
Abstract
Lignin, one of the major chemical constituents of woody biomass, is the second most abundant biopolymer found in nature. The pulp and paper industry has long produced lignin on the scale of millions of tons annually as a by-product of the pulping process, and the dawn of cellulosic ethanol production has further contributed to this amount. Historically, lignin has been perceived as a waste material and burned as a fuel for the pulping process. However, recent research has been geared toward developing cost-effective technologies to convert lignin into valuable commodities. Attributing to the polyphenolic structure of lignin, enzymatic modification of its surface using laccases (benzenediol:oxygen oxidoreductases, EC 1.10.3.2) has demonstrated to be highly successful. The current study aims at developing lignin-core hyperbranched copolymers via the laccase-assisted copolymerization of kraft lignin with methylhydroquinone and a trithiol. Based on the physical properties of the resulting material, it is likely that crosslinking reactions have taken place during the drying process to produce a copolymeric network rather than discrete hyperbranched copolymers, with NMR data providing evidence of covalent bonding between monomers. Preliminary thermal analysis data reveals that the copolymeric material possesses a moderate glass transition temperature and exhibits good thermostability, thus may have potential application as a lignin-based thermoplastic. Scanning electron microscopy images confirm the smooth, glossy surface of the material and that it is densely packed. The presented results are a sustainable, ecofriendly, economic method to create an exciting novel biomaterial from a renewable feedstock while further enhancing lignin valorization.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Modern society has traditionally been based around a linear economy. The vast majority of chemicals and materials used to manufacture consumer products are derived from non-renewable petroleum resources, and once these commodities have been consumed, they are more often than not simply discarded into overflowing landfills or incinerated, the latter being a contributor to greenhouse gas emissions. However, due to the ever-growing concerns about the future welfare of the environment in which we inhabit, there has been a collaborative global effort to develop a more sustainable circular economy. Under this paradigm, raw materials and chemicals used for the manufacture of consumer goods will originate from renewable resources and be designed for reuse, recycle, and biodegradability (Stahel 2016). In this manner, the entire process, from manufacture to degradation, is almost carbon neutral and will thus only contribute a minor portion to society’s carbon footprint.
Lignocellulosic biomass has proved to be a viable alternative to petroleum for the production of fuels, chemicals, and materials due to its renewability and biodegradability. Furthermore, because humans are unable to metabolize lignocellulose, there is no competing demand to use lignocellulose as a food source, making lignocellulosic biomass an exceptionally appealing bioresource. One of the major sources of lignocellulosic biomass is wood, which, depending on its origin, is composed of 35–50% cellulose, 25–30% hemicellulose, and 15–30% lignin (Ragauskas et al. 2006). Lignocellulose bioconversion technologies aim at deconstructing lignocellulosic biomass into its individual components which are then transformed into marketable fuels, chemicals, and materials via downstream processing. While the sugars derived from the cellulose and hemicellulose fractions of woody biomass have found tremendous application, such as for the production of second-generation bioethanol, the valorization of lignin still remains a challenge (Ragauskas et al. 2014).
Lignin is a highly irregular amorphous polyphenol and is the second most abundant biopolymer on terrestrial Earth behind cellulose. Its function in nature is to provide structural support, antimicrobial resistance, and water conductive properties for the tree or plant. Industrial production of lignin occurs on the order of approximately 50 million tons a year globally in the pulping and paper industry as a by-product of the pulping process, while another 60 million tons a year is expected to be produced in the USA alone in the coming years as a result of cellulosic ethanol production (Langholtz et al. 2014). Given the many potential advantages of using lignin as a raw material for the manufacture of consumer products, it is largely underutilized, with only 2% of industrially generated lignin used in the fabrication of marketable products, while the remainder is burned for fuel (Lora and Glasser 2002; Kai et al. 2016). Recently, however, much research has been devoted to the incorporation of lignin into resins used in the production of adhesives and particleboards as a partial replacement of typical phenol-formaldehyde resins (Lora and Glasser 2002; Hüttermann et al. 2001). The incorporation of lignin is not only economically advantageous, but it also provides an end product with comparable performance to products containing traditional phenol-formaldehyde resins while reducing toxic formaldehyde emissions. As another prominent example, lignin has been copolymerized with telechelic polybutadiene for the production of a lignin-based thermoplastic with promising mechanical properties (Saito et al. 2012).
