
Abstract
Five types of sulfate-reducing passive bioreactors with rice bran as substrate were operated at three different mine sites under various operating conditions to investigate and compare the dominant sulfate-reducing bacteria (SRBs) involved in acid mine drainage (AMD) treatment. In all bioreactors, AMD was properly treated under the national effluent standard of Japan when 16 samples in total were taken from different depths of the bioreactors at different sampling times. Analysis of the microbiomes in the five bioreactors by Illumina sequencing showed that Desulfosporosinus spp. were dominant SRBs in all bioreactors (the relative abundances were ~ 26.0% of the total population) regardless of reactor configurations, sizes, and operating conditions. This genus is known to comprise spore-forming, acid-tolerant, and oxygen-resistant SRBs with versatile metabolic capabilities. Microbial populations of AMD water and soil samples (as inocula) from the respective mine sites were also analyzed to investigate the origin of the genus Desulfosporosinus. Desulfosporosinus spp. were detectable in most AMD water samples, even at low relative abundances (0.0025 to 0.0069% of total AMD population), suggesting that the genus Desulfosporosinus is present within the AMD water that flows into the bioreactor. These data strongly imply that the passive treatment system is a versatile and widely applicable process for AMD treatment.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acid mine drainage (AMD) is generated by sulfide oxidation during exposure of mine tailings to oxygen and water in the mining and metallurgical industries and therefore has certain properties such as low pH, high sulfate, and metal concentrations. To avoid detrimental environmental impacts, various types of abiotic and biotic treatment systems have been developed for AMD remediation (Fu and Wang 2011; Kurniawan et al. 2006). Among these systems, passive treatment can offer advantages such as low operating costs and minimal external energy requirements (Skousen et al. 2017). One effective approach in passive treatment is sulfate-reducing bioreactors, in which sulfate-reducing bacteria (SRBs) reduce sulfate (electron acceptor) in AMD to hydrogen sulfide, which is concomitant with oxidation of low molecular carbon sources (electron donors). The resultantly formed hydrogen sulfide immobilizes dissolved metals by precipitating them as metal sulfide. Therefore, SRBs are key microbes in the performance of this AMD treatment process. According to a recent excellent review describing the diversity of SRBs in sulfidogenic bioreactors for AMD treatment, diverse SRB genera including Gram-negative mesophilic SRBs (Phylum: Proteobacteria, Class: Deltaproteobacteria) and Gram-positive spore-forming SRBs (Phylum: Firmicutes, Class: Clostridia) were detected in the bioreactors (Sánchez-Andrea et al. 2014). The important factors affecting the types of SRBs in bioreactors are thought to be the feed substrates, pH, and inocula of the process; however, the details remain unexplained.
Since AMD usually contains a very low organic carbon concentration (< 10 ppm), the addition of electron donors is necessary for both SRB activation and bioreactor performance (Zagury et al. 2006). When fed with ethanol, the dominant SRB genera were reported to include Desulfovibrio spp. and Desulfobacter spp. (Zhao et al. 2017) or Desulfovibrio spp. and Desulfomicrobium spp. (Hiibel et al. 2011). The Desulfovibrio-related operational taxonomic unit (OTU) could be detected in low-pH environments (Sánchez-Andrea et al. 2013), but is known to oxidize the substrate incompletely to acetate (Vita et al. 2015). Hence, additional important SRB species such as Desulfobacter spp. or Desulfomicrobium spp., which can completely oxidize acidified intermediates to CO2 (completely oxidizing SRB), were also enriched in addition to incompletely oxidizing Desulfovibrio spp. (Copeland et al. 2009; Widdel 1992). By contrast, in many sulfate-reducing bioreactors, complex organic substrates are used to promote the long-term activity of SRBs. The added organic substrates are often low cost or locally available materials; the effectiveness of these organic materials for sulfate reduction has been investigated (Skousen et al. 2017). Although the type of dissolved organic carbon from mixed organic substrates is suggested to play an important role in sulfate reduction (Neculita et al. 2011; Zhang and Wang 2014), little is known about the dynamics of SRB communities in passive bioreactors with complex organic substrates compared with those with simple substrates.
Sánchez-Andrea et al. (2014) described the importance of the choice of a suitable inoculum for sulfate reduction to remediate AMD. For example, when a neutrophilic inoculum is used as the source of SRBs in the bioreactor, reactor operation at lower pH values might depend on the adaptability of SRBs in the community (Bijmans et al. 2010). Meanwhile, when using an acidophilic inoculum from naturally extreme acidic environments, metal precipitation can be achieved under controlled low pH (2.2–2.5) (Ňancucheo and Johnson 2012). Therefore, it is important in AMD remediation to predict the origin of the predominant SRBs in the bioreactors.
