shi Q, Ji X, Bao H, Li D, Chen Y (2017) Development of a blocking ELISA using a monoclonal antibody to a dominant epitope in non-structural protein 3A of foot-and-mouth disease virus, as a matching test for a negative-marker vaccine. PLoS One 12(1):e0170560
Gao M, Zhang R, Li M, Li S, Cao Y, Ma B, Wang J (2012) An ELISA based on the repeated foot-and-mouth disease virus 3B epitope peptide can distinguish infected and vaccinated cattle. Appl Microbiol Biotechnol 93(3):1271–1279
Gao Y, Sun SQ, Guo HC (2016) Biological function of foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol J 13(1):107
Höhlich BJ, Wiesmüller KH, Schlapp T, Haas B, Pfaff E, Saalmüller A (2003a) Identification of foot-and-mouth disease virus-specific linear B-cell epitopes to differentiate between infected and vaccinated cattle. J Virol 77(16):8633–8639
Höhlich BJ, Wiesmüller KH, Haas B, Gerner W, Correa R, Hehnen HR, Schlapp T, Pfaff E, Saalmüller A (2003b) Induction of an antigen-specific immune response and partial protection of cattle against challenge infection with foot-and-mouth disease virus (FMDV) after lipopeptide vaccination with FMDV-specific B-cell epitopes. J Gen Virol 84(Pt 12):3315–3324
Hosamani M, Basagoudanavar SH, Tamil Selvan RP, Das V, Ngangom P, Sreenivasa BP, Hegde R, Venkataramanan R (2015) A multi-species indirect ELISA for detection of non-structural protein 3ABC specific antibodies to foot-and-mouth disease virus. Arch Virol 160(4):937–944
Inoue T, Parida S, Paton DJ, Linchongsubongkoch W, Mackay D, Oh Y, Aunpomma D, Gubbins S, Saeki T (2006) Development and evaluation of an indirect enzyme-linked immunosorbent assay for detection of foot-and-mouth disease virus nonstructural protein antibody using a chemically synthesized 2B peptide as antigen. J Vet Diagn Investig 18(6):545–552
Jamal SM, Belsham GJ (2013) Foot-and-mouth disease: past, present and future. Vet Res 44(1):116
Jaworski JP, Compaired D, Trotta M, Perez M, Trono K, Fondevila N (2011) Validation of an r3AB1-FMDV-NSP ELISA to distinguish between cattle infected and vaccinated with foot-and-mouth disease virus. J Virol Methods 178(1-2):191–200
Kumar N, Sharma R, Kakker NK (2007) Non-structural protein 3A for differentiation of foot-and-mouth disease infected and vaccinated animals in Haryana (India). Zoonoses Public Health 54(9-10):376–382
Li C, Liang W, Liu W, Yang D, Wang H, Ma W, Zhou G, Yu L (2016) Identification of a conserved linear epitope using a monoclonal antibody against non-structural protein 3B of foot-and-mouth disease virus. Arch Virol 161(2):365–375
Liu W, Yang B, Wang M, Wang H, Yang D, Ma W, Zhou G, Yu L (2017a) Identification of a conformational neutralizing epitope on the VP1 protein of type A foot-and-mouth disease virus. Res Vet Sci 115:374–381
Liu Z, Shao J, Zhao F, Zhou G, Gao S, Liu W, Lv J, Li X, Li Y, Chang H (2017b) Chemiluminescence immunoassay for the detection of antibodies against the 2C and 3ABC nonstructural proteins induced by infecting pigs with the foot-and-mouth disease virus. Clin Vaccine Immunol 24(8):CVI.00153-17
Liu Z-Z, Zhao F-R, Gao S-D, Shao J-J, Zhang Y-G, Chang H-Y (2018) Development of a chemiluminescence immunoassay using recombinant non-structural epitope-based proteins to accurately differentiate foot-and-mouth disease virus-infected and vaccinated bovines. Transbound Emerg Dis 65(2):338–344
Lu ZJ, Zhang XL, Fu YF, Cao YM, Tian MN, Pu S, Dong L, Liu ZX, Xie QG (2010) Expression of the major epitope regions of 2C integrated with the 3AB non-structural protein of foot-and-mouth disease virus and its potential for differentiating infected from vaccinated animals. J Virol Methods 170(1-2):128–133
Mahajan S, Mohapatra JK, Pandey LK, Sharma GK, Pattnaik B (2015) Indirect ELISA using recombinant nonstructural protein 3D to detect foot and mouth disease virus infection associated antibodies. Biologicals 43(1):47–54