One method that has proved to be highly effective in converting lignin into a useful material is enzymatic functionalization (Kalia et al. 2014). In particular, laccases (benzenediol:oxygen oxidoreductases, EC 1.10.3.2) have shown to be highly applicable in assisting the copolymerization of lignin with a variety of monomers and polymers for the synthesis of novel biomaterials that have found applications in adhesives, fillers, and plastics (Cannatelli and Ragauskas 2015a, 2016). These copper-containing enzymes catalyze the mono-electron oxidation of a variety of organic substrates, including lignin, while concomitantly reducing molecular oxygen to water (Messerschmidt 1997; Solomon et al. 2008), and owing to their wide substrate range and mild operation conditions, they have gained increased industrial attention in recent years. The vast majority of industrially relevant laccases originate from fungi where their main function is to oxidize lignin (Thurston 1994), hence the reason fungal laccases are a prime candidate for successful lignin functionalization.
There has been an increase in research within the field of hyperbranched polymers over the past few years due to their desirable properties, such as high functionality, high solubility, and low viscosity (Zheng et al. 2015). Relating to this, using lignin as a core polymer and branching out on its surface provides a promising strategy to synthesize novel biomaterials from lignin. Kai et al. (2016) have developed a series of lignin-core supramolecular hyperbranched polymers by functionalizing the surface of lignin with poly(ethylene glycol) methyl ether methacrylate (PEGMA) via atom transfer radical polymerization (ATRP) followed by combination with α-cyclodextrin. The produced hydrogels exhibit many promising properties and may have potential application within the biomedical field. The current study aims to develop a lignin-core hyperbranched copolymer (LCHC) by employing laccases to continuously graft small molecules onto the surface of kraft lignin. We envision that the resulting LCHCs may have potential application as a renewable and sustainably produced lignin-based biocomposite plastic. In a previous study utilizing substituted hydroquinones as lignin model compounds, we were able to successfully couple a dithiol to the laccase-generated para-quinones via a laccase-catalyzed thiol-Michael addition (Cannatelli and Ragauskas 2015b). Building on this work and similar work within our group (Cannatelli and Ragauskas 2017), the current study applies this fundamental laccase-catalyzed coupling chemistry for the synthesis of LCHCs by combining kraft lignin with methylhydroquinone and a trithiol mediated by laccase catalysis.
Materials and methods
Materials
Laccase from Trametes villosa was donated by Novo Nordisk Biochem (Franklinton, USA). Southern pine softwood kraft lignin, isolated via the LignoBoost process, was donated by Domtar (Plymouth, USA) and was purified following standard methods developed previously within our group (Froass 1996; Kosa 2012) prior to use (see supplementary material). Endo-N-hydroxy-5-norbornene-2,3-dicarboximide (NHND) was purchased from Alfa Aesar (Tewksbury, USA). All other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, USA) and were used as received, except chloroform and dioxane, which were distilled just prior to use. Tris(2-mercaptoethyl)amine was synthesized based on a combination of two previously published syntheses (Barbaro et al. 1994; Sun et al. 2012) (see supplementary material). Nitrogen gas was purchased from Airgas (Radnor, USA) and was dried using a Drierite™ gas-drying unit (Sigma-Aldrich).
Enzyme assay
Laccase activity was determined according to standard literature procedures which involve the oxidation of 2,2′-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt (ABTS) (Wolfenden and Willson 1982). The oxidation of 3.50 mL of solution consisting of 50 μM ABTS in 0.10 M sodium acetate buffer (pH 5.0) by 8.0 × 10−5 mL laccase solution was observed spectrophotometrically at room temperature (22 °C) via a UV-vis spectrophotometer by following the absorbance increase at 420 nm (ε 420 = 3.6 × 104 M−1 cm−1). Laccase activity is expressed in units (U) per mL where U = μmol ABTS oxidized per minute. The laccase activity was measured to be 1510 U per mL enzyme solution.
Synthesis of LCHCs
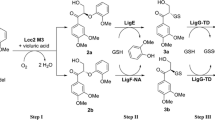
Approximately 100 mg of pure kraft lignin was added to a 50 mL round-bottom flask equipped with a stir bar. 2 mL of dioxane was then added, followed by 8 mL of 0.1 M sodium phosphate buffer pH 8.0, and the mixture was stirred and heated to 50 °C for approximately 10 min to dissolve the lignin. Then, 496.56 mg (4.0 mmol) of methylhydroquinone was added to the mixture followed by 333 μL (394.77 mg, 2.0 mmol) of tris(2-mercaptoethyl)amine. Finally, 200 U of laccase was added and the reaction mixture was allowed to stir for 20 h at 50 °C. After this time, the mixture was allowed to cool to room temperature and centrifuged, the supernatant decanted, and the remaining brown sludge washed with 3 × 15 mL deionized water followed by 3 × 15 mL dioxane. The brown paste was then dried in a vacuum oven at 30 °C for 24 h. Gravimetric yield is 524 mg.