Recently, we demonstrated the stable operation of a vertical down-flow packed bed bioreactor fed with rice bran as an organic material source (designated the Japan Oil, Gas and Metals National Corporation [JOGMEC] process; Sato et al. 2018). Because rice bran contains abundant carbohydrate and other nutrients (generally, 48.8% (w/w) carbohydrate (20.5% fiber), 19.6% fat, 13.4% protein, 4.4% minerals) and is commonly available in Japan as food waste, it is suitable as an organic substrate for the bioreactor. In the JOGMEC process, AMD flows downward through a layer of rice bran, and microbial degradation of the rice bran provides the substrates for SRBs, which are present in the lower layers of the bioreactor. Resultantly, continuous sulfate reduction and metal removal (Zn, Cu, and Cd) from AMD can be achieved for approximately 800 days (Sato et al. 2018; Aoyagi et al. 2018). Also, the effects of hydraulic retention time (HRT) and influent pH on reactor performance were investigated by process monitoring using a combination of chemical analyses and Illumina sequencing of 16S ribosomal RNA (rRNA) genes (Aoyagi et al. 2017). Although the microbial community degrading the rice bran was monitored in laboratory-scale JOGMEC processes (Aoyagi et al. 2017, 2018), a detailed analysis of the SRB community involved in the process has not been performed at pilot or laboratory scales.
The main objectives of this study were (1) to investigate and compare the dominant species of SRBs in the bioreactors at different test sites with varying conditions and (2) to surmise the source of the dominant SRBs in the bioreactors. To address these objectives, pilot- or laboratory-scale sulfate-reducing passive bioreactors following the JOGMEC process were operated at three different test sites, mine sites A, B, and C, with different parameters (e.g., scale, flow rate, pH), and a comparative study of the sulfidogenic microbiota among the bioreactors was performed using high-throughput Illumina MiSeq sequencing methods. Furthermore, the microbial community structures of AMD samples and soil samples around the respective mine sites were also analyzed.
Materials and methods
Sulfate-reducing bioreactors at three test sites and their operational conditions
The basic concept and design of the vertical flow sulfate-reducing bioreactor with rice bran as substrate (the JOGMEC process) was described in our previous report (Sato et al. 2018). Briefly, the reactor was first packed with a mixture of rice husks, limestone (3–20 mm in diameter), and field soil as inoculum. Then, rice bran was placed on top of this layer as an organic matter source, and the reactor was saturated with AMD. The reactor has several sampling ports at different heights in addition to the output port. The study areas of the three abandoned metal mine sites (sites A, B, and C) were located in the northern part of Japan. Table S1 describes the characteristics of the original AMD water and the respective AMD water after treatment under various conditions at each site.
Reactor setup at site A
A square-shaped bioreactor (A: volume 2 m3; Fig. S1A) was operated in which AMD was neutralized with limestone before being fed into the bioreactor. Since the raw AMD used in bioreactor A (leachate, pH 3.7) contains various heavy metals but no Fe (Table S1), no pretreatment system for Fe removal was equipped. The HRT was adjusted to 25 h (input flow rate 400 mL/min). AMD water samples from sampling port 1 and the output port were collected on February 22, 2017, and April 20, 2017 (71 and 128 operation days, respectively; four total samples: two time points and two depths) and used for both chemical and microbial community analyses.
Reactor setup at site B
Two square-shaped bioreactors (B1 and B2: volume 15 m3 each; Fig. S1B) were operated at an HRT of 25 h (input flow rate 2600 mL/min). Table S1 shows the characteristics of the original AMD at site B (mine water, pH 3.5) containing 35–40 mg/L of Fe, which was initially passed through an iron oxidation reactor (Fig. S1B). As a result, approximately 30–35 mg/L of Fe was removed before entering the sulfate-reducing bioreactors (Fig. S1B). In the first period of operation (November 21, 2016, to May 9, 2017), both bioreactors, B1 and B2, were operated under neutral conditions with limestone (pH 6.3). Subsequently, the bioreactors had mechanical issues, and thus were fixed and re-started. In the second period of operation (from May 10, 2017), reactor B1 was operated under acidic conditions using AMD (pH 3.5), while reactor B2 was operated under neutral conditions (pH 6.3). AMD water samples from sampling ports 1 and 4 were collected on March 15, 2017, and May 30, 2017 (day 114 of the first period and day 28 of the second period (after the re-start), respectively; eight total samples: two time points, two conditions, and two depths).
Reactor setup at site C
Two column bioreactors (C1 and C2: volume 0.03 m3 each; Fig. S1C) were operated under neutral conditions. Reactor C1 was operated at an HRT of 12.5 h (input flow rate 13 mL/min); reactor C2 was operated at an HRT of 6 h (26 mL/min). As the raw AMD used in the reactors (raw water; i.e., a mixture of leachate and mine water; pH 3.4) contained only 2–4 mg/L of Fe (Table S1), there was no pretreatment system for Fe removal. AMD samples (pH 3.9) from the upper and lower parts of the reactor were collected on March 27, 2017, after 160 days of continuous operation (four total samples: two HRT conditions and two depths).
