
Abstract
The Escherichia coli (E. coli) expression system has been widely used to produce recombinant proteins. However, in some heterologous expressions, there are still difficulties in large-scale production. The use of fusion partners is one of the strategies for improving the expression levels of proteins in E. coli host. Here, we demonstrate a novel fusion element, the NT11-tag, which enhances protein expression. The NT11-tag was derived from the first 11 amino acid residues within the N-terminal N-half domain of a duplicated carbonic anhydrase (dCA) from Dunaliella species. Previously, we have found that the tag improves expression of the C-half domain of dCA when linked to its N-terminus. To verify its use as a protein production enhancer tag, two kinds of CAs derived from Hahella chejuensis (Hc-CA) and Thermovibrio ammonifican (Ta-CA) and the yellow fluorescent protein (YFP) were used as model proteins to measure their increased expression upon fusion with the NT11-tag. The NT11-tag amplified protein expression in E. coli by 6.9- and 7.6-fold for Ta-CA and YFP, respectively. Moreover, the tag also enhanced the soluble expression of Hc-CA, Ta-CA, and YFP by 1.7-, 5.0-, and 3.2-fold, respectively. Furthermore, protein yield was increased without inhibiting protein function. These results indicate that the use of the NT11-tag is a promising method for improving protein production in E. coli.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The discovery of recombinant DNA technology in the 1970s initiated the era of recombinant protein expression, which has been broadly applied in many fields including enzyme assays/engineering, therapeutics, and agriculture (Jones and Fayerman 1987). Several host organisms have been used to express recombinant proteins. Of them, E. coli is the most widely used expression host because of its rapid growth, high protein yield, ease of culture, well set-up of gene manipulation, and cost-effectiveness (Demain and Vaishnav 2009). However, some recombinant genes are poorly expressed and some proteins, even expressed, become aggregated or insoluble forms. These remain the limitations in using E. coli expression systems (Peti and Page 2007; Terpe 2006).
Several methods have been used to optimize recombinant protein production in E. coli. The strategy of using protein fusion partners has been effective for increasing expression of target proteins and inhibiting inclusion body formation (Esposito and Chatterjee 2006). There are many well-known widely used fusion partners: MBP (maltose-binding protein) (Kapust and Waugh 1999), TrxA (thioredoxin) (Dyson et al. 2004), NusA (N utilization substance A) (Kohl et al. 2008), GST (Glutathione-S-transferase) (Hu et al. 2008), and SUMO (small ubiquitin-related modifier) (Marblestone et al. 2006). Most of them are 20–300 residues in length and sometimes—according to the downstream applications—should be removed from the target protein to prevent interference with the proper structure and function of the target protein (Ramos et al. 2013). Removal of the fusion tag can be difficult and could also induce protein precipitation (Waugh 2011). In addition, short peptide tags have also been developed to improve recombinant protein expressions including poly-Lys, poly-Arg (Terpe 2003), Fh8 and histidine tags (Costa et al. 2013).
In fact, recombinant protein production is controlled by many factors, including the cloning vector types (Rosano and Ceccarelli 2014; Sorensen and Mortensen 2005), interaction of mRNA sequence with the ribosome (Shine and Dalgarno 1975), codon usage, physiological stress and cultivation performance (Chou 2007), translation initiation (Kozak 2005), mRNA stability and initial phase of elongation (Bivona et al. 2010). The translation initiation region (TIR) sequence is also one of the important factors for target protein synthesis because it promotes interaction with rRNA that initiates translation (Allen et al. 2005). Specifically, the folding free energy of the region between − 10 and + 35 has the greatest influences on prokaryotic translation efficiency. Accordingly, it is required to optimize the nucleotide sequences around the TIR for high-level protein production.
Previously, we found an interesting result in recombinant protein expression by designing a chimeric carbonic anhydrase (CA) based on an internally duplicated CA from Dunaliella species (Dsp-CA). Although both N-half/C-half domains of Dsp-CA have structures similar to that of a known CA (PDB ID: 1y7w), only the C-half domain (GenBank: MH636012), termed as Dsp-CA-c, exhibited enzymatic activity, albeit with lower expression. In contrast, the expression level of N-half domain of Dsp-CA, termed as Dsp-CA-n, was high. The first ten amino acid residues of Dsp-CA-c were replaced with the NT11 sequence (VSEPHDYNYEK) of highly expressed Dsp-CA-n. The resulting Dsp-nCA-c construct (GenBank: MH613347) showed a 2-fold increase in soluble expression and enzyme activity compared to the Dsp-CA-c (Ki et al. 2016). These results suggested that NT11 sequence might work as a protein enhancement tag for the CAs expressed in E. coli.
In this study, we investigated whether the NT11 could function as a protein production enhancement tag for other CAs. Specifically, we measured the expression of Hc-CA, a CA from Hahella chejuensis that is highly active in alkaline conditions but is mostly expressed in an insoluble form (Ki et al. 2013), and Ta-CA, one of the most thermostable CAs (Di Fiore et al. 2015) from Thermovibrio ammonificans, which is also poorly expressed in E. coli host system. Moreover, we tested YFP (yellow fluorescent protein—a variant of GFP), the gene codon of which was optimized for mammalian cell expression and that is expressed in low abundance in E. coli. In the NT11-tag fusion expression system, the coding sequence of NT11-tag is located within TIR, which might affect the expression levels of recombinant protein via interacting with the ribosomal interaction region. The resulting recombinant fusion proteins were assessed for their production yields, native structure, and function changes compared to their untagged forms to determine the effects of the NT11-tag.