Meyer RF, Babcock GD, Newman JFE, Burrage TG, Toohey K, Lubroth J, Brown F (1997) Baculovirus expressed 2C of foot-and-mouth disease virus has the potential for differentiating convalescent from vaccinated animals. J Virol Methods 65(1):33–43
Mohapatra JK, Pandey LK, Sanyal A, Pattnaik B (2011) Recombinant non-structural polyprotein 3AB-based serodiagnostic strategy for FMD surveillance in bovines irrespective of vaccination. J Virol Methods 177(2):184–192
Mohapatra AK, Mohapatra JK, Pandey LK, Sanyal A, Pattnaik B (2014) Diagnostic potential of recombinant nonstructural protein 3B to detect antibodies induced by foot-and-mouth disease virus infection in bovines. Arch Virol 159(9):2359–2369
Oem JK, Kye SJ, Lee KN, Park JH, Kim YJ, Song HJ, Yeh M (2005) Development of synthetic peptide ELISA based on nonstructural protein 2C of foot and mouth disease virus. J Vet Sci 6(4):317–325
Pacheco JM, Gladue DP, Holinka LG, Arzt J, Bishop E, Smoliga G, Pauszek SJ, Bracht AJ, O’Donnell V, Fernandez-Sainz I (2013) A partial deletion in non-structural protein 3A can attenuate foot-and-mouth disease virus in cattle. Virology 446(1-2):260–267
Robiolo B, Seki C, Fondevilla N, Grigera P, Scodeller E, Periolo O, Torre JL, Mattion N (2006) Analysis of the immune response to FMDV structural and non-structural proteins in cattle in Argentina by the combined use of liquid phase and 3ABC-ELISA tests. Vaccine 24(7):997–1008
Shourian M, Ghourchian H, Boutorabi M (2015) Ultra-sensitive immunosensor for detection of hepatitis B surface antigen using multi-functionalized gold nanoparticles. Anal Chim Acta 895:1–11
Sørensen KJ, Madsen KG, Madsen ES, Salt JS, Nqindi J, Mackay DK (1998) Differentiation of infection from vaccination in foot-and-mouth disease by the detection of antibodies to the non-structural proteins 3D, 3AB and 3ABC in ELISA using antigens expressed in baculovirus. Arch Virol 143(8):1461–1476
Stenfeldt C, Arzt J, Pacheco JM, Gladue DP, Smoliga GR, Silva EB, Rodriguez LL, Borca MV (2018) A partial deletion within foot-and-mouth disease virus non-structural protein 3A causes clinical attenuation in cattle but does not prevent subclinical infection. Virology 516:115–126
Wang M, Xu Z, Liu W, Li M, Wang H, Yang D, Ma W, Zhou G, Yu L (2019) Identification of a conserved linear epitope using monoclonal antibody against non-structural protein 3A of foot-and-mouth disease virus with potential for differentiation between infected and vaccinated animals. Res Vet Sci 124:178–185
Yang PC, Chu RM, Chung WB, Sung HT (1999) Epidemiological characteristics and financial costs of the 1997 foot-and-mouth disease epidemic in Taiwan. Vet Rec 145:731–734
Yang SZ, Yang JF, Zhang GP, Qiao SL, Wang XN, Zhao D, Li XW, Deng RG, Zhi AM, You LM (2010) Development of a peptide-based immunochromatographic strip for differentiation of serotype O Foot-and-mouth disease virus-infected pigs from vaccinated pigs. J Vet Diagn Investig 22(3):412–415
Funding
This study was supported by grants from the National Key R&D Program of China (2017YFD0500902 and 2016YFE0204100).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All procedures involving animals were approved by the Animal Ethics Committee of LVRI, Chinese Academy of Agricultural Sciences.
Conflict of interest
The authors declare that they have no conflicting interests.
Ethics statement
All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 331 kb)
Rights and permissions
About this article

Cite this article
Liu, W., Shao, J., Chen, D. et al. Identification of three linear B cell epitopes against non-structural protein 3ABC of FMDV using monoclonal antibodies. Appl Microbiol Biotechnol 103, 8075–8086 (2019). https://doi.org/10.1007/s00253-019-10081-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10081-0