DNA extraction and Illumina sequencing
For 16S rRNA gene amplicon sequencing, DNA samples were prepared for 16 samples from two reactor depths (upper and lower) of the three reactors types, six AMD water samples, and three soil samples (as inoculum) around each site. All samples were prepared in triplicate and the sequencing analyses were performed using the triplicate samples. DNA was extracted from each sample using a direct lysis protocol with bead beating (Noll et al. 2005). The V4 region of the 16S rRNA gene was amplified using the universal primers 515F and 806R, both of which were modified to contain an Illumina adapter region, with the latter primer also containing a 12-bp barcode for multiplex sequencing (Caporaso et al. 2012). Following PCR amplification and purification of the resultant amplicons (Hori et al. 2015; Navarro et al. 2015; Sato et al. 2016), the barcode-labeled DNA library and an initial control (PhiX; Illumina, San Diego, CA, USA) were subjected to paired-end sequencing using a 500-cycle MiSeq Reagent kit (Illumina) with a MiSeq sequencer (Illumina).
Sequence data analysis
The paired-end sequences were assessed for quality (i.e., removal of low quality (Q < 30), chimeric, and PhiX sequences) and assembled as described elsewhere (Itoh et al. 2014; Aoyagi et al. 2015). QIIME (Caporaso et al. 2010) was used for taxonomic classification and OTU clustering at the 97% identity level. A single sequence was selected from each respective OTU and taxonomically assigned using the Blast with the NCBI nucleotide database (https://blast.ncbi.nlm.nih.gov). Alpha diversity indices (i.e., Chao1 and Simpson reciprocal) of the Illumina sequence data were determined using QIIME software based on the same number of sequences (n = 33,390) for each reactor sample (Table S3) (Fig. 1). Community variation among the samples was evaluated by principal coordinate analysis (PCoA) of the Illumina sequence data based on weighted UniFrac distances calculated by QIIME software. SRB-related OTUs were extracted using an in-house script. Briefly, OTUs were assigned as SRBs if the binominal name of the OTU was identical to one of the 953 species whose dissimilatory sulfite reductase genes are registered in the RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/). In Fig. 2c, the relative abundances of OTUs with identical taxonomies were merged into one.

Diversity of microbial communities in the pilot- and laboratory-scale bioreactors operated at three different mine sites. The α-diversity indices were calculated based on Illumina sequencing data of 16S ribosomal RNA (rRNA) genes (16 samples) from each depth of the bioreactors. Each diversity index, Chao1 (a) and Simpson reciprocal (b), was calculated based on an equivalent number of sequences (n = 33,390) subsampled from the original libraries. A principal coordinate analysis (PCoA) scatter plot of 16S rRNA sequences (c) based on the weighted UniFrac distances was obtained by Illumina sequencing of the 16 samples. Details of the sampling sites (sites A–C), bioreactors (A, B1, B2, C1, and C2), sampling times (January to June), and respective operation conditions are described in the text

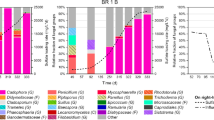
Microbial compositions at class (a) and genus (b) level, and the relative abundances of sulfate-reducing bacteria (SRB)-related operational taxonomic units (OTUs) (c) in the pilot- and laboratory-scale bioreactors operated at three different mine sites. The phylogenetic groups (class or genus) or SRB species names are indicated by color and their taxonomies are shown to the right of the graphs. Details of the sampling sites (sites A–C), bioreactors (A, B1, B2, C1, and C2), sampling times (January to June), and respective operation conditions are described in the text. SRB-related OTUs were selected using an in-house script in which OTUs were assigned one of 953 binominal names of microorganisms possessing a dissimilatory sulfite reductase gene registered in the RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/). SRB-related OTUs with identical taxonomies were merged; Desulfosporosinus meridiei contains OTU 10. Mean values of three replicates were used in all graphs
The nucleotide sequences obtained from the Illumina sequencing analyses based on 16S rRNA genes have been deposited in the DDBJ Sequence Read Archive under accession code DRA007949.
Results
Bioreactor performances
For site A, stable operation of the bioreactor was confirmed in February, showing a sulfate removal ratio of 13% (average value of n = 20), a Zn concentration of 0.54 mg/L in the output port (n = 20), and an oxidation-reduction potential (ORP) value of − 132 mV in the output port (n = 20). By contrast, in April, the sulfate removal ratio (9.2%, n = 20) and Zn concentration in the output port (0.44 mg/L, n = 20) were stable, but the ORP value in the output port was + 153 mV (n = 20), suggesting the possibility that a reducing atmosphere was not maintained in the lower part of the bioreactor.
The average values (n = 20) of sulfate removal ratio, Zn concentration, and ORP for site B are shown in Table S2. Although the operation condition of reactor B1 had been changed to acidic (pH 3.5) from neutral (pH 6.3) after the mechanical issue, both reactors B1 and B2 showed stable performances.
As to site C, stable operation of the bioreactor was confirmed during the period, and the average values of sulfate removal ratio, Zn concentration, and ORP were 20% (n = 11), 0.86 mg/L (n = 11), and − 175 mV (n = 15), respectively, in the lower part.
16S rRNA gene amplicon sequences
Approximately 11.4 million sequences were obtained in total from the 16 reactor samples, corresponding to an average of 210,731 sequences per library (minimum, 130,152; maximum, 967,640; standard deviation, 108,247) (Table S3). By contrast, approximately 7.5 million sequences were obtained in total from the six AMD samples and three soil samples, corresponding to an average of 278,709 sequences per library (minimum, 188,654; maximum, 465,146; standard deviation, 47,174) (Table S4).