Material and methods
Strains, plasmids, and reagents
DH5α and BL21 (DE3) Escherichia coli (Agilent Technologies Inc., Santa Clara, CA, USA) were used as the host cells for the cloning and expression system, respectively. The expression vectors were constructed using the plasmids pET42b(+) and pET22b(+), from Novagen Inc. (Madison, WI, USA). Antibiotics, isopropyl β-d-thiogalactopyranoside (IPTG), p-nitrophenyl acetate (p-NPA), phenylmethylsulfonyl fluoride (PMSF), lysozyme, and DNase were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). EDTA-free protease inhibitor (Halt Protease Inhibitor Cocktail) and protein assay reagents were obtained from Thermo Fisher Scientific Inc. (Rockford, IL, USA). All the reagents used in the experiments were of analytical grade.
Construction of fusion vectors
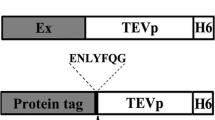
To investigate the influence of the NT11-tag in protein expression, model proteins were selected and designed with an NT11-tag at the N-terminus and a His-tag at the C-terminus (Fig. 1). The Dsp-short CA-c (termed Dsp-sCA-c) was derived from Dsp-nCA-c, in which the NT11-tag at N-terminus was deleted. The Hc-CA gene, which was 50% identical to the other CAs (PDB ID: 1KOP and 1Y7W from Neisseria gonorrhoeae and Dunaliella salina, respectively), was amplified by PCR from pET42b-Hc-CA OPT (Ki et al. 2013) using forward and reverse primers. The cDNA sequences encoding Ta-CA (PDB ID: 4C3T_A) was synthesized (GenScript, Piscataway, NJ, USA). The cDNA sequences encoding Hc-CA and Ta-CA as well as the NT11-tag were used as templates in overlap extension PCR to obtain the NT11-Hc-CA (GenBank: MH636008), and NT11-Ta-CA (GenBank: MH636009) fusion genes. In addition, the cDNA sequence of YFP was amplified by PCR from pcDNA3YFP that was a gift from Doug Golenbock (Addgene plasmid no. 13033; http://n2t.net/addgene:13033; RRID:Addgene_13033) and NT11-YFP gene (GenBank: MH636010) was also amplified by PCR using the cDNA sequence of YFP as a template. All the PCR reactions were carried out in a standard method for 30 cycles (cycling parameters: denaturation at 94 °C for 30 s, annealing at 64 °C for 60 s, and extension at 72 °C for 60 s), and final extension was carried out at 72 °C for 6 min. The primers used in the PCR reactions are listed in Table 1. The signal sequences of CAs were removed for maximal cytoplasmic expression in E. coli.

Linear diagrams of target proteins with or without the N-terminal NT11-tag (dark upward diagonal box) and with the C-terminal His-tag (black box)
Finally, the PCR products were cloned into the T-easy vector to produce the recombinant plasmids and transformed into DH5α E. coli for amplification. The presence of the cloned genes was confirmed by automated DNA sequencing (Cosmogen Tech. Co., Seoul, Korea). The fragments resulting from digestion of the T-vector with NdeI/HindIII (Dsp-sCA-c, Hc-CAs, and YFPs) and NdeI/XhoI (Ta-CAs) were subcloned into the digested pET42b and pET22b vectors, respectively, to construct the prokaryotic expression vectors, pET42b/Dsp-sCA-c, pET42b/Hc-CA, pET42b/NT11-Hc-CA, pET42b/YFP, pET42b/NT11-YFP, pET22b/Ta-CA, and pET22b/NT11-Ta-CA. E. coli BL21 (DE3) was transformed with the expression vectors, and the transformants were selected on LB agar plates supplemented with kanamycin (25 μg/mL) for pET42b vectors or ampicillin (100 μg/mL) for pET22b vectors.
Expression and purification of recombinant proteins
All recombinant proteins were expressed in BL21 (DE3) E. coli and purified using nickel immobilized metal affinity chromatography as described previously (Jo et al. 2014; Ki et al. 2016; Min et al. 2016). To express Dsp-CAs or YFPs, BL21 (DE3) E. coli containing the expression vector was grown to 0.6–0.8 optical density (OD) at 600 using a UV-Vis spectrophotometer (Mecasys Co Ltd., Daejeon, Korea), followed by the addition of IPTG to a final concentration of 0.1 mM and incubated overnight at 20 °C. The expression of Hc-CAs or Ta-CAs was the same as described above except that the cells were cultured at 37 °C after IPTG addition. The cell pellet was dissolved in lysate buffer, and cell disruption was performed by sonication (Branson Digital Sonifier 250, Connecticut, USA) with an amplitude of 30%, processing time 5 min, ON time 1 s, OFF time 3 s, and the sample was kept on ice during sonicating. The proteins were purified using a HisPur Ni-NTA Superflow Agarose column (Thermo Fisher Scientific, Rockford, IL, USA), as previously reported, and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE using 10% polyacrylamide gels). To compare total expression levels on SDS-PAGE between the proteins with and without NT11, we loaded each sample with the same volume (10 μL) from 20 mL of cell lysate recovered from 200 mL culture broths incubated under the same conditions. In case of soluble fractions, each purified protein pooled in the same volume of elution buffer and of which 10 μL was used. The proteins were transferred onto polyvinylidene fluoride (PVDF) microporous membrane (Millipore Cor., Merck, Darmstadt, Germany) by a semi-dry transfer (HorizBLOT 2 M, Atto Co., Osaka, Japan). The recombinant proteins with His-tag on membrane were probed with a mouse monoclonal His-tag antibody (Millipore Cor., Merck, Darmstadt, Germany) and IR-dye 800CW-conjugated goat anti-mouse IgG as secondary antibody. The detection of antibody reactivity was accomplished by the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA). Protein concentration was determined using the Bradford method (Thermo Fisher Scientific, Rockford, IL, USA).
Native PAGE and SEC
The oligomerization states of proteins were determined by native PAGE using 10% gels in the absence of reducing agents or denaturing detergents. Proteins were dissolved in a sample buffer without heating. For size-exclusion chromatography (SEC), the purified protein using Ni-NTA was injected onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare, Chicago, IL, USA), by a 500 μL loop at 0.5 mL min−1 using an ÄKTA Prime Plus FPLC system (GE Healthcare, Chicago, IL, USA). The mobile phase was 20 mM Tris–SO4, 0.15 M NaCl, pH 7.6.