Elemental analysis
Elemental analysis of the LCHCs was conducted by Atlantic Microlab, Inc. (Norcross, USA). C, H, N, and S were determined as percent weight via combustion. The results of elemental analysis are as follows: 48.86% C, 6.15% H, 4.64% N, and 30.17% S. Based on the nitrogen content, the tris(2-mercaptoethyl)amine mass component of the copolymeric material was calculated as follows:
Assuming that all nitrogen in the copolymeric material originates from tris(2-mercaptoethyl)amine, then
Thus, the remaining 35% of the material is composed of lignin and methylhydroquinone; however, it is not possible to distinguish between the two based on elemental analysis.
Molecular weight determination
The molecular weight distribution of the pure kraft lignin was determined via gel permeation chromatography (GPC) using an Agilent Technologies SECcurity GPC system equipped with ultraviolet (UV) and refractive index (RI) detectors. Tetrahydrofuran (THF) was used as the mobile phase, and the pure kraft lignin sample was prepared with a concentration of 1 mg/mL. The LCHCs could not be analyzed by GPC due to insolubility in all tested solvents.
Structural characterization
Both pure kraft lignin and LCHCs were dried in a vacuum oven at 30 °C for 48 h prior to analysis. Fourier transform infrared (FTIR) spectroscopy experiments were performed on a PerkinElmer Spectrum 100 FTIR spectrometer using the attenuated total reflectance (ATR) method with 128 scans. All nuclear magnetic resonance (NMR) spectroscopy experiments were carried out on a Bruker 400 MHz spectrometer. 1H, 13C, 13C distortionless enhancement by polarization transfer (DEPT)-135, and 1H-13C heteronuclear multiple bond correlation (HMBC) NMR experiments were conducted at 50 °C using deuterated dimethyl sulfoxide (DMSO-d 6) as the solvent. All chemical shifts are given in ppm relative to trimethylsilane (TMS), and multiplicities are designated as singlet (s), doublet (d), triplet (t), and multiplet (m). Approximately 75 mg of pure kraft lignin or LCHCs was used for the analyses; however, it must be noted that the LCHCs needed to be ground up and vigorously stirred in the NMR solvent at 50 °C for 6 h to obtain a dark cloudy mixture suitable for NMR analysis. Quantitative 31P NMR analysis was conducted on the pure kraft lignin only. The method and solvent preparation are based on experiments developed by Granata and Argyropoulos (1995) and Zawadzki and Ragauskas (2001) (see supplementary material).
Thermal analyses
Thermal analysis of the purified kraft lignin and LCHCs was conducted on a PerkinElmer Pyris 1 thermogravimetric analyzer (TGA). Approximately 15 mg of sample was loaded into the sample holder, and the experiments were conducted under nitrogen (flow rate of 50 mL/min) with the following temperature profile: heat from 50 to 600 °C at a rate of 20 °C/min and holding at 600 °C for 5 min. Data was recorded as mass % of starting material. Glass transition (T g) data was collected via a TA Instruments Q200 differential scanning calorimeter (DSC). Approximately 5 mg of sample was placed in the sample pan, and the experiments were conducted under nitrogen (flow rate of 50 mL/min) with the following temperature profile: hold at 0 °C for 5 min, heat from 0 to 160 °C at 20 °C/min, hold at 160 °C for 5 min, cool to 0 °C at a rate of 20 °C/min, hold at 0 °C for 5 min, heat from 0 to 160 °C at 20 °C/min, hold at 160 °C for 5 min, and cool to 25 °C at a rate of 20 °C/min. Data was collected during the second heating cycle.
Surface characterization
Surface morphology of pure kraft lignin and LCHCs was analyzed on a Zeiss LEO 1530 Gemini field emission scanning electron microscope (SEM). The samples were sputter coated with gold prior to analysis to provide a thin conducting layer on the sample surface.
Results
Pure kraft lignin molecular weight data and structural characterization
Molecular weight distribution data for the purified kraft lignin was obtained via GPC. The number average molecular weight (M n) and weight average molecular weight (M w) were quite small, with values of 993 and 2352 g/mol, respectively, while the polydispersity index (PDI) was also quite narrow, yielding a value of 2.37.
