
Abstract
The short-chain dehydrogenase/reductase (SDR) family, the largest family in dehydrogenase/reductase superfamily, is divided into “classical,” “extended,” “intermediate,” “divergent,” “complex,” and “atypical” groups. Recently, several open reading frames (ORFs) were characterized as intermediate SDR aldehyde reductase genes in Saccharomyces cerevisiae. However, no functional protein in the atypical group has been characterized in S. cerevisiae till now. Herein, we report that an uncharacterized ORF YLL056C from S. cerevisiae was significantly upregulated under high furfural (2-furaldehyde) or 5-(hydroxymethyl)-2-furaldehyde concentrations, and transcription factors Yap1p, Hsf1p, Pdr1/3p, Yrr1p, and Stb5p likely controlled its upregulated transcription. This ORF indeed encoded a protein (Yll056cp), which was grouped into the atypical subgroup 7 in the SDR family and localized to the cytoplasm. Enzyme activity assays showed that Yll056cp is not a quinone or ketone reductase but an NADH-dependent aldehyde reductase, which can reduce at least seven aldehyde compounds. This enzyme showed the best Vmax, Kcat, and Kcat/Km to glycolaldehyde, but the highest affinity (Km) to formaldehyde. The optimum pH and temperature of this enzyme was pH 6.5 for reduction of glycolaldehyde, furfural, formaldehyde, butyraldehyde, and propylaldehyde, and 30 °C for reduction of formaldehyde or 35 °C for reduction of glycolaldehyde, furfural, butyraldehyde, and propylaldehyde. Temperature and pH affected stability of this enzyme and this influence varied with aldehyde substrate. Metal ions, salts, and chemical protective additives, especially at high concentrations, had different influence on enzyme activities for reduction of different aldehydes. This research provided guidelines for study of more uncharacterized atypical SDR enzymes from S. cerevisiae and other organisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The dehydrogenase/reductase superfamily, consisted of long-chain dehydrogenase/reductase (LDR) (~360–550 amino acids), medium-chain dehydrogenase/reductase (MDR) (~350 amino acids), and short-chain dehydrogenase/reductase (SDR) (~250 amino acids) families, contains tens of thousands of enzymes (Kavanagh et al. 2008; Persson et al. 1991, 2008). The classification of these three dehydrogenases/reductase families is determined by multiple factors, including protein chain length, distinct sequence motifs, mechanistic features, and structural comparisons (Jörnvall et al. 1995; Nordling et al. 2002; Persson et al. 1991). The SDR family, as the largest protein family covering almost all biological organisms from prokaryotes up to eukaryotes, has received most extensive attention (Jörnvall et al. 1999; Kallberg and Persson 2006). SDRs display catalytic activities on a wide spectrum of substrates, such as alcohols, aldehydes, ketones, steroids, polycyclic aromatic hydrocarbons, and retinoids (Kavanagh et al. 2008). Alcohol dehydrogenases from insects and sorbitol dehydrogenases from bacteria, which belong to the SDR family, were initially reported in the early 1970s and 1980s (Jörnvall et al. 1981; Schwartz and Jörnvall 1976; Thatcher and Sawyer 1980). A lot of SDR enzymes have been constantly discovered since then. Till now, over 140,000 SDR members have been identified and deposited in the sequence databases (Jörnvall et al. 2015). The SDRs can be divided into several kinds of types, including “classical,” “extended,” “intermediate,” “divergent,” “complex,” and “atypical” groups, according to a structurally conserved Rossmann fold (alpha/beta folding pattern with a central beta-sheet), an NAD(P)H-binding region, and a diverse C-terminal region (Supplementary material Table S1) (Kavanagh et al. 2008; Marchler-Bauer et al. 2013, 2015).