Enzyme activity assay
The esterase activity of the CA was determined spectrophotometrically using p-NPA, a chromogenic substrate at 348 nm using a standard modification (Verpoorte 1967). The reaction was initiated by adding 100 μL of freshly prepared 3 mM p-NPA to 200 μL of 20 mM Tris–SO4 buffer (pH 7.6) containing CA (catalyzed reaction) or a buffer control (uncatalyzed reaction) and continuously monitored steadily for 10 min using a microplate reader (Infinite M200 PRO, TECAN, Austria). The activity was calculated from the amount of released p-nitrophenol (p-NP) from p-NPA, and one enzyme unit is defined as the formation of 1 nmol of p-NP per min.
The CO2 hydration activity of CA was determined by monitoring the time required for the pH of the reaction mixture to change from 8.3 to 7.0. Four micrograms of CA (1–3 μL) was added to 0.6 mL of ice-cold 20 mM Tris–SO4 buffer with phenol red 0.004% (pH 8.3). The reaction was initiated by adding 0.4 mL of ice-cold CO2-saturated water. The pH was monitored every 2 s during the 100-s incubation period. CA activity is expressed in Wilbur-Anderson unit (WAU) per mg of protein used (Wilbur and Anderson 1948). WAU is defined as (t0−t) / t, where t0 and t are recorded as the time required for the pH to decrease from 8.3 to 7.0 in the buffered control (uncatalyzed reaction) and CA solution (catalyzed reaction), respectively.
FACS and fluorescence spectrometry
The E. coli (BL21) expressing Hc-CA was used as a non-fluorescence control. The culture broths of E. coli (BL21) expressing YFP, NT11-YFP, and Hc-CA grown under the same conditions described above were pelleted, washed twice with PBS (pH 7.4), resuspended in PBS, and then transferred to a 5-mL round bottom tube, respectively. Fluorescence-activated cell sorting (FACS) analysis was carried on a MoFlo™ XDP Cell Sorter (Beckman Coulter, IN, USA). The events (5 × 104) were counted, but only 80% of the main population of cells was analyzed. The cells passed through a 488-nm laser beam, and the emission signal was filtered using a 529 ± 14-nm FL1 band pass filter. The light signals emitted from the cells were converted to a voltage value; a minimum voltage value (400 V) was used. The data were analyzed using the Summit software, version 5.3.
The fluorescence proteins were diluted to 0.01 mg/mL in 50 mM Tris–SO4 (pH 7.6), and their fluorescence intensities were measured using a fluorescence spectrometer (Cary Eclipse Fluorescence Spectrophotometer, Agilent Technology, CA, USA). The emission intensity was measured from 490 to 700 nm for YFP and NT11-YFP at an excitation wavelength of 400 nm.
RNA secondary structure prediction and analysis
The free energy of mRNA structure with + 200 mRNA sequence from transcription mRNA after T7 promoter was analyzed and compared by mRNA structure analyzer (http://rna.urmc.rochester.edu/RNAstructureWeb; Bellaousov et al. 2013) in 293.15 or 310.15 K, which is the induction conditions of Dsp-CAs and YFPs (20 °C), or those of Hc-CAs and Ta-CAs (37 °C), respectively. We also estimated ΔGUTR values, which show the ribosome binding ability with mRNA − 25 of the untranslated region from AUG and + 35 from N-terminus of coding sequences, by UTR designer (https://sbi.postech.ac.kr/utr_library/; Seo et al. 2013).
Results
Expression and purification of CA
In our previous work, the replacement of the first ten residues of Dsp-CA-c with NT11 sequence was found to show an expression enhancement, which also leads to the increase in the soluble protein yield, enzyme activity, and thermostability (Ki et al. 2016). We tested whether the NT11 sequence could act as an additional peptide tag for other CA gene expression enhancement. Several sequences of α-CAs were investigated by amino sequence alignment analysis (Fig. S1 of supplementary materials). We selected Hc-CA (a CA from Hahella chejuensis) and Ta-CA (a CA from Thermovibrio ammonificans) as model target CA proteins. Hc-CA is highly active in alkaline conditions but is mostly expressed in an insoluble form (Ki et al. 2013), and Ta-CA is highly thermostable CAs (Di Fiore et al. 2015), but it is also poorly expressed in E. coli host system. In the sequence alignment with Dsp-sCa-c (Fig. S2 of supplementary materials), Hc-CA and Ta-CA showed several regions with high conservation, including the Zn-binding site and the active site, though their identity was not high. Dsp-sCA-c and Hc-CA exhibit 22% identity, Dsp-sCA-c and Ta-CA exhibit 25% identity, and Hc-CA and Ta-CA exhibit 47% identity.
NT11-tag was fused to the N-termini of Hc-CA and Ta-CA to form NT11-Hc-CA and NT11-TaCA, respectively. The Ta-CA genes were inserted into pET22b(+), while all the other CA genes were inserted into pET42b(+). All constructs were successfully expressed in BL21 (DE3) E. coli. To determine the effects of the NT11-tag on the biochemical properties of CA, cloning and expressing of target genes should follow almost the same system using Dsp-nCA-c [pET42b(+), BL21 (DE3)] containing a C-terminal poly His-tag as was done in the previous study.
The induction conditions were optimized to obtain the highest protein expression levels. The pellets were dissolved in lysis buffer (50 mM Tris-sulfate, 300 mM NaCl, and 1% glycerol, pH 7.6). Protein constructs were purified from the soluble fraction obtained from sonication and centrifugation using HisPurTM Nickel resin according to the manufacturer’s protocol (Thermo Fisher Scientific Inc., Waltham, MA, USA).