Comparison of microbial diversity among the bioreactors at different test sites under various conditions
Microbial diversity of each sample was evaluated by calculating alpha diversity indices and weighted UniFrac distances for PCoA. Although the values of Chao1 indices, which denote the predicted species richness, tended to increase according to the site order, from A to C, few differences were observed among the different sites and conditions (Fig. 1a and b). The PCoA scatter plot indicated the shifts in microbial communities associated with depth direction from the upper part to the lower part (Fig. 1c). For example, in site A (both February and April), the plots for port 1 were clustered at the left side, but shifted to the middle for the lower part of the reactor (output port). The distance between the top and output ports was longer in site A than in sites B and C, implying that the microbial communities in these reactor positions changed greatly (Fig. 1c). By contrast, at sites B and C, the plots shifted from the upper part to the lower part of the plot with the depth direction of each reactor, and the distance between the ports was relatively shorter, compared with that in site A (Fig. 1c). These results indicate that the shifting trends were only similar within the same site, while differing among sites.
Microbial community structures in the bioreactors
The microbial compositions of the respective bioreactors were compared with class (Fig. 2a) and genus levels (Fig. 2b). Although the bioreactors at the different mine sites were operated under various conditions in terms of reactor type, scale, pH, and AMD content, the microbial compositions at the class level appeared to be similar in most samples. The classes Clostridia (phylum Firmicutes) and Bacteroidia (phylum Bacteroidetes) highly dominated, accounting for 10.4% and 48.6% of the total population at site A; 31.7% and 81.9% at site B; and 24.7% and 49.6% at site C, respectively (Fig. 2a). In addition to Clostridia and Bacteroidia, Gammaproteobacteria were also dominant, accounting for 51.8% and 59.4% of the total populations at sites A and C, respectively.
The six most dominant OTUs in bioreactors A, B1, B2, C1, and C2 are listed in Table 1. At site A, three of the six dominant OTUs belonged to the class Bacteroidia, which is often observed within reactors of the JOGMEC process (Aoyagi et al. 2017, 2018; Sato et al. 2018). In bioreactors B1 and B2, all six dominant OTUs belonged to the class Bacteroidia or Clostridia. Similarly, in bioreactors C1 and C2, the six most dominant OTUs were related to the classes Bacteroidia, Clostridia, or Gammaproteobacteria.
By contrast, the most dominant OTU at site A was related to a Sulfuricurvum kujiense (Epsilonproteobacteria; abundance 35.2% in February and 14.6% in April) and the fifth dominant OTU was a Pseudomonas sp. (Gammaproteobacteria). S. kujiense (OTU 1) is capable of sulfur oxidation, which is the reverse reaction of sulfate reduction, the key metabolic pathway in the passive treatment process (Han et al. 2012). As the S. kujiense dominated only in site A, in which the reactor performance became unstable (increased ORP value), the predominance of sulfur-oxidizing bacteria may indicate an insufficiency in the reactor performance. On the other hand, Pseudomonas spp. (OTU 5) are known to secret siderophores that bind not only Fe3+ but also Co2+, Zn2+, Mn2+, Ni2+, and Ga3+ (Ferreira et al. 2018), and are capable of biofilm formation, which is a major strategy for resisting heavy metal toxicity (Giovanella et al. 2017). This OTU was presumed to play roles in the detoxification of metal ions.
The community structures of SRB species in the bioreactors at respective mine sites were revealed by the detailed analysis on sequence data, demonstrating that the relative abundances of SRBs varied among test sites and operational conditions (Fig. 2c). In all samples analyzed, the most dominant SRB-like OTU was related to Desulfosporosinus meridiei (> 97% sequence identity; determined using the Greengenes database), accounting for 27.3 to 87.0% of each total SRB-like population. In addition to the genus Desulfosporosinus, the genera Clostridium, Ruminococcus, Desulfovibrio, and Desulfobulbus were also detected; these five genera accounted for 100% of the total SRB-like population in the bioreactors. The OTU belonging to genus Desulfosporosinus (OTU 10, Desulfosporosinus fructosivorans) was also found as the sixth most dominant OTU at site B (Table 1), which accounted for 5.7% in March and 4.2% in June at site B1, and 2.8% in March and 4.6% in June at site B2. Similarly, OTU 10 was detected as the third most dominant species at cite C (relative abundances 3.2% at C1 and 9.0% at C2)
Microbial community structure in AMD and soil samples at each test site
Since the SRB communities in the JOGMEC process are thought to be derived mainly from AMD (acidic environment) and/or soil (neutral environment) samples, it is important to predict the origins of SRB inocula for AMD remediation. Alpha diversity indices (i.e., Chao1 and Simpson reciprocal) of the Illumina sequence data were determined based on the same number of sequences (n = 36,560) for AMD and soil samples (Table S4). The diversity indices varied among the respective AMD samples at each site, but the microbial communities of the soil samples were clearly more diverse than those of the AMD samples, especially in terms of community evenness (Fig. 3a and b, Simpson reciprocal). Lower diversity of AMD samples compared with soil samples might reflect the extreme environmental conditions of AMD (approximately pH 3.5; organic carbon concentration < 10 ppm). In the PCoA scatter plots, the AMD and soil samples were separated from each other, but all plots of soil sample communities at the three different sites were located close together (Fig. 3c). By contrast, the microbial community structures of AMD samples were suggested to differ from one another (Fig. 3c).