Structural features of the purified kraft lignin were elucidated via various spectroscopic techniques. The 1H and 13C NMR spectra (Fig. S1a and S2a) show significant amounts of methoxyl protons and carbons at chemical shifts of 3.79 and 55.64 ppm, respectively, which are typical of softwood kraft lignins (Sealey and Ragauskas 1998). The 1H NMR spectrum revealed signals for aromatic protons centered around 6.77 ppm and a broad peak for phenolic protons centered around 8.64 ppm. The FTIR spectrum (Fig. S3a) also displays a prominent O–H stretching absorption in the range of 3150–3550 cm−1. Quantitative 31P NMR spectroscopy is another invaluable tool to analyze the abundance of different types of hydroxyl groups present in lignin. The quantitative 31P NMR spectrum is displayed in Fig. S4, and the various types of hydroxyl groups have been quantified using NHND as an internal standard (Table 1). As can be seen in Table 1, the guaiacyl hydroxyl group is the most abundant with 2.06 mmol/g lignin, while there are also substantial amounts of C5 condensed and aliphatic hydroxyl groups, which are characteristic of alkali-processed softwood lignins (Cateto et al. 2008).
Structural characterization of LCHCs
NMR spectroscopic studies provided insight into the possible structure of the copolymeric material, which is presented in Fig. 1. Comparing the 1H NMR spectrum of the copolymeric material (Fig. S1d) with the 1H NMR spectra of its constituents (i.e. pure kraft lignin, methylhydroquinone, and tris(2-mercaptoethyl)amine (Fig. S1)), the structural features of each component can be identified and a full assignment of the spectrum can be achieved. Furthermore, a broadening of the peaks in the 1H NMR spectrum, particularly of the phenolic protons dispersed around 8.31 ppm, aromatic protons in the range of 6.45–6.70 ppm, the methylene protons at 2.85 ppm, and the methyl protons between 2.05 and 2.10 ppm, is indicative of polymerization. The methoxy protons originating from lignin are still present at 3.72 ppm, albeit at a rather low intensity, while the S–H peak at 2.33 ppm in Fig. S1c is unable to be identified, which is likely due to the decreased ratio of free S–H to CH2 groups in the LCHCs compared to the unreacted tris(2-mercaptoethyl)amine, indicating the successful Michael addition of the thiol group.

Proposed structure of lignin-core hyperbranched copolymers
Analysis of the 13C NMR data further aids in confirming the various structural features of the copolymeric material. As can be seen from the 13C NMR spectrum in Fig. S2d, and by comparison with the 13C NMR spectra of the individual components (Fig. S2), all the structural features of the various constituents can be identified and assigned. The degree of substitution of the carbon atoms and therefore their origin were confirmed via 13C DEPT-135 NMR analysis (Fig. S2e).
Covalent linkage between each individual component, an indication of successful copolymerization, was analyzed via 1H-13C HMBC NMR. The HMBC spectrum of the copolymeric material (Fig. S5a) displays a signal that correlates the proton peak at 2.91 ppm and the carbon peak at 121.80 ppm, which we attribute to a 3 J correlation between methylene protons of tris(2-mercaptoethyl)amine with a quaternary aromatic carbon of methylhydroquinone. Thus, this signal provides evidence of a covalent linkage between tris(2-mercaptoethyl)amine and methylhydroquinone. This assignment was further supported by evidence from the 1H-13C HMBC spectrum of the product of the reaction of tris(2-mercaptoethyl)amine with methyl-1,4-quinone in the absence of laccase (Fig. S5b), which displays the same signal. Correlations representing covalent bonding between lignin and either of the other two components could not be unambiguously identified in the HMBC spectrum.
Thermal analysis
Thermal analysis of the copolymeric material was accomplished by TGA and DSC methods and the data compared to the data obtained for pure kraft lignin. The TGA curves for both the copolymeric material and pure kraft lignin are presented in Fig. 2 and the pertinent data extracted from the curves is given in Table 2. Based on the decomposition temperature, T d, and peak derivative temperature, T p (temperature at which greatest mass loss occurs), it can be inferred that the copolymeric material possesses greater thermostability compared to pure kraft lignin, with T d and T p values of 238 and 266 °C, respectively, for the copolymeric material, which are approximately 100 °C higher than those obtained for pure kraft lignin. The mass percent remaining at 500 °C (i.e. char yield) was significantly less for the copolymeric material compared to the pure kraft lignin, 22% as opposed to 52%, which is expected due to the lower lignin content in the copolymeric material. The T g range for the copolymeric material obtained from the DSC curve (Fig. S6) was measured to be 50–60 °C, while no T g range could be determined accurately for pure kraft lignin based on DSC measurements, which is not surprising as it has been documented that while lignin in its native state within wood exhibits a T g range, kraft lignin in its purified form does not exhibit a distinctive T g (Lora and Glasser 2002).

