
Abstract
Methanotrophs have recently gained interest as biocatalysts for mitigation of greenhouse gas emission and conversion of methane to value-added products; however, their slow growth has, at least partially, hindered their industrial application. A rapid isolation technique that specifically screens for the fastest-growing methanotrophs was developed using continuous cultivation with gradually increased dilution rates. Environmental samples collected from methane-rich environments were enriched in continuously stirred tank reactors with unrestricted supply of methane and air. The reactor was started at the dilution rate of 0.1 h−1, and the dilution rates were increased with an increment of 0.05 h−1 until the reactor was completely washed out. The shifts in the overall microbial population and methanotrophic community at each step of the isolation procedure were monitored with 16S rRNA amplicon sequencing. The predominant methanotrophic groups recovered after reactor operations were affiliated to the gammaproteobacterial genera Methylomonas and Methylosarcina. The methanotrophic strains isolated from the reactor samples collected at their respective highest dilution rates exhibited specific growth rates up to 0.40 h−1; the highest value reported for methanotrophs. The novel isolation method developed in this study significantly shortened the time and efforts needed for isolation of methanotrophs from environmental samples and was capable of screening for the methanotrophs with the fastest growth rates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Methane (CH4) is a potent greenhouse gas with ~ 28 times higher global warming potential (GWP) than carbon dioxide (CO2) if released to the atmosphere (Pachauri et al. 2015), while highly concentrated CH4 collected as biogas and natural gas is a valuable resource utilizable as energy source or feedstock for chemical industry (Conrado and Gonzalez 2014; Fei et al. 2014; Olah et al. 2009). The recent resurgence in natural gas production due to the global shale gas boom has rekindled worldwide interest in the biological and chemical processes that would maximize value creation from CH4 (Conrado and Gonzalez 2014). Chemical or biological syntheses of value-added products from methane inevitably involve partial oxidation of methane to methanol as its first step (Olah et al. 2009). Aerobic methanotrophs, the unique groups of bacteria utilizing CH4 as their sole carbon and energy source, cope with this difficult task by expressing and utilizing two distinct forms of methane monooxygenases, i.e., membrane-bound particulate methane monooxygenases (pMMO) and/or cytoplasmic soluble methane monooxygenases (sMMO) (Semrau et al. 2010). Thus, methanotrophs have recently attracted significant scientific attention as potential biocatalysts for methane transformation (Haynes and Gonzalez 2014; Kalyuzhnaya et al. 2013; Strong et al. 2015; Strong et al. 2016).
The majority of aerobic methanotrophs belong to either of two phylogenetic groups, Alpha- or Gammaproteobacteria (Semrau et al. 2010). The two groups of methanotrophs both utilize pMMO or sMMO for initial oxidation of CH4 to methanol (CH3OH), but have dissimilar carbon assimilation pathways. Gammaproteobacterial methanotrophs utilize ribulose monophosphate (RuMP) pathway, while alphaproteobacterial methanotrophs utilize the serine cycle for assimilation of C1 compounds resulting from oxidation of CH4. Utilization of more efficient RuMP pathway may explain higher growth rates, carbon conversion efficiencies, and methane utilization rates generally observed for the Gammaproteobacterial methanotrophs, which are often viewed to be more attractive for industrial applications (Hoefman et al. 2012; Kalyuzhnaya et al. 2013); however, alphaproteobacterial methanotrophs certainly have their own advantages, as their metabolic diversity would allow for production of broader range of chemical products (Crombie and Murrell 2014; Semrau et al. 2011; Strong et al. 2015). The number of available methanotrophic isolates is too limited and physiological studies have been too biased to a selected group of laboratory strains. Thus, expansion of the methanotroph inventory is warranted to enhance understanding of their physiological diversity and stretch their industrial potential.
One of the bottlenecks in biotechnological applications of methanotrophs is their slow generation time. Dated literatures report specific growth rates as high as ~ 0.4 h−1 for moderately thermophilic Methylococcus genus; however, such high specific growth rate was not reproduced in recent studies and the highest specific growth rate reported since was 0.33 h−1 of Methylomarinum vadi and Methylomarinovum caldicuralii isolated from marine environments (Hirayama et al. 2014; Hirayama et al. 2013). As genetic manipulations are necessary for redirection of metabolic pathways towards production of value-added chemicals, the slow growth is a definite drawback in developing methanotrophs as microbial refineries (Yan et al. 2016). The failure in finding methanotrophs with faster growth may be due to the reliance of the isolation technique on the half-a-century-old traditional method—batch enrichment of environmental samples with CH4 and O2 followed by repetitions of streaking and single-colony-picking on agar plates or dilution-to-extinction (Hoefman et al. 2012; Whittenbury et al. 1970; Wise et al. 1999). These traditional isolation procedures are rigorous and time-taking and lack the capability to specifically select for methanotrophs with the highest growth rates if the fastest-growing methanotrophs exist as unseen minority and/or have a long lag period.