The classical SDRs are typically about 250 residues long, and have a TGxxx[AG]xG co-factor binding motif and a YxxxK catalytic active site motif, while the extended SDRs have an additional 100-residue domains in the C-terminal region and typically have a TGxxGxxG co-factor binding motif (Kavanagh et al. 2008). The intermediate SDRs are closely rated to the classical SDRs in forms but differ from the classical SDRs mainly within the Gly-rich co-factor binding region. The divergent SDRs are characterized by both an irregular co-factor binding motif (GxxxxxSxA) and a catalytic active site motif (YxxMxxxK), while the complex SDRs have a GGxGxxG co-factor binding motif and an altered catalytic active site motif of YxxxN (Kavanagh et al. 2008). The atypical SDRs are more complex than the five SDR groups mentioned above, and then further classified into at least 12 subgroups (Marchler-Bauer et al. 2013, 2015). Some atypical SDRs have lost their catalytic activities or have an unusual NAD(P)-binding motif, and miss or have unusual catalytic active site residues. Meanwhile, during research of the co-factor binding region, a majority of the extended SDRs are predicted to be NAD(H)-binding rather than NADP(H)-binding (Kallberg et al. 2002). However, the co-factor specificity might also depend on other factors than the sequence patterns defined thus far. Some enzymes show dual co-factor specificity and might bind alternative co-factors in different tissues and/or cellular compartments (Liu et al. 2008; Peralba et al. 1999; Wang et al. 2016).
The yeast Saccharomyces cerevisiae, as the simplest and model eukaryote, has been completely sequenced for 20 years (Goffeau et al. 1996). According to the sequence of 12,157 kilobases from the genome of the S. cerevisiae, 5907 potential protein-encoding genes and 425 potential RNA genes were annotated (http://www.kegg.jp/kegg/) (Giaever et al. 2002). Many genes or open reading frames (ORFs) encoded by S. cerevisiae have been characterized extensively since then. Nevertheless, there are still about 1000 uncharacterized ORFs, putative proteins, or proteins with unknown function existed in S. cerevisiae (http://www.yeastgenome.org/), which need to be identified further. In recent years, uncharacterized ORFs of YGL157W, YML131W, YNL134C, YDR541C, and YGL039W were cloned, and their expression proteins were successfully obtained by overexpression in yeast (Li et al. 2015; Liu and Moon 2009; Moon and Liu 2015; Zhao et al. 2015). Enzyme activity assays showed that all of them encode proteins with aldehyde reductase activity. Yml131wp and Ynl134cp are grouped into the non-zinc-containing MDR group, while Ygl157wp (Ari1p), Ydr541cp, and Ygl039wp are grouped into the intermediate SDR group. These new findings suggest that, most probably, some uncharacterized dehydrogenase/reductase enzymes still exist in S. cerevisiae.
In our previous study, we found that an uncharacterized ORF YLL056C, like YGL157W, YML131W, and YNL134C, was greatly upregulated under HMF (5-hydroxymethylfurfural, or 5-(hydroxymethyl)-2-furaldehyde) stress conditions in S. cerevisiae (Ma and Liu 2010). The phenomenon of this upregulated expression was also observed under furfural (2-furaldehyde) stress conditions by other researchers (Heer et al. 2009). BLAST (Basic Local Alignment Search Tool) analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi) of the protein deduced from YLL056C indicates it is a SDR protein and is grouped into atypical subgroup 7 (SDR_a7). However, the functions of all the proteins in the SDR_a7 subgroup remain unknown till now. Based on the evidences mentioned above, we speculate that YLL056C most probably encodes a SDR protein involved in the reduction of aldehyde, and/or quinone and ketone in S. cerevisiae. The objective of this study are as follows: (a) to confirm the transcriptional response of YLL056C to furfural and HMF by standard methods of qRT-PCR; (b) to test the enzyme activity of its encoded protein Yll056cp for reduction of furfural, HMF, other selected aldehydes, quinones, and ketones; (c) to further examine the enzymatic characteristics of Yll056cp on its catalytic reactions; and (d) to analyze its phylogenetic properties and predict proteins likely possessing the same enzymatic activities toward reduction of aldehydes in S. cerevisiae and other species of microorganisms.