Diversity of microbial communities in acid mine drainage (AMD) water and soils at three different mine sites. The α-diversity indices were calculated based on Illumina sequencing data of 16S rRNA genes (nine samples) from respective AMD and soil samples. Each diversity index, Chao1 (a) and Simpson reciprocal (b), was calculated based on an equivalent number of sequences (n = 36,560) subsampled from the original libraries. A PCoA scatter plot of 16S rRNA sequences (c) based on the weighted UniFrac distances was obtained by Illumina sequencing of the nine samples. In the graph of the 10 most predominant OTUs of respective AMD and soil samples (d), phylogenetic information (with sequence identities) of the closest related species are shown to the right. Mean values of three replicates were used in panels a, b, and d, while the respective three values were plotted in panel c. Bars indicate standard deviation of the replicates in panels a and b.
The dominant members of the microbial communities of the respective AMD and soil samples at the OTU level were compared. The 10 most dominant OTUs are shown in Fig. 3d. Although the respective AMD samples were obtained from different sites and locations, the autotrophic iron-oxidizing genus Gallionella highly dominated, accounting for 14.2% of the total population in AMD samples at site A, 88.3% at site B, and 30.6% at site C. By contrast, in AMD2 samples at site A, Herminiimonas spp., belonging to the family Oxalobacteraceae, accounted for approximately 34.3% of the total.
To investigate whether the dominant SRB in the JOGMEC process, Desulfosporosinus sp., was derived from AMD or soil harvested around the respective mine sites, the relative abundances of the genus in both samples were analyzed (Table 2). Except for the AMD2 sample from site B, OTU 10 and OTU 17, both of which were related to Desulfosporosinus spp., were detected in most AMD samples (relative abundance 0.0025–0.0069%). The Blast analysis revealed that OTU 10 and OTU 17 shared 100% identity with Desulfosporosinus fructosivorans strain 63.6F and Desulfosporosinus sp. strain 063, respectively.
Discussion
Microbial communities in the five sulfate-reducing passive bioreactors operated at three different mine sites were investigated using Illumina sequencing. The classes Clostridia and Bacteroidia were found to highly dominate most of the microbial communities. Previously, we reported that the phylum Firmicutes and Bacteroidetes, in which Clostridia and Bacteroidia affiliated, respectively, accumulated in lower parts of the bioreactor during stable operation of the JOGMEC process when treating both neutral and acidic AMD. Because they are capable of degrading complex organic matters, e.g., rice bran, their predomination might lead to the metabolic activation of SRBs by providing low-molecular-weight organic substances and to resultant stable reactor performance conditions (Aoyagi et al. 2017). In this study, relatively stable bioreactor operations were observed at respective sampling times, except for site A in April. Therefore, the microbial community data may reflect the satisfactory performances of the respective bioreactors.
As for SRB communities, even though SRBs are usually not a major component of the total population in the AMD bioreactors (approximately < 2% of the total) (Baldwin et al. 2016), accumulation of 10 to 30% SRBs was observed in some samples in this study (e.g., Fig. 2c, sample B1-Jun-1). Baldwin et al. (2016) reported that SRBs in bioreactors with a local pulp mill for AMD remediation were classified into the orders Desulfobacterales, Desulfovibrionales, Desulfarculales, and Desulfuromonadales, all of which belong to the class Deltaproteobacteria. However, in the JOGMEC process at the three mine sites under different conditions, the genera Desulfosporosinus, Clostridium, and Ruminococcus, which belong to the class Clostridia, were commonly found to be highly dominant. Although Desulfosporosinus spp. were detected in some anaerobic bioreactors treating AMD (Sánchez-Andrea et al. 2014), this is the first report on sulfate-reducing passive bioreactors highly accumulated by the genus.