Continuous cultivation has been traditionally used for selective enrichment or isolation of microorganisms. As chemostats can provide natural growth conditions that cannot be simulated with batch cultivation (e.g., consistent supply of the limiting substrate), chemostats have been employed to select for organisms that thrive under substrate limitations (Hanke et al. 2014; van den Berg et al. 2015). Chemostats were also used to enrich organisms that utilize cytotoxic compounds as their growth substrates or generate suicidal compounds, as the concentration of the toxic but necessary substrates can be maintained below the toxicity threshold concentrations (Spain and Nishino 1987). A chemostat reactor would be an ideal tool also for screening of the fastest-growing organism in an environmental sample, as the fundamental property of a chemostat (the specific growth of a steady-state chemostat culture equals the dilution rate) implies that the organisms with specific growth rates lower than the dilution rate of the chemostat would eventually be washed out; however, such utilization of a chemostat has yet to be explored. In this study, a novel isolation method for specific selection of the fastest-growing methanotrophs in an environmental sample was proposed applying the chemostat principle. A chemostat reactor supplied with a continuous stream of gas mixture (80% air and 20% CH4) was operated with gradually increased aqueous phase dilution rates until complete washout was observed. The shift in methanotrophic population through each enrichment process was monitored with 16S rRNA gene amplicon sequencing using MiSeq sequencing technology. The cell suspension sample extracted from the chemostat at the highest dilution rate (before washout) was subjected to the subsequent isolation procedure, which was completed within 2 weeks. The entire isolation procedure was completed within a month. The isolated strains, both Gammaproteobacteria, exhibited the fastest specific growth rates ever reported for mesophilic methanotrophs with specific growth rates measured up to 0.4 h−1 at 30 °C.
Materials and methods
Sampling sites description
The stream sediment (SS) sample used in this study was collected from the north bank of Gapcheon stream and the anaerobic digester effluent (AD) from Daejeon municipal wastewater treatment plant (Daejeon, Korea; 36° 21′ 37.1” N 127° 21′ 15.1″ E and 36° 23′ 03.1” N 127° 24′ 34.7″ E, respectively). As the coexistence of methane and oxygen is essential for methanotrophic metabolism and growth, the SS sample was collected from the sediment ~ 20 cm below the ground surface, and the AD sample was collected from the water surface to target the oxic-anoxic interface. Plant biomass was carefully removed from the SS sample. The samples were stored in sealed sterile Mason jars at 4 °C before use. The pH of both SS and AD samples were circumneutral, with the values ranging from 6.8 to 7.2.
Growth conditions for methanotrophs
A typical nitrate mineral salts (NMS) medium pre-defined for cultivation of methanotrophs was used for all experiments performed in this study (Whittenbury et al. 1970). The NMS medium contained, per liter, 1 g MgSO4∙7H2O (4.06 mM), 1 g KNO3 (9.89 mM), 0.2 g CaCl2∙2H2O (1.36 mM), 0.1 mL of 3.8% (w/v) Fe-EDTA solution, 0.5 mL of 0.1% (w/v) Na2MoO4∙2H2O solution, and 1 mL of the trace element solution. To avoid precipitation, a phosphate buffer stock solution adjusted to pH 6.8 was prepared separately and added to the medium to the final concentration of 5 mM after autoclaving. CuCl2∙2H2O was added from a stock solution to the final concentration of 10 μM. The ×200 vitamin stock solution was prepared aseptically as previously defined, and 5.0 mL of the stock solution was added per liter of culture medium (Whittenbury et al. 1970). All culture bottles were incubated at 30 °C and CH4 was provided as the sole electron donor and carbon source in all experiments performed in this study.
Batch enrichment
Duplicate batch enrichment cultures were prepared by adding 10 g (wet weight) slurries of the SS sample or 1-mL aliquots of the AD sample into 50 mL of NMS medium dispensed in 160-mL serum bottles (Wheaton, Millville, NJ). The serum bottles were sealed with butyl rubber stoppers (Geo-Microbial Technologies, Ochelata, OK) and aluminum crimp seals. After sealing, 20% of the headspace (22 of 110 mL headspace) was replaced with 99.999% CH4 (Deokyang Co., Ulsan, Korea) using a sterile syringe connected to a 0.2-μm syringe filter (Advantec INC, Tokyo, Japan). Samples were placed horizontally in a shaking thermostat incubator and incubated in dark with shaking at 140 rpm. The mixing ratio of CH4 in the headspace was monitored until it dropped to 10% (v/v). One milliliter of each enrichment culture was transferred to a fresh NMS medium and incubated identically to the initial enrichment. After CH4 consumption was verified, 5 mL of the enrichment culture was extracted from one of the duplicate bottles and used as the inoculum for the chemostat reactor.