TGA curve of pure kraft lignin (blue) and copolymeric material (red) (Color figure online)
Surface characterization
A sample of the copolymeric material is displayed in Fig. 3, which shows the smooth, glossy surface of the material. It can also be noticed that the material takes on the characteristic dark brown color of lignin. The surface morphology of the pure kraft lignin sample and the copolymeric material was examined via SEM; a few representative images are provided in Fig. 3. Inspection of the pure kraft lignin sample in Fig. 3b exposes the porous, rock-like structure of lignin. The copolymeric material, on the other hand, has a rather smooth surface, which is visible to the naked eye, and confirmed by SEM images (Fig. 3). A top view of the copolymeric material (Fig. 3c, d) shows the smooth surface with crater-like structures dispersed throughout, while an extreme close-up view reveals the perfectly spherical shape of one such crater (Fig. 3d), which seems to be protruding from the surface. Furthermore, the material was fractured and examination of the fracture line (Fig. 3e, f) also reveals a smooth surface and demonstrates that the material is densely packed throughout.

a Image of the copolymeric material. b–f SEM images of pure kraft lignin (b) and copolymeric material—top view (c, d) and fracture line (e, f)
Discussion
The use of laccases for the copolymerization of lignin with both low molecular weight compounds and other polymers has proved to be a successful strategy for lignin functionalization in recent years (Cannatelli and Ragauskas 2016). Furthermore, the use of lignin as a core macromolecular scaffold for which hyperbranched polymers can be constructed upon has only just been established in the past few years and presents an exciting new route for lignin valorization (Li et al. 2014; Kai et al. 2016). With both these in mind, we viewed this as a great opportunity to synthesize LCHCs via laccase-assisted grafting of small molecules onto lignin.
For this study to be successful, it was important to select an appropriate lignin that would be conducive to laccase-assisted functionalization. We decided on an industrial softwood kraft lignin for two main reasons: (1) kraft lignin is highly abundant as it is produced in the millions of tons by the pulp and paper industry as a by-product of the kraft pulping process, and (2) softwood lignins are composed of primarily guaiacyl units (Vanholme et al. 2010), which, unlike syringyl units found in hardwood lignins, contain a vacant position on the aromatic ring, which is essential for significant grafting to occur. The obtained industrial softwood kraft lignin was treated prior to use to remove any trace metals, extractives, and remaining carbohydrates. 31P NMR analysis of the purified lignin using NHND as internal standard revealed guaiacyl hydroxyl groups as the most abundant hydroxyl group (Table 1), which is reasonable for softwood kraft lignins (Cateto et al. 2008). This is desirable as it infers that there is an ample amount of positions on the lignin macromolecule for laccase-mediated grafting of small molecules to take place. Molecular weight distribution data revealed a rather low M w of 2352 g/mol, which equates to approximately 13 monolignol units per lignin polymer.
Based on previous research conducted in our group on laccase-catalyzed coupling chemistry, we identified thiols as good nucleophiles for the Michael addition reaction with laccase-generated para-quinones (Cannatelli and Ragauskas 2015b). Thus, we attempted to synthesize LCHCs utilizing this fundamental laccase-catalyzed coupling chemistry. Methylhydroquinone was selected as the quinone precursor because it contains three vacant positions for potential nucleophilic addition and the methyl group allows for facile detection of the hydroquinone monomer within the copolymer via NMR spectroscopic analysis. A trithiol was selected as a prime bridging reagent for the synthesis of LCHCs because the thiol groups add rapidly and reliably to in situ-generated para-quinones. Thus, the synthesis of LCHCs proceeded using purified kraft lignin, methylhydroquinone, and tris(2-mercaptoethyl)amine as the components and laccases as the catalysts.
A proposed mechanism for the formation of LCHCs is provided in Fig. 4. As can be seen, laccases are capable of oxidizing both lignin and methylhydroquinone to generate phenoxy radicals, which can couple with one another via either carbon-carbon (C–C) or carbon-oxygen (C–O) bond formation. This is followed by laccase-catalyzed oxidation of the methylhydroquinone moiety to produce a para-quinone, which subsequently undergoes nucleophilic addition by tris(2-mercaptoethyl)amine. The remaining free thiols are then capable of reacting with laccase-generated para-quinones that may be present in the reaction mixture, and the laccase-catalyzed polymerization continues. While the presented reaction mechanism represents an idealized scenario, it must be mentioned that other mechanistic routes may occur, such as radical-radical C–C or C–O couplings between methylhydroquinone radicals or sulfur-sulfur couplings between laccase-mediator-generated sulfur radicals.