Materials and methods
Strains, plasmids, media, and reagents
The S. cerevisiae yeast strains BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) from Open Biosystems (Huntsville, AL, USA) and INVSc1 (his3Δ1/his3Δ1 leu2/leu2 trp1-289/trp1-289 ura3-52/ura3-52) from Invitrogen (Carlsbad, CA, USA) were used for amplification and overexpression of YLL056C, respectively. For regular cloning and recombinant DNA manipulation, the Escherichia coli strain DH5α from Sangon Biotech (Shanghai, China) was used as the host. The shuttle plasmid pYES2/NT B (Invitrogen) and pDDGFP-2 (Newstead et al. 2007) were applied to construct the recombinant plasmid for overexpression of Yll056cp for enzymatic study and localization study in cells by fused green fluorescent protein (GFP) labeling, respectively. To obtain yeast cells of the BY4742 for genomic DNA isolation, a yeast peptone dextrose (YPD) medium (w/v, 1% yeast extract, 2% peptone, and 2% glucose) was prepared. A synthetic complete medium without uracil (SC-U) supplemented with 2% (w/v) glucose was used for selection of the recombinant yeast strains containing the newly constructed recombinant plasmid, and a SC-U medium supplemented with 2% (w/v) galactose and 1% (w/v) raffinose was used for induced expression of Yll056cp (Ma et al. 2013). A Luria-Bertani (LB) medium (w/v, 0.5% yeast extract, 1% tryptone, 1% NaCl, pH 7.0) supplemented with ampicillin (100 mg/l) was used to cultivate E. coli DH5α for selection of the recombinant plasmid. Solid medium was prepared by adding 2.0% (w/v) agar. Media ingredients, NADH, and NADPH were purchased from Sangon Biotech, Difco (Detroit, MI, USA) or Sigma-Aldrich (St. Louis, MO, USA). All of the chemical reagents used in this study were purchased from Best-Reagent (Chengdu, Sichuan, China).
Evaluation of the transcriptional response to furfural and HMF
The S. cerevisiae strain BY4742 was pre-cultivated in YPD medium at 30 °C with shaking at 200 rpm overnight. Fresh yeast cells collected from the pre-culture by centrifugation (4000×g, 2 min at room temperature) were inoculated into fresh YPD medium and cultivated at 30 °C with shaking at 200 rpm overnight. When cell density of the cultures, measured by reading the optical density at 600 nm (OD600), reached approximately 1.0 using a UV-2802 spectrophotometer (Unico, NJ, USA), furfural or HMF were added into the culture at final concentrations of 10, 30, and 50 mM, respectively. The time when furfural or HMF were added into YPD medium was designated as 0 h in this study. Cell samples were harvested at 0 and 2 h, respectively, by centrifugation at the same conditions as described above. Control cell samples from cultures without furfural or HMF addition were also collected at the same time points. The harvested cell samples were immediately frozen in liquid nitrogen to stop cell activity, and then transferred to a freezer for storage at −80 °C until use. Using an acid phenol:chloroform method as described previously (Liu and Slininger 2007), total RNA was extracted from the cell samples and further purified using an RNAclean Kit (Tiangen Biotech Co., Ltd., Beijing, China). The concentration and quality of the purified RNA were measured using a NanoDrop 2000C fluorescence spectrophotometer (Thermo-Fisher Scientific, Wilmington, DE, USA) and then verified by agarose gel electrophoresis. Using the online software of primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/), we designed the primer pair qYLL056C_F and qYLL056C_R (Supplementary material Table S2), based on the DNA sequence of YLL056C (accession no. NM_001181876), for qRT-PCR assay. For reverse transcription reactions, a FastQuant RT Kit (with gDNase) was used. A calibrated messenger RNA (mRNA) control mix from Z. Lewis Liu (NCAUR-ARS, US Department of Agriculture, Peoria, USA) as a reference was integrated into the reverse transcription reactions (Liu and Slininger 2007). For quantitative PCR reactions, a Real Master Mix (SYBR Green) was used, and the reactions were performed on a Mastercycler® EP Realplex system (Eppendorf, Hamburg, Germany) following the procedures reported previously (Liu and Slininger 2007). The experiments were performed in biological triplicate and technical triplicate for each condition. Acquired data was analyzed using the method and procedure developed by Liu et al. (2009b).