Selection of fast-growing methanotrophs using a chemostat reactor
A continuously stirred tank reactor (CSTR) was constructed with a 500-mL Duran® pyrex glass bottle (DWK Life Sciences GmbH, Wertheim am Main, Germany) sealed with a GLS 80® cap and a Masterflex® dual-channel peristaltic pump (Cole-Parmer Instrument Co., Chicago, IL) (Fig. 1). The volume of NMS medium in the CSTR was maintained at 250 mL, as the rate of inflow of fresh NMS medium was the same as the rate of outflow of waste medium from the reactor. The mixed gas (~ 20% CH4 and ~ 80% air) supplied to the CSTR was prepared by passing separate streams of compressed air (Deokyang Co., Ulsan, Korea) and 99.999% CH4 through an empty 500-mL pyrex bottle. The flowrates of the compressed air and CH4 were maintained at 0.8 and 0.2 mL min−1, respectively. The mixed gas exiting the pyrex bottle was diffused into the liquid phase of CSTR through a ball-shaped gas diffuser (Thermo Fisher Scientific, Waltham, MA). Neither CH4 nor O2 was likely the limiting substrate for growth of methanotrophs in the chemostat reactor under any examined culturing condition, as the CH4 mixing ratio of the gas effluent was > 15% at all times. The CSTR was submerged in a water bath maintained at 30 °C.

A schematic diagram of the CSTR used for isolation of fast-growing methanotrophs
After inoculation, the chemostat was operated in a fed-batch mode with the peristaltic pump turned off and the gas stream continuously feeding CH4 and O2 into the reactor. The increase in cell density was monitored by measuring OD600 values with 1-mL samples extracted from the CSTR. After growth halted, the peristaltic pump was turned on and the reactor was operated as a chemostat with the dilution rate initially set to 0.1 h−1. The dilution rate was gradually increased with an increment of 0.05 h−1 after passing through five reactor volumes of fresh NMS medium at each dilution rate. The maximum dilution rates at which steady states were established were 0.3 h−1 for the SS sample and 0.45 h−1 for the AD sample, as higher dilution rates resulted in washout. At each dilution rate, 15 mL of the reactor effluent was collected and stored at 4 °C for isolation of methanotrophs and another 15-mL aliquot was stored at − 20 °C for analysis of the microbial community.
Analytical methods
The methane concentration in the headspace was measured with a gas chromatograph equipped with GC-Alumina column (30 m length × 0.53 mm inner diameter, 1.5-μm film thickness) and a flame ionization detector (YL instrument, Gyeonggi, Korea). Injector, oven, and detector temperatures were set to 250, 100, and 275 °C, respectively. Helium was used as the carrier gas and a gas split ratio of 50:1 was used. For each sampling event, 150 μL of headspace was sampled using a 1700-series gas-tight syringe (Hamilton Company, Reno, NV) and 100 μL was manually injected into the gas chromatograph. The syringe was flushed three times with sterile pressurized N2 gas before use to remove residual CH4 in the syringe.
DNA extraction and microbial community analysis
Each enrichment process was monitored with 16S rRNA amplicon sequencing of the samples collected at different enrichment stages. The DNA extracted from the original environmental sample, the batch enrichment after initial incubation with CH4 and O2, the reactor culture after fed-batch incubation, and the steady-state chemostat enrichment at different dilution rates were analyzed. DNA extraction was performed with 0.25 g (wet weight) of a sediment sample or 1.5 mL of an aqueous sample. Power Soil DNA Isolation Kit (MoBio Laboratories, Hilden, Germany) was used for DNA extraction, according to the protocol provided by the manufacturer. The extracted DNA was stored at − 20 °C before use. The hypervariable V6-V8 region of 16S rRNA gene was amplified with the 926F (5’-AAACTYAAAKGAATTGRCGG-3′)/1392R (5’-ACGGGCGGTGTGTRC-3′) primer pair (with barcodes attached to the 5′ end of 926F) and the amplicons were sequenced by Macrogen (Seoul, Korea) using MiSeq sequencing platform (Illumina, Inc., San Diego, CA) (Matsuki et al. 2002). The raw sequence data supporting the conclusions of this article have been deposited in the NCBI SRA database, https://www.ncbi.nlm.nih.gov/sra (accession numbers: SRP115081 and SRP115083 for the SS and AD samples, respectively).
Non-16S rRNA sequences were screened from the raw sequence datasets using SortMeRNA v2.0 (Kopylova et al. 2012). The resulting datasets were processed using the QIIME pipeline v 1.9.1 with options set to default values (Caporaso et al. 2010). After demultiplexing of the raw reads, ambiguous reads or reads with quality scores lower than the default cutoff value (Q20) were filtered out and the chimeric reads were removed. The filtered reads were then clustered into operational taxonomic units (OTUs) with USEARCH algorithm against the Greengenes v13.8 database and de novo clustering (for sequences with no matching sequence in the database) using 97% as the cutoff value (Schloss et al. 2009). The OTUs were assigned to taxa using the RDP classifier against the Greengenes V13.8 database. The reads that remained unclassified after assignment were discarded.