Proposed reaction mechanism for the laccase-catalyzed synthesis of lignin-core hyperbranched copolymers
The product that precipitated out of the reaction mixture was in the form of a thick brown paste, which was subsequently washed with deionized water and then dioxane to remove any unreacted lignin that may have also precipitated. Following drying in a vacuum oven at 30 °C for 24 h, the paste had hardened into a solid glossy material that was insoluble in organic solvents. It is likely that curing took place during the drying process, possibly due to the presence of methylquinone moieties as well as reactive free thiol end groups, which has been known to occur for hyperbranched polymers synthesized with tris(2-mercaptoethyl)amine and ethylene glycol diacrylate (Sun et al. 2012). Thus, it is likely that a network of LCHCs exists, such as that illustrated in Fig. 5, rather than discrete LCHCs. This is the likely cause of the lack of solubility of the synthesized LCHCs in organic solvents, which is not uncommon for lignin-containing copolymers (Saito et al. 2012; Sivasankarapillai et al. 2012; Li et al. 2014). It is due to this insolubility that molecular weight data for the synthesized LCHCs could not be obtained via GPC analysis.

Schematic of lignin-core hyperbranched copolymer network
The lack of solubility of the copolymeric material in any organic solvent tested posed a serious challenge for structural analysis at the molecular level. However, upon grinding up the material and vigorously stirring in DMSO-d 6 at 50 °C for 6 h, we were able to obtain a dark cloudy mixture that was suitable for NMR analysis. Comparison of the 1H and 13C NMR spectra of the copolymeric material (Fig. S1d and S2d, respectively) with the individual 1H and 13C NMR spectra of the three components—lignin, methylhydroquinone, and tris(2-mercaptoethyl)amine (Fig. S1 and S2)—established that all three components are present in the copolymeric material, with 13C assignments confirmed via 13C DEPT-135 NMR analysis (Fig. S2e). Furthermore, a broadening of the peaks in the 1H NMR spectrum of the copolymeric material suggested that successful polymerization of the constituent monomers had occurred. It must also be mentioned that FTIR spectra of the kraft lignin and the copolymeric material were obtained (Fig. S3); however, no fine structural details could be elucidated.
While the 1D NMR results provide evidence that all three components exist in polymer form within the copolymeric material, they do not provide any information about covalent linkages between each component, which is important to decipher whether the material is just a simple blend of polymers or if it is a true copolymer. Therefore, it was necessary to analyze the copolymeric material via correlation spectroscopy, such as 1H-13C HMBC NMR. Indeed, the 1H-13C HMBC NMR spectrum of the copolymeric material (Fig. S5a) displayed a signal that correlates methylene protons of tris(2-mercaptoethyl)amine with a quaternary aromatic carbon of methylhydroquinone, which was supported by a similar signal for the product of the uncatalyzed reaction of tris(2-mercaptoethyl)amine with methyl-1,4-quinone (Fig. S5b). Thus, a covalent linkage between two of the components (i.e. tris(2-mercaptoethyl)amine and methylhydroquinone) had been identified. Unfortunately however, no covalent linkages between lignin and either of the other two components could be identified from the 1H-13C HMBC NMR spectrum, possibly due to the low amount of lignin in the copolymeric material, or the 1H and 13C correlations were too great a range to be detected in an observable quantity by the HMBC experiment.
Thermal analysis of the copolymeric material via TGA not only provides useful information regarding the thermal properties of the material but, in this scenario, may also provide structural insight. By inspecting the TGA curves in Fig. 2, it can be noticed that the curve for the copolymeric material is very clean, in that the material exhibits a distinct onset temperature for thermal degradation. A clean curve such as this would imply that the material is a uniform copolymer network with covalent linkages rather than a blend of polymers, as the latter would most likely display multiple degradation temperatures for each of the individual components. Furthermore, the curve for kraft lignin and the curve for the copolymeric material are quite different, and no sign of any free lignin within the copolymeric material can be detected.
Based on the TGA data, it was determined that the copolymeric material possesses greater thermostability compared to the kraft lignin, with a T p value 100 °C higher (Table 2). Comparison of these values with other LCHCs synthesized elsewhere, such as lignin copolymerized with PEGMA, reveals that our copolymeric material is less thermally stable, with T p values for the lignin-PEGMA hyperbranched copolymers approximately 150 °C higher than the value obtained for our material (Kai et al. 2016). However, it must be noted that the M w of the starting lignin used for the synthesis of lignin-PEGMA hyperbranched copolymers is an order of magnitude larger than the M w of the lignin used in this study, which may be a likely cause for the increased thermostability of the lignin-PEGMA hyperbranched copolymers. The T g range obtained for the copolymeric material (50–60 °C) is comparable to T g values obtained for lignin-based thermal-responsive elastomers that were synthesized by copolymerizing lignin with hyperbranched poly(ester-amine-amide) (Li et al. 2014).