Desulfosporosinus spp. form spores, enabling survival for months under low pH, dryness, and oxic conditions (Widdel 1992; Kimura et al. 2006), and are considered to be important players in sulfate reduction in acidic mine wastes (Mardanov et al. 2016). Several studies have detected Desulfosporosinus in acidic mine environments, and notably, Desulfosporosinus was the only phylotype with the known capability to reduce sulfate in the oxidized mine waste materials at the Berikul site (Karnachuk et al. 2009). Among the four complete and four draft genomes of Desulfosporosinus reported so far (Abicht et al. 2011; Pester et al. 2012; Abu et al. 2015; Petzsch et al. 2015), only Desulfosporosinus sp. strain I2 has been analyzed with respect to its metabolism and biological properties in detail based on the genome information (Mardanov et al. 2016). Although sulfate reduction was thought to occur under anoxic conditions, this acidophilic and copper-tolerant Desulfosporosinus strain was isolated from oxidized layers of gold mine tailings, and multiple oxygen detoxification systems were identified in its genome, such as superoxide dismutase and catalase genes that are typically found in aerobic bacteria (Mardanov et al. 2016). Interestingly, the genes encoding components of cytochrome c oxidase, a key system for aerobic respiration, were also found in the Desulfosporosinus genome. In the JOGMEC processes operated at the three mine sites, especially the two pilot-scale passive bioreactors at sites A and B, oxygen can be contaminated in the upper part of the reactors because the processes have no rigid lids, as necessitated for their easier handling at the respective sites. One possible reason for the dominance of the genus Desulfosporosinus involves such unique properties of sulfate reduction that may occur beyond strictly anaerobic conditions. In addition, in the JOGMEC processes using rice bran as a substrate, various metabolites can be produced from rice bran degradation within the bioreactors. Hence, another possible reason for the dominance of the genus Desulfosporosinus involves its metabolically versatile lifestyle, as the genus can use a wide variety of substrates as electron donors for sulfate reduction (Mardanov et al. 2016). Recently, other group has also reported that although abundance was low, Desulfosporosinus spp. were detected in various semi-passive bioreactors (Rezadehbashi and Baldwin 2018). Furthermore, Desulfosporosinus spp. have been demonstrated to contribute to sulfate reduction at high rates even though their relative abundance was quite low (0.006%) (Pester et al. 2010). Considering their high abundances in the bioreactors presented here, contributions of Desulfosporosinus to sulfate reduction in the JOGMEC process would also be vital.
In this study, microbial communities in AMD and soil samples at each test site were also investigated. The dominant OTUs include metal-oxidizing bacteria such as Gallionella sp. and Herminiimonas sp. (Fig. 3d), which are well-known iron oxidizer and arsenite oxidizer, respectively (Volant et al. 2014; Koh et al. 2017). While investigation of the relative abundances of the Desulfosporosinus-related OTUs in both AMD and soil samples demonstrated that Desulfosporosinus was present in most of AMD samples (Table 2). Actually, several members of Desulfosporosinus have been detected in low pH, metal-rich environments (Sánchez-Andrea et al. 2011, 2014; Rezadehbashi and Baldwin 2018), supporting our findings. By contrast, the genus Desulfosporosinus could not be detected in most soil samples at our sequencing depth, possibly due to the more diverse microbiota in soils compared with AMD water. However, Desulfosporosinus sp. seems exist in soil samples also, as it was found in the soil sample at site B (Table 2). Altogether, we could not conclude whether the Desulfosporosinus spp. in the bioreactors were mainly derived from AMD or soil samples. However, the results may indicate that the sulfate-reducing bioreactor with rice bran as substrate is a versatile and widely applicable process for AMD treatment even without any inoculum, because the AMD samples analyzed in this study contained Desulfosporosinus spp. surviving under low pH and oxic conditions, which is the key SRB of the process during AMD treatment. Detection of the genus in the bioreactor can be indicative of stable operation of the process.
In conclusion, pilot- and laboratory-scale sulfate-reducing bioreactors with rice bran as substrate were operated at three different mine sites with various reactor sizes and under different conditions, and the high dominance of the genus Desulfosporosinus indicates that Desulfosporosinus spp. is key SRBs in the bioreactors during AMD treatment (< 160-day operation). This is likely because the genus has such properties as acid tolerance, oxygen resistance, and versatile metabolism. In addition, Desulfosporosinus spp. were also detected in most AMD samples, suggesting that an SRB inoculum may not be necessary if the type of AMD water used in this study flows into the bioreactor. Periodic fine-scale investigations of the SRB community structure during longer-term operations (> 160 days) will be necessary to reveal the changes in the types and roles of key SRBs in the JOGMEC process.