Isolation and determination of the growth rates
The samples extracted from the CSTRs operated at the highest dilution rates were used as the starting points for isolation of the fastest-growing methanotrophs. One milliliter of the reactor effluent sample stored at 4 °C was inoculated into fresh NMS medium prepared as described above. After the culture was grown to an OD600 value of ~ 0.4 with CH4 as the sole carbon source, 100 μL of the enrichment culture was spread onto NMS agar plates, which were then incubated in a BBL® GasPak® 150 anaerobic jar (Becton Dickinson, Franklin Lakes, NJ) filled with 80% air and 20% CH4. The jar was placed in a dark thermostat incubator set to 30 °C. Single colonies from the agar plates were transferred to fresh agar plates. The process was repeated three times and the single colonies from the fourth transfer culture were taken and suspended into fresh NMS medium and grown with CH4. For each isolate, 16S rRNA gene and pmoA gene were amplified with 27f/1492r and A189f/mb661r primer sets, respectively, and cloned using TOPO TA cloning kit (Invitrogen, San Diego, CA). Ten 16S rRNA clones and ten pmoA clones were sequenced for confirmation of purity and identification of each isolate.
The growth rates of the isolated methanotrophs and a reference strain of methanotroph (Methylosinus trichosporium strain OB3b) were determined by monitoring the OD600 values of the batch cultures incubated with CH4. Five milliliters of the preculture at exponential phase (OD600 ~ 0.25) grown with 80% air and 20% CH4 in the headspace was inoculated to 45 mL of fresh NMS medium in a 160-mL serum bottle. The serum bottles were incubated at 30 °C with shaking at 250 rpm. At each sampling time point, 1 mL of the culture was extracted to measure the OD600 value. The experiment was performed in triplicates.
Results
Characterization of the overall microbial communities and methanotrophic populations in the environmental samples
The indigenous methanotrophic population was identified in the environmental samples prior to enrichment, using MiSeq amplicon sequencing targeting the V6–V8 hypervariable region of 16S rRNA genes (Table S1). The samples collected from the Gapcheon stream sediment and the anaerobic digester effluent varied greatly in the compositions of microbial communities and indigenous methanotrophic populations (Table 1, Fig. 2, Table S1). The most abundant OTUs in the SS and AD samples were assigned to the uncultured DA101 genus of Chthoniobacteraceae family and the uncultured T78 genus of Anaerolinaceae family, respectively. In both SS and AD samples, the bacterial OTUs assigned to genera previously identified as alpha- and gammaproteobacterial methanotrophs constituted minor populations. The OTU assigned to genus Crenothrix was the most abundant methanotrophic population identified in the SS sample before enrichment (0.246% of the total reads). Other methanotrophs identified in the SS sample included the OTUs assigned to the genera Methylosinus (0.072%), Methylocaldum (0.030%), Methylomonas (0.016%), Methylomicrobium (0.002%), and Methylococcus (0.002%) and unidentified genera (or unable to be unambiguously assigned to a specific genus) in the Methylocystaceae and Methylococcaceae families (0.025 and 0.011%, respectively). The most abundant methanotrophic OTU in the AD sample was assigned to the genus Methylosinus (0.210%). Other methanotrophs included the OTUs affiliated to the genera Methylosarcina (0.019%), Methylocaldum (0.012%), Methylomonas (0.002%), Crenothrix (0.001%), and Methylocella (0.001%) and an unidentified genus in the Methylocystaceae family (0.019%). The fractions of indigenous methanotrophic populations in the SS and AD samples were 0.42 and 0.27% of the total microbial populations, respectively.

The shift in the methanotrophic communities of a SS and b AD samples upon enrichment as monitored with 16S amplicon sequencing. The relative abundances of methanotrophic genera at different stages of the enrichment procedures are presented. The inserted figures are blowups of the data with the same label(s) in the main Figs. EB: the environmental samples; IB: the samples collected after initial batch cultivation; FB: the samples collected after fed-batch cultivation; and HD: the samples collected from the chemostats operated at the highest viable dilution rates (0.30 h−1 for the SS sample and 0.45 h−1 for the AD sample)
Monitoring of the population shifts at different stages of enrichment
The shift in microbial community was monitored at different enrichment stages using MiSeq 16S rRNA amplicon sequencing (Table S1). Although methanotrophs were significantly enriched after the initial batch enrichment, the proportions of the methanotrophic populations remained at 2.61 and 12.6% of the total microbial population in the batch enrichments of the SS and AD samples, respectively. The OTUs affiliated to the genus Crenothrix spp. and Methylomonas spp. were the most abundant methanotrophs in the SS and AD enrichments (68.9 and 70.7% of the methanotrophic population), respectively.
Extended period of incubation with unrestricted supply of CH4 and O2 in the reactor operated at a fed-batch mode specifically favored enrichment of gammaproteobacterial methanotrophs. In the reactor inoculated with SS batch enrichment, the OTUs assigned to the genus Methylomonas constituted 97.7% of the methanotrophic population and 18.3% of the overall microbial population. The OTUs assigned to the genus Methylomonas was also the dominant population in the reactor inoculated with AD batch enrichment (60.2% of the methanotrophic population and 9.95% of the overall microbial population). In the reactor fed with the SS sample, the washout of the chemostat culture occurred at the dilution rate of 0.35 h−1. Methylomonas spp. remained as the dominant microbial group that constituted 53.8% of the total bacterial/archaeal population and 99.2% of the methanotrophic population. As for the AD chemostat, the microbial community of the suspension collected at the highest dilution rate, 0.45 h−1, contained Methylosarcina as the dominant group of microorganism (36.8% of the bacterial/archaeal population and 94.1% of the total methanotrophic population). Notably, the dominating methanotrophs at the fed-batch mode, Methylomonas spp. was nearly washed out (0.402% of the total microbial population) upon chemostat cultivation. In both reactor operations, the methanotrophic OTUs observed to be the most abundant in the original environmental sample and initial batch enrichments, e.g., Clenothrix spp. and Methylosinus spp., were recovered only at very low relative abundances.