At the conditions for optimum protein expression, the yield of proteins with and without NT11-tag was calculated (Table 2), as shown in Fig. 2. Under denaturing and reducing conditions, all the model proteins were observed at a molecular weight identical to their expected sizes (Table 3). The presence of the NT11-tag on the protein molecular weight was negligible owing to the very small size of the NT11-tag (1.38 kDa).

SDS-PAGE analysis of CAs expressed in E. coli (BL21) with and without the NT11-tag and comparison of their total and soluble protein yields: total protein yield (a, c) and soluble protein yield (b, d). M: marker; lane 1: Dsp-sCA-c; lane 2: NT11-Dsp-CA-c; lane 3: Hc-CA; lane 4: NT11-Hc-CA; lane 5: Ta-CA; lane 6: NT11-TaCA
The total yield increase of Dsp-sCA-c was 1.5-fold a little lower than that of Dsp-nCA-c. Interestingly, the NT11-tag significantly enhanced the expression of Ta-CA up to 6.9-fold. However, the difference in protein yield was not large between Hc-CA with and without the tag. To further analyze the effects of the NT11-tag, the expression levels of soluble proteins were also measured. Expression of the soluble forms of Dsp-sCA-c, Hc-CA, and Ta-CA is difficult to detect using small volumes of culture (2 mL). Thus, 200 mL of culture was used to determine the soluble protein yields of all proteins. Dsp-sCA-c was completely insoluble, while Dsp-nCA-c showed a high soluble yield of 21.7 mg/L. The NT11-tag clearly increased the expression of soluble Dsp-sCA-c in E. coli. Likewise, the NT11-tag also increased the expression of soluble Hc-CA and Ta-CA. From the same volume of bacterial culture, the yield of soluble NT11-Hc-CA was 1.7-fold higher than that of untagged Hc-CA, even though the ratio of the soluble to the insoluble fraction was low compared to a previously reported value (Min et al. 2016). Probably the difference in expression vector affected Hc-CA yield, this study used pET42b(+), while the previous study used the pETDuet vector. Here, the soluble yield of Ta-CA was low (approximately 6 mg/L) and consistent with values reported previously (Jo et al. 2014), it was about 6 mg/L. However, the soluble expression was changed up to 5-fold by fusion of the NT11-tag.
Structure of fusion CA
Regarding the native structure of the proteins, native PAGE analysis revealed that the Hc-CAs primarily exist as multimeric forms (Fig. 3a). As previously reported, Ta-CA forms a tetrameric complex that is stabilized by intermolecular disulfide bonds (James et al. 2014), which inhibits protein migration in native gels. Under non-denaturing condition, Ta-CAs could not be visualized using either 10 or 7% native gels. However, immunoblot analysis of purified Ta-CAs showed a predominant band indicative of a monomer and another band indicative of the dimeric form in the presence of β-mercaptoethanol (Fig. 3b). Furthermore, the oligomeric states of Ta-CA and NT11-Ta-CA were identical in the absence of β-mercaptoethanol. In this condition, two types of monomers were observed: one contains the intramolecular disulfide bond (between Cys28 and Cys183 in Ta-CA sequence) which only partially formed in the structure (James et al. 2014), while the other does not. Size-exclusion chromatography suggested that the dimeric state is the predominant form for Ta-CAs (Fig. 3c).

Analysis of protein structure. a Native PAGE of Hc-CA protein using a 10% gel (M: marker, lane 1: Hc-CA, lane 2: NT11-Hc-CA). b Immunoblotting of Ta-CA with His-tag antibody. The intrasubunit disulfide bond was formed in monomer 1, but not in monomer 2. Lanes 1 and 2: Ta-CA and NT11-Ta-CA in the presence of β-mercaptoethanol (MeSH). Lanes 3 and 4: Ta-CA and NT11-Ta-CA in the absence of β-mercaptoethanol (No MeSH). c Size-exclusion chromatography for the multimerized form of Ta-CA with and without the NT11-tag
Enzyme activity of CA
To assess whether the NT11-tag interferes with the active site of CA and alters their enzymatic activities, the activities of the CA were measured by esterase and CO2 hydration assays. The results were analyzed and are shown in Table 2 and Fig. 4. Approximately, 80% of CO2 hydration activity was retained in NT11-TaCA. The NT11-tag increased the esterase activity of Hc-CA and Ta-CA, as well as the hydration reaction of CO2 in Hc-CA.

Effect of the NT11-tag on the enzyme activity of CA. a Esterase activity. b CO2 hydration
Biochemical properties of YFP and NT11-YFP
YFP gene from mammalian expression vector pcDNA3YFP was inserted into E. coli expression vector, pET42b. In fact, the gene codon of YFP used here was not optimized for E. coli but optimized for mammalian cell expression. As expected, YFP in pET42b exhibited a relatively low expression level compared to YFP whose gene codon had been optimized for E. coli expression. We tested whether NT11 could increase the expression level of non-optimized YFP gene in E. coli by fusing NT11 to the YFP. FACS cytometry analysis of YFP and NT11-YFP recombinant E. coli indicated a higher fluorescent signal for NT11-YFP than YFP. The data displayed that small amount of YFP recombinant E. coli cells were induced and produced a small amount of YFP protein, while the others were not and only produced non-fluorescent proteins, like in the control sample (Fig. 5a). Interestingly, we found that the total protein expression of NT11-YFP was significantly amplified up to 7.6-fold compared to that of YFP (Fig. 5b–d). Accordingly, the NT11-YFP had a 3.2-fold higher soluble protein yield than that of YFP under the same conditions.