By visual inspection of the copolymeric material, it can be seen that the material possesses a glossy surface (Fig. 3a), which has also been observed for other lignin-containing copolymers (Sivasankarapillai et al. 2012; Li et al. 2014). Delving further into the fine details of the surface structure, examination of the SEM images (Fig. 3) uncovers a smooth homogeneous surface, which seems to extend throughout the bulk material, as witnessed by SEM images of the fracture line (Fig. 3e, f). This is interesting given the porous nature of the kraft lignin (Fig. 3b), which correlates well with the role of native lignin in wood as a means for water conduction. The lack of resemblance of any porous structure within the copolymeric material may be due to the low overall lignin content, which, given that tris(2-mercaptoethyl)amine accounts for 65% of the material’s mass (based on elemental analysis), realistically may only account for about 10% of the material’s mass. The morphology of the material, which lacks any order or orientation, is in contrast to a laccase-generated lignin-isocyanate copolymer, which shows a distinct fibrillar structure with the fibers oriented in a uniform direction (Milstein et al. 1994).
This study demonstrates the potential to synthesize LCHCs via laccase-catalyzed coupling chemistry. It has been stated previously that the vast majority of hyperbranched polymers synthesized to date are of laboratory interest rather than commercial usage, but value-added products are gradually being developed (Zheng et al. 2015). The current study is a preliminary study, and future work aimed at developing practical materials utilizing this chemistry may focus on different monomer selection so as to produce a material with desirable thermal or mechanical properties. Nonetheless, the material synthesized in this study possesses a relatively moderate T g (50–60 °C), which is sustained throughout multiple heating cycles, thus, combined with its resistance to thermal degradation (based on a high T d value), may present as a potential lignin-based thermoplastic. Addition of an external plasticizer into the formulation, such as polybutadiene, which has been combined with lignin in the past for the production of lignin-based thermoplastic, is one potential method that could be used to improve the physical properties of the material (Saito et al. 2012). Also, given that the material synthesized in this study exists as a paste before drying, which then presumably undergoes crosslinking reactions, it may be worthwhile evaluating its potential use as a lignin-based adhesive in particleboard manufacture, as it has been demonstrated in the past that lignin copolymerized with other small molecules or polymers via laccase catalysis produces resins that can be used as adhesives for particleboards and wool floor coverings (Ibrahim et al. 2013; Aracri et al. 2014). Nevertheless, this study provides evidence that laccases can be employed for the synthesis of LCHCs under sustainable conditions and provides yet another novel route for lignin valorization.
References
Aracri E, Diaz Blanco C, Tzanov T (2014) An enzymatic approach to develop a lignin-based adhesive for wool floor coverings. Green Chem 16(5):2597–2603
Barbaro P, Bianchini C, Scapacci G, Masi D, Zanello P (1994) Dioxomolybdenum (VI) complexes stabilized by polydentate ligands with NO3, N2O2, and NS2 donor-atom sets. Inorg Chem 33(14):3180–3186
Cannatelli MD, Ragauskas AJ (2015a) Value added biomaterials via laccase-mediated surface functionalization. J Biotechnol Biomater 5(1):1–2
Cannatelli MD, Ragauskas AJ (2015b) Laccase-catalyzed synthesis of 2,3-ethylenedithio-1,4-quinones. J Mol Catal B Enzym 119:85–89
Cannatelli MD, Ragauskas AJ (2016) Conversion of lignin into value-added materials and chemicals via laccase-assisted copolymerization. Appl Microbiol Biotechnol 100(20):8685–8691
Cannatelli MD, Ragauskas AJ (2017) Two decades of laccases: advancing sustainability in the chemical industry. Chem Rec 17(1):122–140
Cateto CA, Barreiro MF, Rodrigues AE, Brochier-Salon MC, Thielemans W, Belgacem MN (2008) Lignins as macromonomers for polyurethane synthesis: a comparative study on hydroxyl group determination. J Appl Polym Sci 109(5):3008–3017
Froass PM (1996) Structural changes in lignin during kraft pulping and chlorine dioxide bleaching. Dissertation, Institute of Paper Science and Technology, Atlanta, USA
Granata A, Argyropoulos DS (1995) 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J Agric Food Chem 43(6):1538–1544