The shift in the composition of non-methanotrophic population was evident in both SS and AD samples at different stages of enrichment (Table 1, Table S1). The OTUs affiliated to the genera or families under the Flavobacteriales order of the Bacteroidetes phylum (the genera Flavobacterium and Leadbetterella or the Flavobacteriaceae family) were commonly enriched in both chemostats (> 10% relative abundances). The composition of the enriched non-methanotrophic organisms outside Flavobacteriaceae family varied greatly between the two chemostats, likely due to the differences in the quantities and/or compositions of organic compounds released by the major methanotrophic populations. The obligate methylotrophs (genera Methylotenera and Methylobacillus and Methylophilacaea family) constituted a substantial fraction of the microbial population in the SS reactor but were negligible in the AD reactor, suggesting that the composition of the exuded organic compounds from the Methylomonas strain enriched in the SS chemostat and the Methylosarcina strain enriched in the AD chemostat may differ substantially.
Isolation and initial characterization of fast-growing methanotrophs
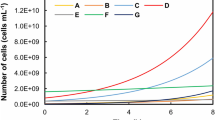
In order to isolate and obtain pure cultures of fast-growing methanotrophs, the sample from the chemostat operated at the highest dilution rate was enriched and spread onto NMS agar plates. Single colonies appeared within 2 days of spreading and were transferred to fresh NMS agar plates on the third day. For both SS and AD chemostat enrichments, nine single colonies were picked after four transfers and subjected to confirmation of methane oxidation activities. Seven of the nine SS enrichment cultures turned turbid and depleted 20% CH4 in the headspace within 2 days of inoculation (with an average final OD600 value of ~ 0.4). Three of the seven microbial cultures were randomly selected and their purity and identity were confirmed by sequencing ten 16S rRNA gene clones and pmoA clones each. The consensus 16S rRNA gene amplicon sequence was identical to the amplicon sequence of the dominant Methylomonas-affiliated OTU, and unexpectedly, the amplicon sequences of 16S rRNA gene and pmoA were both 100% identical at nucleotide sequence level to Methylomonas sp. LW13 (GenBank accession number AF150792.1 and AF150793.1), previously isolated from Lake Washington (Auman et al. 2000). Two of nine single colonies from AD enrichment chemostat grew on CH4 and O2 and were identified as pure cultures of Methylosarcina spp., based on amplicon sequencing of 16S rRNA and pmoA genes. The 16S rRNA and pmoA gene sequences of the isolate were both > 99% identical to those of Methylosarcina fibrata (GenBank accession number NR_025039.1 and WP_020564881.1) at nucleotide sequence level. These results were rather unexpected, as neither Methylomonas sp. LW13 nor strains belonging to Methylosarcina fibrata had been previously recognized for high growth rates. The exponential growth rates of the isolated strains affiliated to Methylosarcina and Methylomonas were measured to be 0.31 ± 0.02 h−1 and 0.40 ± 0.04 h−1, respectively, and were significantly higher than the exponential growth rates of the reference methanotrophic strain M. trichosporium strain OB3b (0.16 ± 0.01 h−1) determined using the identical method (Fig. 3).

The growth curves of the methanotrophs isolated from the SS sample (open squares), the AD sample (closed circles), and a reference strain, M. trichosporium strain OB3b (closed triangles). The data points are the averages of triplicate measurements (error bars are not presented, as the standard deviations of the triplicate OD600nm measurements were negligible)
Discussion
Methanotrophs have great potential as environmentally friendly biocatalysts in the chemical industry, mainly due to their capability to transform CH4, a potent greenhouse gas, to various value-added organic chemical products via CH3OH. Such biochemical transformations have been found to be feasible in laboratory-scale experiments (Demidenko et al. 2017; Dong et al. 2017; Henard et al. 2016; Pieja et al. 2012). Success of methanotroph biotechnology depends primarily on rapid isolation of metabolically diverse fast-growing methanotrophs that are amenable to rapid genetic modifications. The novel isolation technique proposed in this study was able to significantly shorten the time required for isolation of methanotrophs to within a month of sample collection. This novel technique screened out most of the slow-growing methanotrophs before plating, saving rigorous efforts necessary for screening arbitrary isolates for high specific growth rates. The fast-growing methanotrophs isolated using this method will still require extensive characterization before being adopted as platform organisms; nevertheless, our novel technique would significantly enhance the throughput of the upstream screening process without the necessity for any sophisticated equipment or procedure.