Biochemical properties of YFP and NT11-YFP. a Expression levels of YFP and NT11-YFP in E. coli as assessed by FACS. Black: control (Hc-CA recombinant E. coli cells); Orange: YFP recombinant E. coli cells; Green: NT11-YFP recombinant E. coli cells. SDS-PAGE analysis of YFP expression in E. coli (BL21): b total protein and c purified-soluble protein (M: marker; lane 1: YFP; lane 2: NT11-YFP). d Comparison of YFP and NT11-YFP protein yields. e Native PAGE of YFP and NT11-YFP using a 10% gel (M: marker; lane 1: YFP; lane 2: NT11-YFP). f Spectroscopic intensity measurements of YFP (black line) and NT11-YFP (green line) in 50 mM Tris–HCl, 150 mM NaCl, pH 8.0
Regarding the native structures of the proteins, YFP with and without the NT11-tag were both dimeric and exhibited no substantial change in size (Fig. 5e). Moreover, they displayed the same fluorescence emission spectra with the peak at 530 nm by scanning range from 490 to 700 nm at an excitation wavelength of 400 nm (Fig. 5f).
Discussion
In previous work, we designed a chimeric CA by replacing the first ten amino acid residues of active Dsp-CA-c with the NT11 sequence (VSEPHDYNYEK) of Dsp-CA-n. The resulting Dsp-nCA-c construct showed a 2-fold increase in protein expression and enzyme activity compared to the Dsp-CA-c (Ki et al. 2016). From the results, we initiated this study to investigate whether the NT11 could function as a protein production enhancement tag for the other CAs expressed in E. coli.
To develop an efficient enzymatic carbon sequestration process, CAs, in particular highly active and stable at high pH and temperatures, have been considered as prominent biocatalysts. Therefore, the recombinant production of such CAs in large quantities should be achieved. Among the CAs, Hc-CA was reported as highly active in alkaline conditions (Ki et al. 2013), whereas Ta-CA exhibited high thermostability and activity, making them ideal biocatalysts (Di Fiore et al. 2015; James et al. 2014). Hc-CA and Ta-CA were both chosen as model proteins in this study since both CAs are expressed poorly with a low yield in E. coli. Meanwhile, as another model protein, we selected YFP originated from mammalian expression vector, pcDNA3YFP. The YFP cloned into pET42b shows low expression level in E. coli, since its codon is not optimized for E. coli. We tested whether NT11 could increase the expression level of non-optimized YFP gene by fusing NT11 to the YFP.
To improve the expression levels of the target proteins, we focused on the − 10 to + 35 region in the bacterial ribosomal binding region. As observed for the chimeric protein, Dsp-nCA-c, the nucleotide segments that encode for the initial 11 amino acid residues could contribute to their high expression levels. Removal of the NT11-tag from Dsp-nCA-c (resulting in Dsp-sCA-c) decreased the protein yield from 185 to 120.7 mg/L, and all protein of Dsp-sCA-c was insoluble.
When the NT11-tag was introduced to the model proteins, the total expression levels of all model proteins were increased. The NT11-tag not only enhanced expression of the α-type CA, such as Dsp-sCA-c, Hc-CA, and Ta-CA, but also that of YFP. Since the NT11-tag would not disrupt the native structure or soluble enzyme activity, there might be no need for cleavage of the tag. The SDS-PAGE, native PAGE, immune-blotting, and SEC results indicated that a negligible difference between the structures of the proteins with and without the NT11-tag. Moreover, the fluorescence intensity of NT11-YFP was consistent with that of the untagged YFP. These data confirmed that the NT11-tag does not have any severe effect on YFP structure. In short, the structures of the model proteins might not be altered by the NT11-tag.
The expression levels of Ta-CA and YFP were amplified 6.9- and 7.6-fold by inclusion of the NT11-tag. These data suggest that the NT11-tag can be used to produce large amounts of protein. This is especially useful for production of proteins needed for industrial applications and structural analyses, such as nuclear magnetic resonance (NMR) and X-ray crystallography, which often require high concentrations of protein (Christendat et al. 2000; Yee et al. 2003). In addition, the soluble expression levels were also increased. Even though some proteins might be insoluble forms, the large amount of the expressed proteins has advantages in the refolding process. There are several refolding strategies, such as dialysis (Tsumoto et al. 2003; Umetsu et al. 2003), addition of amino acids (Kudou et al. 2011; Ohtake et al. 2011; Reddy et al. 2005), glycerol (Kohyama et al. 2010; Timasheff 2002), or cyclodextrins (Sharma and Sharma 2001; Vandevenne et al. 2011) and use of microfluidic chips (Yamaguchi et al. 2010).
Recently, a quantitative prediction method such as UTR Designer can be used to optimize the nucleotide sequences around the TIR for high-level protein production based on the calculated ∆GUTR value (Seo et al. 2013). Therefore, at the nucleotide level, the ΔGUTR values of all the fusion proteins calculated by UTR designer were − 8.89 (Table 3.). This value suggests that the secondary structure of the mRNA has higher flexibility and could interact more readily with the ribosome during translation initiation and extension. Also, it is worth noting that the shift in the ΔGUTR value caused by the addition of the NT11-tag might be related with whether this strategy should be used for enhancing both the total yields of proteins. As observed for Dsp-sCA-c, Ta-CA, and YFP, the shifts in the ΔGUTR value were 3.2, 4.5, and 4.55, respectively (Table 3), which may be involved in such significantly improved total and yields of NT11-tagged proteins. In contrast, the shift in the ΔGUTR value for Hc-CA (1.45) caused by the addition of the tag was insufficient, not inducing a remarkable increase in expression of NT11-Hc-CA.
The NT11 tag also enhanced the soluble expression of Hc-CA, Ta-CA, and YFP by 1.7-, 5.0-, and 3.2-fold, respectively. The sequence contains nine hydrophilic amino acids. Considering the low pI (4.42) and grand average of hydropathy (GRAVY) value (− 1.99), the NT11 tag is an acidic peptide. It is possible that the formation of a large net-negative charge around the tag increases electrostatic repulsion, resulting in inhibition of protein aggregation (Su et al. 2007; Zhang et al. 2004). In addition, the GRAVY values of all target proteins would become more negative, indicating higher hydrophilicity. In addition, the mRNA free energy of Dsp-nCA-c, NT11-Hc-CA, NT11-TaCA, and NT11-YFP systems are − 70.4, − 51.7, − 56.9 and − 87.2, respectively (Table 3), indicating that the mRNA structure of NT11-tagged proteins is more unstable than the NT11-untagged ones. This means that the transcript of NT11-tagged genes could be more linearized form, which is favorable for subsequent protein translation (Kudla et al. 2009). However, further researches are required to reveal how the NT11-tag promotes the soluble fraction in the expression of fusion protein in E. coli.