Hüttermann A, Mai C, Kharazipour A (2001) Modification of lignin for the production of new compounded materials. Appl Microbiol Biotechnol 55(4):387–394
Ibrahim V, Mamo G, Gustafsson PJ, Hatti-Kaul R (2013) Production and properties of adhesives formulated from laccase modified kraft lignin. Ind Crop Prod 45:343–348
Kai D, Low ZW, Liow SS, Karim AA, Ye H, Jin G, Li K, Loh XJ (2016) Development of lignin supramolecular hydrogels with mechanically responsive and self-healing properties. ACS Sustain Chem Eng 3(9):2160–2169
Kalia S, Thakur K, Kumar A, Celli A (2014) Laccase-assisted surface functionalization of lignocellulosics. J Mol Catal B Enzym 102:48–58
Kosa M (2012) Direct and multistep conversion of lignin to biofuels. Dissertation, Georgia Institute of Technology, Atlanta, USA
Langholtz M, Downing M, Graham R, Baker F, Compere A, Griffith W, Boeman R, Keller M (2014) Lignin-derived carbon fiber as a co-product of refining cellulosic biomass. SAE Int J Mater Manf 7(1):115–121
Li H, Sivasankarapillai G, McDonald AG (2014) Lignin valorization by forming thermally stimulated shape memory copolymeric elastomers—partially crystalline hyperbranched polymer as crosslinks. J Appl Polym Sci 131(22):41103/1–41103/10
Lora JH, Glasser WG (2002) Recent industrial applications of lignin: a sustainable alternative to nonrenewable materials. J Polym Environ 10(1):39–48
Messerschmidt A (1997) Multi-copper oxidases. World Scientific, Singapore
Milstein O, Hüttermann A, Fründ R, Lüdemann HD (1994) Enzymatic co-polymerization of lignin with low-molecular mass compounds. Appl Microbiol Biotechnol 40(5):760–767
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) The path forward for biofuels and biomaterials. Science 311(5760):484–489
Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis MF, Davison BH, Dixon RA, Gilna P, Keller M, Langan P, Naskar AK, Saddler JN, Tschaplinski TJ, Tuskan GA, Wyman CE (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344(6185):1246843
Saito T, Brown RH, Hunt MA, Pickel DL, Pickel JM, Messman JM, Baker FS, Keller M, Naskar AK (2012) Turning renewable resources into value-added polymer: development of lignin-based thermoplastic. Green Chem 14(12):3295–3303
Sealey J, Ragauskas AJ (1998) Residual lignin studies of laccase-delignified kraft pulps. Enzym Microb Technol 23(7–8):422–426
Sivasankarapillai G, McDonald AG, Li H (2012) Lignin valorization by forming toughened lignin-co-polymers: development of hyperbranched prepolymers for cross-linking. Biomass Bioenergy 47:99–108
Solomon EI, Augustine AJ, Yoon J (2008) O2 reduction to H2O by the multicopper oxidases. Dalton Trans 30:3921–3932
Stahel WR (2016) Circular economy. Nature 531(7595):435–438
Sun M, Hong CY, Pan CY (2012) A unique aliphatic tertiary amine chromophore: fluorescence, polymer structure, and application in cell imaging. J Am Chem Soc 134(51):20581–20584
Thurston CF (1994) The structure and function of fungal laccases. Microbiology 140(1):19–26
Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W (2010) Lignin biosynthesis and structure. Plant Physiol 153(3):895–905
Wolfenden BS, Willson RL (1982) Radical-cations as reference chromogens in kinetic studies of one electron transfer reactions: pulse radiolysis studies of 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulphonate). J Chem Soc Perkin Trans II 7:805–812
Zawadzki M, Ragauskas AJ (2001) N-hydroxy compounds as new internal standards for the 31P-NMR determination of lignin hydroxy functional groups. Holzforschung 55(3):283–285
Zheng Y, Li S, Weng Z, Gao C (2015) Hyperbranched polymers: advances from synthesis to applications. Chem Soc Rev 44(12):4091–4130
Acknowledgments
The authors are thankful for a student fellowship supported by the Renewable Bioproducts Institute at Georgia Institute of Technology. The authors would also like to thank Dr. Y. Berta for her assistance in gathering SEM images and Brett Hester for his help with TGA experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was funded by a student fellowship supported by the Renewable Bioproducts Institute at Georgia Institute of Technology.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 775 kb)
Rights and permissions
About this article

Cite this article
Cannatelli, M.D., Ragauskas, A.J. Laccase-mediated synthesis of lignin-core hyperbranched copolymers. Appl Microbiol Biotechnol 101, 6343–6353 (2017). https://doi.org/10.1007/s00253-017-8325-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8325-2