Initial batch cultivation of the environmental samples with limited amounts of CH4 and O2 enriched multiple phylogenetic groups of methanotrophs, which included both alpha- and gammaproteobacterial methanotrophs; however, further enrichment with continuous supply of CH4 exclusively enriched gammaproteobacterial methanotrophs of the genera Methylomonas and Methylosarcina. The genera Crenothrix and Methylosinus, with large abundance in the original environmental samples and the initial batch enrichments, were outcompeted and washed out upon chemostat cultivation. These results were in support of the general perception that the gammaproteobacterial methanotrophs have higher specific growth rates than alphaproteobacterial or verrucomicrobial methanotrophs under CH4- and O2-rich growth conditions (Demidenko et al. 2017; Hoefman et al. 2012). That Methylosarcina spp. outcompeted Methylomonas spp. in the AD chemostat, while Methylomonas was the dominating genera in the SS chemostat suggested fast growth is not a genus- or species-specific characteristic. In fact, the fastest growth rate recorded for any methanotroph prior to this study was observed with Methylomicrobium buryantense strain 5GB1, and none of previously isolated Methylomonas or Methylosarcina strains exhibited growth rates exceeding 0.20 h−1 (or, in other words, doubling times lower than ~ 3.5 h). Although 16S rRNA and pmoA sequences of the fast-growing isolates were nearly identical to Methylomonas sp. LW13 and Methylosarcina fibrata, the specific growth rates of the Methylomonas and Methylosarcina isolates were at least 1.5-fold higher than the reported growth rates of any isolates affiliated to these genera. Genome sequencing and annotation of the Methylomonas and Methylosarcina isolates are currently in progress and the search for specific genomic features that allow for this extraordinarily fast growth may be an interesting topic for future research. Identification of such genomic features may provide insights necessary to resolve the long-standing enigma that methanotrophic Proteobacteria have surprisingly low growth rates compared to heterotrophic Proteobacteria even when the system is not limited by mass transfer of CH4 or O2.
Isolation of methanotrophs is often complicated by methylotrophs and heterotrophs attached to the methanotrophs in what appears as single colonies (Hoefman et al. 2012). Even when CH4 is provided as the sole source of carbon and electron that can be utilized only by organisms possessing pMMO or sMMO (in oxic condition), methanotrophs are known to exude one-carbon, e.g., CH3OH and formate (HCOOH), and multi-carbon organic compounds (e.g., fatty acids and exopolymeric substances) utilizable to non-methanotrophic methylotrophs and heterotrophs (Hilger et al. 2000; Oshkin et al. 2014; Sheets et al. 2016). The chemostat cultures, even at the highest dilution rates, were not completely devoid of the methylotrophs and heterotrophs; however, the proportions of the non-methylotrophic organisms were notably lower in the fully operational chemostat than in the initial batch enrichment or the reactor operated in the fed-batch mode. The removal of methylotrophs and heterotrophs from the enrichment culture significantly facilitated the downstream isolation procedures, so that axenic cultures could be obtained without difficulty after three or four rounds of plating and single-colony picking.
Several unexpected phylogenetic groups of microorganisms were enriched in the chemostat reactors. In the chemostat inoculated with the SS sample, Legionella spp., a group of organisms that cause Legionnaires’ disease in mammals, were enriched as one of the organismal groups with the highest relative abundances. Legionella spp. are facultative intracellular bacteria rarely found to replicate in the free-living form due to their unique nutrient requirement (Feeley et al. 1979; Pine et al. 1979). In the chemostat reactor inoculated with the AD sample, an OTU affiliated to the Opitutaceae family, a Verrucomicrobia group known to be notoriously difficult to enrich or isolate, was one of the organismal groups with greatly increased relative abundance upon chemostat operation (Chin et al. 2001). Verrucomicrobia, including Opitutaceae family, have evaded cultivation or enrichment although microbial community analyses have suggested their abundance in agricultural soils and presumed importance in the biogeochemical cycles (Hugenholtz et al. 1998; Joseph et al. 2003). Several recent studies investigated non-methanotropic microbial communities developing around methanotrophs; however, neither of these culture-resistant organismal groups was previously identified to associate with gammaproteobacterial methanotrophs (Ho et al. 2016; Oshkin et al. 2014).
Cross-feeding is the most intuitive mechanism of association between methanotrophs and non-methanotrophs, as methanotrophs often release intermediates of CH4 metabolism, e.g., CH3OH and organic acids, attracting non-methanotrophic methylotrophs, and heterotrophs (Kalyuzhnaya et al. 2013; Oshkin et al. 2014). The enriched Opitutacea and Legionella could have certainly benefitted from such organic exudates from the fast-growing methanotrophs; however, this rationale is not sufficient to explain their enrichment in the chemostat, which require the cells to grow at a rate higher than 0.30 h−1 for survival. The fast-growing methanotrophs may have produced and exuded micronutrients essential for these culture-resistant organisms that are absent in synthetic culture media. Such commensal association was previously observed between cobalamin-producing organisms and vitamin B12 auxotrophs (Yan et al. 2013). An interesting future research would involve identification of such micronutrients in the spent media of the isolated gammaproteobacterial methanotrophs that may facilitate enrichment and isolation of these culture-resistant organisms with potential clinical or environmental significance.