A major disadvantage for using expression enhancement tags is that they can interfere with the structure of the target protein, causing unexpected effects on oligomerization. Therefore, they should be removed for structural and functional applications (Waugh 2005; Young et al. 2012). Tag removal has some drawbacks (Butt et al. 2005; Esposito and Chatterjee 2006; Li 2011; Waugh 2011), particularly resulting in decreased protein yield because of precipitation and aggregation after cleavage. Regarding the esterase activity of Hc-CAs and Ta-CAs, both displayed values consistent with previous reports (Jo et al. 2014; Ki et al. 2013). Interestingly, the NT11-tag not only retained but also increased esterase activity of the model proteins. Owing to its small size, the tag did not interfere with passenger proteins, since there was no effect on the active sites containing zinc ions. Similarly, the CO2 hydration ability of NT11-tagged proteins was negligibly different compared to that of their non-tagged counterparts. Ta-CA is an excellent candidate for CO2 capture at the industrial scale because of its high activity and thermostability (James et al. 2014; Jo et al. 2014). In such view point, NT11-Ta-CA could be a promising choice for use in enzymatic CO2 capture process development owing to its high expression, CO2 hydration activity, and thermostability.
Up to now, a wide variety of protein expression tags have been used, such as MBP, GST, SUMO, mysB, and NusA. Most of them are larger than the NT11-tag in size, and their GRAVY value is higher than that of NT11-tag (Table 4). Among the fusion partners reported by Su’s group (Su et al. 2007), msyB (a 14 kDa acidic protein from E. coli) was comparable to the well-known, 55 kDa, acidic solubility enhancer, NusA (Costa et al. 2014). By fusing the partners to two target proteins (enterokinase EK and GFP), the acidity was found to greatly contribute to the enhancement of fusion protein solubility (Su et al. 2007). Nonetheless, the retention of native structure and function were not measured in that study. In addition, both model proteins were acidic, whereas our study applied an acidic fusion partner (the NT11-tag) to both acidic proteins (Dsp-sCA-c, Hc-CA, and YFP) and also a basic protein (Ta-CA). It has also been reported that EspA (20.6 kDa E. coli secreted protein A) is an effective fusion partner because owing to the high affinity between EspA and EspA-specific monoclonal antibody, this tag is convenient for protein purification. Nevertheless, it must be removed from the protein by enterokinase, because of its size and high immunogenic property. While EspA can increase the solubility of GFP from 40 to 90% (Cheng et al. 2010), the NT11-tag increased expression of YFP by 760%.
Short peptide tags have also been reported, such as poly-Lys, poly-Arg, Fh8, and H tag. In comparison, the size and GRAVY value of the NT11-tag are less than those of the Fh8 tag (8 kDa, − 0.773, respectively) (Costa et al. 2013). Based on its size, Fh8 can lead to an overoptimistic assessment in its effect on soluble protein expression. The NT11, acidic short peptide tag, shows higher GRAVY value than poly-Arg and poly-Lys, though their sizes are similar one another. Interestingly, poly-Lys and poly-Arg tags have also been reported to function as protein solubility enhancement factors for insoluble proteins (Kato et al. 2007); however, they are basic peptides and also their influence on protein activity has not been fully investigated. Further comparative studies could be interesting, in particular, to reveal how each short tag promotes the soluble fraction in the expression of fusion protein in E. coli.
In conclusion, we investigated NT11 as an effective fusion partner for improving total protein expression yields of recombinant proteins in E. coli. The NT11-tag with 11 amino acids possesses an appropriate acidity, and not only enhances protein expression but also maintains the structural stability and enzyme activity of the proteins without cleavage. The enzyme activity of the NT11-tag fused CAs was increased slightly. The native structure and function of the fusion proteins were carefully evaluated. The NT11-tag on model CAs did not cause a severe change in conformation or enzyme activity and had no effect on the fluorescence intensity of YFP. Owing to its small size and lack of influence on the biochemical properties of the target proteins, the tag can remain on the proteins in further experiments. The NT11-tag is an ideal candidate for enhancing recombinant protein expression in E. coli.