References
Abicht HK, Mancini S, Karnachuk OV, Solioz M (2011) Genome sequence of Desulfosporosinus sp. OT, an acidophilic sulfate reducing bacterium from copper mining waste in Norilsk, Northern Siberia. J Bacteriol 193:6104–6105
Abu LN, Tan B, Dao A, Foght J (2015) Draft genome sequence of uncultured Desulfosporosinus sp. strain Tol-M, obtained by stable isotope probing using [13C6] toluene. Genome Announc 3(1):e01422-14
Aoyagi T, Kimura M, Yamada N, Navarro RR, Itoh H, Ogata A, Sakoda A, Katayama Y, Takasaki M, Hori T (2015) Dynamic transition of chemolithotrophic sulfur-oxidizing bacteria in response to amendment with nitrate in deposited marine sediments. Front Microbiol 6:426
Aoyagi T, Hamai T, Hori T, Sato Y, Kobayashi M, Sato Y, Kobayashi M, Sato Y, Inaba T, Ogata A, Habe H, Sakata T (2017) Hydraulic retention time and pH affect the performance and microbial communities of passive bioreactor for treatment of acid mine drainage. AMB Express 7:142
Aoyagi T, Hamai T, Hori T, Sato Y, Kobayashi M, Sato Y, Inaba T, Ogata A, Habe H, Sakata T (2018) Microbial community analysis of sulfate-reducing passive bioreactor for treating acid mine drainage under failure conditions after long-term continuous operation. J Environ Chem Eng 6:5795–5800
Baldwin SA, Mattes A, Rezadehbashi M, Taylor J (2016) Seasonal microbial population shifts in a bioremediation system treating metal and sulfate-rich seepage. Minerals 6:36
Bijmans MFM, de Vries E, Yang CH, Buisman CJN, Lens PLN, Dopson M (2010) Sulfate reduction at pH 4.0 for treatment of process and wastewaters. Biotechnol Prog 26:1029–1037
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Copeland A, Spring S, Göker M, Schneider S, Lapidus A, Del Rio TG, Tice H, Cheng JF, Chen F, Nolan M, Bruce D, Goodwin L, Pitluck S, Ivanova N, Mavrommatis K, Ovchinnikova G, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CC, Meincke L, Sims D, Brettin T, Detter JC, Han C, Chain P, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP, Lucas S (2009) Complete genome sequence of Desulfomicrobium baculatum type strain (XT). Stand Genomic Sci 1:29–37
Ferreira ML, Ramirez SA, Vullo DL (2018) Chemical characterization and ligand behavior of Pseudomonas veronii 2E siderophores. World J Microbiol Biotechnol 34:134
Fu F, Wang Q (2011) Removal of heavy metal ions from wastewaters: A review. J Environ Manag 92:407–418
Giovanella P, Cabral L, Costa AP, de Oliveira Camargo FA, Gianello C, Bento FM (2017) Metal resistance mechanisms in Gram-negative bacteria and their potential to remove Hg in the presence of other metals. Ecotoxicol Environ Saf 140:162–169
Han C, Kotsyurbenko O, Chertkov O, Held B, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, Goodwin LA, Pitluck S, Liolios K, Pagani I, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Brambilla EM, Rohde M, Spring S, Sikorski J, Göker M, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP, Detter JC (2012) Complete genome sequence of the sulfur compounds oxidizing chemolithoautotroph Sulfuricurvum kujiense type strain (YK-1T). Stand Genomic Sci 6:94–103
Hiibel SR, Pereyra LP, Riquelme Breazeal MV, Reisman DJ, Reardon KF, Pruden A (2011) Effect of organic substrate on the microbial community structure in pilot-scale sulfate-reducing biochemical reactors treating mine drainage. Environ Eng Sci 28:563–571
Hori T, Aoyagi T, Itoh H, Narihiro T, Oikawa A, Suzuki K, Ogata A, Friedrich MW, Conrad R, Kamagata Y (2015) Isolation of microorganisms involved in reduction of crystalline iron(III) oxides in natural environments. Front Microbiol 6:386
Itoh H, Navarro R, Takeshita K, Tago K, Hayatsu M, Hori T, Kikuchi Y (2014) Bacterial population succession and adaptation affected by insecticide application and soil spraying history. Front Microbiol 5:457
Karnachuk OV, Gerasimchuk AL, Banks D, Frengstad B, Stykon GA, Tikhonova ZL, Kaksonen A, Puhakka J, Yanenko AS, Pimenov NV (2009) Bacteria of the sulfur cycle in the sediments of gold mine tailings, Kuznetsk Basin, Russia. Microbiology 78:483–491
Kimura S, Hallberg KB, Johnson DB (2006) Sulfidogenesis in low pH (3.8–4.2) media by a mixed population of acidophilic bacteria. Biodegradation 17:159–167
Koh H-W, Hur M, Kang M-S, Ku Y-B, Ghai R, Park S-J (2017) Physiological and genomic insights into the lifestyle of arsenite-oxidizing Herminiimonas arsenitoxidans. Sci Rep 7:15077
Kurniawan TA, Chan GYS, Lo W-H, Babel S (2006) Physico-chemical treatment technologies for wastewater laden with heavy metals. Chem Eng J 118:83–98
Mardanov AV, Panova IA, Beletsky AV, Avakyan MR, Kadnikov VV, Antsiferov DV, Banks D, Frank YA, Pimenov NV, Ravin NV, Karnachuk OV (2016) Genomic insights into a new acidophilic, copper-resistant Desulfosporosinus isolate from the oxidized tailings area of an abandoned gold mine. FEMS Microbiol Ecol 92:fiw11