References
Auman AJ, Stolyar S, Costello AM, Lidstrom ME (2000) Molecular characterization of methanotrophic isolates from freshwater lake sediment. Appl Environ Microbiol 66:5259–5266. https://doi.org/10.1128/aem.66.12.5259-5266.2000
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chin KJ, Liesack W, Janssen PH (2001) Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division ‘Verrucomicrobia’ isolated from rice paddy soil. Int J Syst Evol Microbiol 51:1965–1968. https://doi.org/10.1099/00207713-51-6-1965
Conrado R, Gonzalez R (2014) Envisioning the bioconversion of methane to liquid fuels. Science 343:621–623. https://doi.org/10.1126/science.1246929
Crombie AT, Murrell JC (2014) Trace-gas metabolic versatility of the facultative methanotroph Methylocella silvestris. Nature 510:148–151. https://doi.org/10.1038/nature13192
Demidenko A, Akberdin IR, Allemann M, Allen EE, Kalyuzhnaya MG (2017) Fatty acid biosynthesis pathways in Methylomicrobium buryatense 5G(B1). Front Microbiol 7:2167. https://doi.org/10.3389/fmicb.2016.02167
Dong T, Fei Q, Genelot M, Smith H, Laurens LML, Watson MJ, Pienkos PT (2017) A novel integrated biorefinery process for diesel fuel blendstock production using lipids from the methanotroph, Methylomicrobium buryatense. Energ Convers Manage 140:62–70. https://doi.org/10.1016/j.enconman.2017.02.075
Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB (1979) Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J Clin Microbiol 10:437–441
Fei Q, Guarnieri MT, Tao L, Laurens LML, Dowe N, Pienkos PT (2014) Bioconversion of natural gas to liquid fuel: opportunities and challenges. Biotechnol Adv 32:596–614. https://doi.org/10.1016/j.biotechadv.2014.03.011
Hanke A, Hamann E, Sharma R, Geelhoed JS, Hargesheimer T, Kraft B, Meyer V, Lenk S, Osmers H, Wu R, Makinwa K, Hettich RL, Banfield JF, Tegetmeyer HE, Strous M (2014) Recoding of the stop codon UGA to glycine by a BD1-5/SN-2 bacterium and niche partitioning between Alpha- and Gammaproteobacteria in a tidal sediment microbial community naturally selected in a laboratory chemostat. Front Microbiol 5:231. https://doi.org/10.3389/fmicb.2014.00231
Haynes CA, Gonzalez R (2014) Rethinking biological activation of methane and conversion to liquid fuels. Nat Chem Biol 10:331–339. https://doi.org/10.1038/nchembio.1509
Henard CA, Smith H, Dowe N, Kalyuzhnaya MG, Pienkos PT, Guarnieri MT (2016) Bioconversion of methane to lactate by an obligate methanotrophic bacterium. Sci Rep 6:21585. https://doi.org/10.1038/srep21585
Hilger HA, Cranford DF, Barlaz MA (2000) Methane oxidation and microbial exopolymer production in landfill cover soil. Soil Biol Biochem 32:457–467. https://doi.org/10.1016/S0038-0717(99)00101-7
Hirayama H, Abe M, Miyazaki M, Nunoura T, Furushima Y, Yamamoto H, Takai K (2014) Methylomarinovum caldicuralii gen. nov., sp. nov., a moderately thermophilic methanotroph isolated from a shallow submarine hydrothermal system, and proposal of the family Methylothermaceae fam. nov. Int J Syst Evol Microbiol 64:989–999. https://doi.org/10.1099/ijs.0.058172-0
Hirayama H, Fuse H, Abe M, Miyazaki M, Nakamura T, Nunoura T, Furushima Y, Yamamoto H, Takai K (2013) Methylomarinum vadi gen. nov., sp. nov., a methanotroph isolated from two distinct marine environments. Int J Syst Evol Microbiol 63:1073–1082. https://doi.org/10.1099/ijs.0.040568-0
Ho A, Angel R, Veraart AJ, Daebeler A, Jia Z, Kim SY, Kerckhof F-M, Boon N, Bodelier PLE (2016) Biotic interactions in microbial communities as modulators of biogeochemical processes: methanotrophy as a model system. Front Microbiol 7:1285. https://doi.org/10.3389/fmicb.2016.01285
Hoefman S, van der Ha D, De Vos P, Boon N, Heylen K (2012) Miniaturized extinction culturing is the preferred strategy for rapid isolation of fast-growing methane-oxidizing bacteria. Microbial Biotechnol 5:368–378. https://doi.org/10.1111/j.1751-7915.2011.00314.x
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Joseph SJ, Hugenholtz P, Sangwan P, Osborne CA, Janssen PH (2003) Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl Environ Microbiol 69:7210–7215. https://doi.org/10.1128/aem.69.12.7210-7215.2003
Kalyuzhnaya MG, Yang S, Rozova ON, Bringel F, Smalley NE, Clubb J, Konopka M, Orphan VJ, Beck D, Trotsenko YA, Vuilleumier S, Khmelenina VN, Lidstrom ME (2013) Highly efficient methane biocatalysis revealed in methanotrophic bacterium. Nat Commun 4:2785. https://doi.org/10.1038/ncomms3785
Kopylova E, Noé L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217. https://doi.org/10.1093/bioinformatics/bts611
Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, Oyaizu H, Tanaka R (2002) Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol 68:5445–5451. https://doi.org/10.1128/aem.68.11.5445-5451.2002
Olah GA, Goeppert A, Prakash GKS (2009) Production of methanol: from fossil fuels and bio-sources to chemical carbon dioxide recycling. In: Olah GA, Goeppert A, Prakash S (eds) Beyond oil and gas: the methanol economy, 2nd edn. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, pp 233–278
Oshkin IY, Beck DAC, Lamb AE, Tchesnokova V, Benuska G, McTaggart TL, Kalyuzhnaya MG, Dedysh SN, Lidstrom ME, Chistoserdova L (2014) Methane-fed microbial microcosms show differential community dynamics and pinpoint taxa involved in communal response. ISME J 9:1119–1129. https://doi.org/10.1038/ismej.2014.203
Pachauri RK, Meyer L, Pattner G-K, Stocker T (2015) IPCC, 2014: climate change 2014: synthesis report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change
Pieja AJ, Sundstrom ER, Criddle CS (2012) Cyclic, alternating methane and nitrogen limitation increases PHB production in a methanotrophic community. Bioresour Technol 107:385–392. https://doi.org/10.1016/j.biortech.2011.12.044
Pine L, George JR, Reeves MW, Harrell WK (1979) Development of a chemically defined liquid medium for growth of Legionella pneumophila. J Clin Microbiol 9:615–626
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/aem.01541-09
Semrau JD, DiSpirito A, Yoon S (2010) Methanotrophs and copper. FEMS Microbiol Rev 34:496–531. https://doi.org/10.1111/j.1574-6976.2010.00212.x
Semrau JD, DiSpirito AA, Vuilleumier S (2011) Facultative methanotrophy: false leads, true results, and suggestions for future research. FEMS Microbiol Lett 323:1–12. https://doi.org/10.1111/j.1574-6968.2011.02315.x
Sheets JP, Ge X, Li Y-F, Yu Z, Li Y (2016) Biological conversion of biogas to methanol using methanotrophs isolated from solid-state anaerobic digestate. Bioresour Technol 201:50–57. https://doi.org/10.1016/j.biortech.2015.11.035
Spain JC, Nishino SF (1987) Degradation of 1,4-dichlorobenzene by a Pseudomonas sp. Appl Environ Microbiol 53:1010–1019
Strong PG, Xie S, Clarke WP (2015) Methane as a resource: can the methanotrophs add value? Environ Sci Technol 49:4001–4018. https://doi.org/10.1021/es504242n
Strong PJ, Kalyuzhnaya M, Silverman J, Clarke WP (2016) A methanotroph-based biorefinery: potential scenarios for generating multiple products from a single fermentation. Bioresour Technol 215:314–323. https://doi.org/10.1016/j.biortech.2016.04.099
van den Berg EM, van Dongen U, Abbas B, van Loosdrecht MCM (2015) Enrichment of DNRA bacteria in a continuous culture. ISME J 9:2153–2161. https://doi.org/10.1038/ismej.2015.26
Whittenbury R, Phillips KC, Wilkinson JF (1970) Enrichment, isolation and some properties of methane-utilizing bacteria. Microbiology 61:205–218. https://doi.org/10.1099/00221287-61-2-205
Wise MG, McArthur JV, Shimkets LJ (1999) Methanotroph diversity in landfill soil: isolation of novel type I and type II methanotrophs whose presence was suggested by culture-independent 16S ribosomal DNA analysis. Appl Environ Microbiol 65:4887–4897
Yan J, Im J, Yang Y, Löffler FE (2013) Guided cobalamin biosynthesis supports Dehalococcoides mccartyi reductive dechlorination activity. Phil Trans Royal Soc Lond B 368:20120320. https://doi.org/10.1098/rstb.2012.0320
Yan X, Chu F, Puri AW, Fu Y, Lidstrom ME (2016) Electroporation-based genetic manipulation in type I methanotrophs. Appl Environ Microbiol 82:2062–2069. https://doi.org/10.1128/aem.03724-1
Funding
This research was financially supported by the National Research Foundation of Korea (NRF) (grant no. 2015M3D3A1A01064881) and the “R&D Center for Reduction of Non-CO2 Greenhouse Gases” (grant no. 2017002420002) funded by Korea Ministry of Environment (MOE) as “Global Top Environment R&D Program”. The authors were also financially supported by Korea Ministry of Land, Infrastructure and Transport (MOLIT) as U-City Master and Doctor Course Grant Program and the Brain Korea 21 Plus Project (grant no. 21A20132000003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(XLSX 134 kb)
Rights and permissions
About this article

Cite this article
Kim, J., Kim, D.D. & Yoon, S. Rapid isolation of fast-growing methanotrophs from environmental samples using continuous cultivation with gradually increased dilution rates. Appl Microbiol Biotechnol 102, 5707–5715 (2018). https://doi.org/10.1007/s00253-018-8978-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8978-5