References
Allen GS, Zavialov A, Gursky R, Ehrenberg M, Frank J (2005) The cryo-EM structure of a translation initiation complex from Escherichia coli. Cell 121(5):703–712. https://doi.org/10.1016/j.cell.2005.03.023
Bellaousov S, Reuter JS, Seetin MG, Mathews DH (2013) RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41(Web Server issue):W471–W474. https://doi.org/10.1093/nar/gkt290
Bivona L, Zou ZC, Stutzman N, Sun PD (2010) Influence of the second amino acid on recombinant protein expression. Protein Expr Purif 74(2):248–256. https://doi.org/10.1016/j.pep.2010.06.005
Butt TR, Edavettal SC, Hall JP, Mattern MR (2005) SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif 43(1):1–9. https://doi.org/10.1016/j.pep.2005.03.016
Cheng Y, Gu JA, Wang HG, Yu S, Liu YQ, Ning YL, Zou QM, Yu XJ, Mao XH (2010) EspA is a novel fusion partner for expression of foreign proteins in Escherichia coli. J Biotechnol 150(3):380–388. https://doi.org/10.1016/j.jbiotec.2010.09.940
Chou CP (2007) Engineering cell physiology to enhance recombinant protein production in Escherichia coli. Appl Microbiol Biotechnol 76(3):521–532. https://doi.org/10.1007/s00253-007-1039-0
Christendat D, Yee A, Dharamsi A, Kluger Y, Savchenko A, Cort JR, Booth V, Mackereth CD, Saridakis V, Ekiel I, Kozlov G, Maxwell KL, Wu N, McIntosh LP, Gehring K, Kennedy MA, Davidson AR, Pai EF, Gerstein M, Edwards AM, Arrowsmith CH (2000) Structural proteomics of an archaeon. Nat Struct Biol 7(10):903–909. https://doi.org/10.1038/82823
Costa SJ, Almeida A, Castro A, Domingues L, Besir H (2013) The novel Fh8 and H fusion partners for soluble protein expression in Escherichia coli: a comparison with the traditional gene fusion technology. Appl Microbiol Biotechnol 97(15):6779–6791. https://doi.org/10.1007/s00253-012-4559-1
Costa SJ, Almeida A, Castro A, Domingues L (2014) Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: the novel Fh8 system. Front Microbiol 5:63. https://doi.org/10.3389/fmicb.2014.00063
Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27(3):297–306. https://doi.org/10.1016/j.biotechadv.2009.01.008
Di Fiore A, Alterio V, Monti SM, De Simone G, D'Ambrosio K (2015) Thermostable carbonic anhydrases in biotechnological applications. Int J Mol Sci 16(7):15456–15480. https://doi.org/10.3390/ijms160715456
Douette P, Navet R, Gerkens P, Galleni M, Levy D, Sluse FE (2005) Escherichia coli fusion carrier proteins act as solubilizing agents for recombinant uncoupling protein 1 through interactions with GroEL. Biochem Bioph Res Co 333(3):686–693. https://doi.org/10.1016/j.bbrc.2005.05.164
Dyson MR, Shadbolt SP, Vincent KJ, Perera RL, McCafferty J (2004) Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression. BMC Biotechnol 4:32. https://doi.org/10.1186/1472-6750-4-32
Esposito D, Chatterjee DK (2006) Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol 17(4):353–358. https://doi.org/10.1016/j.copbio.2006.06.003
Hu J, Qin H, Sharma M, Cross TA, Gao FP (2008) Chemical cleavage of fusion proteins for high-level production of transmembrane peptides and protein domains containing conserved methionines. Biochim Biophys Acta 1778(4):1060–1066. https://doi.org/10.1016/j.bbamem.2007.12.024
James P, Isupov MN, Sayer C, Saneei V, Berg S, Lioliou M, Kotlar HK, Littlechild JA (2014) The structure of a tetrameric alpha-carbonic anhydrase from Thermovibrio ammonificans reveals a core formed around intermolecular disulfides that contribute to its thermostability. Acta Crystallogr D Biol Crystallogr 70(Pt 10):2607–2618. https://doi.org/10.1107/S1399004714016526
Jo BH, Seo JH, Cha HJ (2014) Bacterial extremo-alpha-carbonic anhydrases from deep-sea hydrothermal vents as potential biocatalysts for CO2 sequestration. J Mol Catal B-Enzym 109:31–39. https://doi.org/10.1016/j.molcatb.2014.08.002
Jones MD, Fayerman JT (1987) Industrial applications of recombinant-DNA technology. J Chem Educ 64(4):337–339. https://doi.org/10.1021/ed064p337
Kapust RB, Waugh DS (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci 8(8):1668–1674. https://doi.org/10.1110/ps.8.8.1668
Kato A, Maki K, Ebina T, Kuwajima K, Soda K, Kuroda Y (2007) Mutational analysis of protein solubility enhancement using short peptide tags. Biopolymers 85(1):12–18. https://doi.org/10.1002/bip.20596
Ki MR, Min K, Kanth BK, Lee J, Pack SP (2013) Expression, reconstruction and characterization of codon-optimized carbonic anhydrase from Hahella chejuensis for CO2 sequestration application. Bioprocess Biosyst Eng 36(3):375–381. https://doi.org/10.1007/s00449-012-0788-z
Ki MR, Nguyen TKM, Kim SH, Kwon I, Pack SP (2016) Chimeric protein of internally duplicated alpha-type carbonic anhydrase from Dunaliella species for improved expression and CO2 sequestration. Process Biochem 51(9):1222–1229. https://doi.org/10.1016/j.procbio.2016.05.013
Kohl T, Schmidt C, Wiemann S, Poustka A, Korf U (2008) Automated production of recombinant human proteins as resource for proteome research. Proteome Sci 6:4. https://doi.org/10.1186/1477-5956-6-4
Kohyama K, Matsumoto T, Imoto T (2010) Refolding of an unstable lysozyme by gradient removal of a solubilizer and gradient addition of a stabilizer. J Biochem 147(3):426–431. https://doi.org/10.1093/jb/mvp184
Kozak M (2005) Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361:13–37. https://doi.org/10.1016/j.gene.2005.06.037
Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324(5924):255–258. https://doi.org/10.1126/science.1170160
Kudou M, Yunmioka R, Ejima D, Arakawa T, Tsumoto K (2011) A novel protein refolding system using lauroyl-L-glutamate as a solubilizing detergent and arginine as a folding assisting agent. Protein Expr Purif 75(1):46–54. https://doi.org/10.1016/j.pep.2010.08.011
Li YF (2011) Self-cleaving fusion tags for recombinant protein production. Biotechnol Lett 33(5):869–881. https://doi.org/10.1007/s10529-011-0533-8
Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR (2006) Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci 15(1):182–189. https://doi.org/10.1110/ps.051812706
Min KH, Son RG, Ki MR, Choi YS, Pack SP (2016) High expression and biosilica encapsulation of alkaline-active carbonic anhydrase for CO2 sequestration system development. Chemosphere 143:128–134. https://doi.org/10.1016/j.chemosphere.2015.07.020
Ohtake S, Kita Y, Arakawa T (2011) Interactions of formulation excipients with proteins in solution and in the dried state. Adv Drug Deliv Rev 63(13):1053–1073. https://doi.org/10.1016/j.addr.2011.06.011
Peti W, Page R (2007) Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif 51(1):1–10. https://doi.org/10.1016/j.pep.2006.06.024
Ramos R, Moreira S, Rodrigues A, Gama M, Domingues L (2013) Recombinant expression and purification of the antimicrobial peptide magainin-2. Biotechnol Prog 29(1):17–22. https://doi.org/10.1002/btpr.1650
Reddy RC, Lilie H, Rudolph R, Lange C (2005) L-arginine increases the solubility of unfolded species of hen egg white lysozyme. Protein Sci 14(4):929–935. https://doi.org/10.1110/ps.041085005
Rosano GL, Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5(172). https://doi.org/10.3389/fmicb.2014.00172
Seo SW, Yang JS, Kim I, Yang J, Min BE, Kim S, Jung GY (2013) Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng 15:67–74. https://doi.org/10.1016/j.ymben.2012.10.006
Sharma L, Sharma A (2001) Influence of cyclodextrin ring substituents on folding-related aggregation of bovine carbonic anhydrase. Eur J Biochem 268(8):2456–2463. https://doi.org/10.1046/j.1432-1327.2001.02125.x
Shine J, Dalgarno L (1975) Determinant of cistron specificity in bacterial ribosomes. Nature 254(5495):34–38. https://doi.org/10.1038/254034a0
Sorensen HP, Mortensen KK (2005) Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol 115(2):113–128. https://doi.org/10.1016/j.jbiotec.2004.08.004
Su Y, Zou ZR, Feng SY, Zhou P, Cao LJ (2007) The acidity of protein fusion partners predominantly determines the efficacy to improve the solubility of the target proteins expressed in Escherichia coli. J Biotechnol 129(3):373–382. https://doi.org/10.1016/j.jbiotec.2007.01.015
Terpe K (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 60(5):523–533. https://doi.org/10.1007/s00253-002-1158-6
Terpe K (2006) Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72(2):211–222. https://doi.org/10.1007/s00253-006-0465-8
Tessema M, Simons PC, Cimino DF, Sanchez L, Waller A, Posner RG, Wandinger-Ness A, Prossnitz ER, Sklar LA (2006) Glutathione-S-transferase-green fluorescent protein fusion protein reveals slow dissociation from high site density beads and measures free GSH. Cytometry A 69(5):326–334. https://doi.org/10.1002/cyto.a.20259
Timasheff SN (2002) Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry 41(46):13473–13482. https://doi.org/10.1021/bi020316e
Tsumoto K, Ejima D, Kumagai I, Arakawa T (2003) Practical considerations in refolding proteins from inclusion bodies. Protein Expr Purif 28(1):1–8. https://doi.org/10.1016/S1046-5928(02)00641-1
Umetsu M, Tsumoto K, Hara M, Ashish K, Goda S, Adschiri T, Kumagai I (2003) How additives influence the refolding of immunoglobulin-folded proteins in a stepwise dialysis system—spectroscopic evidence for highly efficient refolding of a single-chain FV fragment. J Biol Chem 278(11):8979–8987. https://doi.org/10.1074/jbc.M212247200
Vandevenne M, Gaspard G, Belgsir el M, Ramnath M, Cenatiempo Y, Marechal D, Dumoulin M, Frere JM, Matagne A, Galleni M, Filee P (2011) Effects of monopropanediamino-beta-cyclodextrin on the denaturation process of the hybrid protein BlaPChBD. Biochim Biophys Acta 1814(9):1146–1153. https://doi.org/10.1016/j.bbapap.2011.05.007
Verpoorte JA (1967) Esterase activities of human carbonic anhydrases B and C. J Biol Chem 242(18):4221–4229
Waugh DS (2005) Making the most of affinity tags. Trends Biotechnol 23(6):316–320. https://doi.org/10.1016/j.tibtech.2005.03.012
Waugh DS (2011) An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif 80(2):283–293. https://doi.org/10.1016/j.pep.2011.08.005
Wilbur KM, Anderson NG (1948) Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 176(1):147–154
Yamaguchi H, Miyazaki M, Briones-Nagata MP, Maeda H (2010) Refolding of difficult-to-fold proteins by a gradual decrease of denaturant using microfluidic chips. J Biochem 147(6):895–903. https://doi.org/10.1093/jb/mvq024
Yee A, Pardee K, Christendat D, Savchenko A, Edwards AM, Arrowsmith CH (2003) Structural proteomics: toward high-throughput structural biology as a tool in functional genomics. Acc Chem Res 36(3):183–189. https://doi.org/10.1021/ar010126g
Young CL, Britton ZT, Robinson AS (2012) Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J 7(5):620–634. https://doi.org/10.1002/biot.201100155
Zhang YB, Howitt J, McCorkle S, Lawrence P, Springer K, Freimuth P (2004) Protein aggregation during overexpression limited by peptide extensions with large net negative charge. Protein Expr Purif 36(2):207–216. https://doi.org/10.1016/j.pep.2004.04.020
Funding
This work was supported by the Basic Core Technology Development Program for the Oceans and the Polar Regions of the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning, Korea (NRF-2015M1A5A1037054) and a Marine Biomaterials Research Center grant from the Marine Biotechnology Program funded by the Ministry of Oceans and Fisheries, Korea. This work was also supported by Research Fellow Funding grant funded by the Ministry of Education, Korea (NRF-2014R1A1A2008088) and BK21 plus.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 643 kb)
Rights and permissions
About this article

Cite this article
Nguyen, T.K.M., Ki, M.R., Son, R.G. et al. The NT11, a novel fusion tag for enhancing protein expression in Escherichia coli. Appl Microbiol Biotechnol 103, 2205–2216 (2019). https://doi.org/10.1007/s00253-018-09595-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-09595-w