Ňancucheo I, Johnson DB (2012) Selective removal of transition metals from acidic mine waters by novel consortia of acidophilic sulfidogenic bacteria. Microb Biotechnol 5:34–44
Navarro RR, Aoyagi T, Kimura M, Itoh H, Sato Y, Kikuchi Y, Ogata A, Hori T (2015) High-resolution dynamics of microbial communities during dissimilatory selenate reduction in anoxic soil. Environ Sci Technol 49:7684–7691
Neculita CM, Yim G-J, Lee G, Ji S-W, Jung JW, Park H-S, Song H (2011) Comparative effectiveness of mixed organic substrates to mushroom compost for treatment of mine drainage in passive bioreactors. Chemosphere 83:76–82
Noll M, Matthies D, Frenzel P, Derakshani M, Liesack W (2005) Succession of bacterial community structure and diversity in a paddy soil oxygen gradient. Environ Microbiol 7:382–395
Pester M, Bittner N, Deevong P, Wagner M, Loy A (2010) A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J 4:1591–1602
Pester M, Brambilla E, Alazard D, Rattei T, Weinmaier T, Han J, Lucas S, Lapidus A, Cheng JF, Goodwin L, Pitluck S, Peters L, Ovchinnikova G, Teshima H, Detter JC, Han CS, Tapia R, Land ML, Hauser L, Kyrpides NC, Ivanova NN, Pagani I, Huntmann M, Wei CL, Davenport KW, Daligault H, Chain PS, Chen A, Mavromatis K, Markowitz V, Szeto E, Mikhailova N, Pati A, Wagner M, Woyke T, Ollivier B, Klenk HP, Spring S, Loy A (2012) Complete genome sequences of Desulfosporosinus orientis DSM765T, Desulfosporosinus youngiae DSM17734T, and Desulfosporosinus acidiphilus DSM22704T. J Bacteriol 194:6300–6301
Petzsch P, Poehlein A, Johnson DB, Daniel R, Schlömann M, Mühling M (2015) Genome sequences of the moderately acidophilic sulfate-reducing Firmicute Desulfosporosinus acididurans (strain M1T). Genome Announc 3(4):e00881–e00815
Rezadehbashi M, Baldwin SA (2018) Core sulphate-reducing microorganisms in metal-removing semi-passive biochemical reactors and the co-occurrence of methanogens. Microorganisms 6:16
Sánchez-Andrea I, Rodríguez N, Amils R, Sanz JL (2011) Microbial diversity in anaerobic sediments at Rio Tinto, a naturally acidic environment with a high heavy metal content. Appl Environ Microbiol 77:6085–9603
Sánchez-Andrea I, Stams AJM, Amils R, Sanz JL (2013) Enrichment and isolation of acidophilic sulfate-reducing bacteria from Tinto River sediments. Environ Microbiol Rep 5:672–678
Sánchez-Andrea I, Sanz JL, Bijmans MFM, Stams AJM (2014) Sulfate reduction at low pH to remediate acid mine drainage. J Hazard Mater 269:98–109
Sato Y, Hori T, Navarro RR, Habe H, Ogata A (2016) Functional maintenance and structural flexibility of microbial communities perturbed by simulated intense rainfall in a pilot-scale membrane bioreactor. Appl Microbiol Biotechnol 100:6477–6456
Sato Y, Hamai T, Hori T, Habe H, Kobayashi M, Sakai T (2018) Year-round performance of the passive sulfate-reducing bioreactor using rice bran as an organic substrate for treating acid mine drainage. Mine Water Environ 37:586–594
Skousen J, Zipper CE, Rose A, Ziemkiewicz PF, Nairn R, McDonald LM, Kleinmann RL (2017) Review of passive systems for acid mine drainage treatment. Mine Water Environ 36:133–153
Vita N, Valette O, Brasseur G, Lignon S, Denis Y, Ansaldi M, Dolla A, Pieulle L (2015) The primary pathway for lactate oxidation in Desulfovibrio vulgaris. Front Microbiol 6:606
Volant A, Bruneel O, Desoeuvre A, Héry M, Casiot C, Brun N, Delpoux S, Fahy A, Javerliat F, Bouchez O, Duran R, Bertin PN, Elbaz-Poulichet F, Lauga B (2014) Diversity and spatiotemporal dynamics of bacterial communities: physicochemical and other drivers along an acid mine drainage. FEMS Microbiol Ecol 90:247–263
Widdel F (1992) The genus Desulfotomaculum. In: Balows A, Triiper HG, Dworkin M, Harder W, Schleifer KH (eds) The prokaryotes. Springer-Verlag, New York, pp 1792–1799
Zagury GJ, Kulnieks VJ, Neculia CM (2006) Characterization and reactivity assessment of organic substrates for sulfate-reducing bacteria in acid mine drainage treatment. Chemosphere 64:944–954
Zhang M, Wang H (2014) Organic wastes as carbon sources to promote sulfate reducing bacterial activity for biological remediation of acid mine drainage. Miner Eng 69:81–90
Zhao J, Fang D, Zhang P, Zhou L (2017) Long-term effects of increasing acidity on low-pH sulfate-reducing bioprocess and bacterial community. Environ Sci Pollut Res 24:4067–4076
Acknowledgments
We thank Yan-Jie Zhao, Mayumi Matsushita, Maki Yanagisawa, and Yumiko Kayashima for technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary materials
ESM 1
(PDF 5300 kb)
Rights and permissions
About this article

Cite this article
Sato, Y., Hamai, T., Hori, T. et al. Desulfosporosinus spp. were the most predominant sulfate-reducing bacteria in pilot- and laboratory-scale passive bioreactors for acid mine drainage treatment. Appl Microbiol Biotechnol 103, 7783–7793 (2019). https://doi.org/10.1007/s00253-019-10063-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10063-2