Protein localization analysis
Using a Yeast DNA Kit (Omega Bio-Tek, Norcross, GA, USA), the genomic DNA of the S. cerevisiae BY4742 was isolated. Based on homologous recombination in vivo, the recombinant plasmid pDDGFP-2 containing YLL056C was constructed following a protocol described previously (Drew et al. 2008) with slight modification. Briefly, YLL056C was amplified using the genomic DNA of S. cerevisiae BY4742 as a template with primers YLL056C_GFP_F and YLL056C_GFP_R (Supplementary material Table S2). The PCR reactions include 1 cycle of 5 min at 94 °C, 35 cycles of 1 min at 94 °C, 30 s at 55 °C and 1 min at 72 °C, and a final extension cycle of 10 min at 72 °C. The target PCR product was purified using a Gel Extraction Kit (Omega Bio-Tek) after cutting from an agarose gel, and verified by DNA sequencing (Sangon Biotech). The purified PCR product together with linearized pDDGFP-2 by SmaI were transformed into the S. cerevisiae INVSc1 by the lithium acetate method (Gietz et al. 1995) to realize the homologous recombination. Then, transformants were selected on SC-U medium supplemented with 2% glucose, and positive clones were verified by diagnostic PCR. The positive transformants were incubated in SC-U medium with 2% glucose at 30 °C overnight for cell growth. Fresh yeast cells were harvested and transferred to SC-U medium with 2% galactose and 1% raffinose, and then incubated at 30 °C for inductive expression of Yll056c-GFP protein. Twenty-four hours after induction, cell suspensions were delivered for green fluorescence analysis with a ×100 objective lens using the GFP filter lens of Axio Imager A2 (Carl Zeiss AG, Oberkochen, Germany). To estimate gross localization of the Yll056c-GFP protein in yeast cells, DIC filter lens was used to observe the cell structure.
Gene cloning and expression plasmid construction
The DNA sequence of YLL056C was amplified by PCR reaction from the genomic DNA of the S. cerevisiae BY4742 with primers YLL056C_F and YLL056C_R (Supplementary material Table S2) designed using the PrimerSelect program 7.1 (DNAStar Inc., Madison, WI, USA). Restriction endonuclease sites of NotΙ and XhoΙ were included in the forward primer and reverse primer, respectively. PCR reactions were carried out as described above, and the target PCR product was purified and verified using the same methods mentioned above. After digestion with NotΙ and XhoΙ (Takara, Dalian, Liaoning, China) and purification using a Cycle-Pure Kit (Omega Bio-Tek), the DNA fragment was ligated into the pYES2/NT B vector at the same restriction endonuclease sites and then transformed into the E. coli DH5α. Positive recombinant plasmid extracted from the clones of E. coli DH5α was identified using the diagnostic PCR method and restriction endonuclease analysis.
Protein expression, purification, and characterization
The newly constructed recombinant plasmid was transformed into the S. cerevisiae INVSc1 strain by the lithium acetate method (Gietz et al. 1995). Positive transformants selected from SC-U medium with 2% glucose at 30 °C were verified by the diagnostic PCR method. A yeast transformant containing empty pYES2/NT B without YLL056C insert served as a control. For protein expression, yeast cells from the transformants initially grew in SC-U medium with 2% glucose at 30 °C overnight. Fresh yeast cells were collected and transferred to induction medium for 24-h incubation. Then, the yeast cells were harvested and lysed using a One Step Yeast Active Protein Extraction Kit (Sangon Biotech) for purification of Yll056cp. The target protein with a His-tag was purified from the crude cell lysates using a Ni-NTA Sefinose™ Kit (Sangon Biotech). Concentration and molecular weight of the purified protein were determined using the methods described previously (Zhao et al. 2015). In order to obtain reliable results, the purified protein samples were immediately used for enzyme activity assays within 48 h.
Enzyme activity assay
Enzyme activity was determined by measuring the decrease of the co-factor NADH or NADPH in absorbance at 340 nm using a Lambda 35 UV/VIS Spectrophotometer (PerkinElmer Inc., Fremont, CA, USA). The enzyme activity assays were carried out in 500 μl reaction mixtures, which contain a final concentration of 10 mM substrate, 100 μM co-factor of NADH or NADPH, and 10 μl (~1.0 μg) of the purified protein sample in 100 mM potassium phosphate buffer, pH 7.0. Concentration of NADH was increased by 2–3-fold when glycolaldehyde was used as substrate for enzyme activity test due to its much higher activity. The protein samples were kept on ice and all reagents were warmed at 30 °C in a water bath before use for enzyme reaction. On the basis of the Michaelis-Menten equation in a double-reciprocal Lineweaver-Burk transformation, kinetic parameters for reduction of aldehydes were estimated or calculated. To determine the optimum pH and temperature for reduction of each aldehyde, enzyme activity was measured at pH levels of 4.5–9.0 (with 0.5 as an interval) and at temperatures of 20–60 °C (with 5 °C as an interval) in 100 mM potassium phosphate, respectively. All enzyme assays were run in technical triplicate. Enzyme unit and specific activity defined by Liu et al. (2008) were used in this study.
Thermal stability and pH stability
To determine thermal stability of Yll056cp, protein samples were incubated at 25–45 °C with 5 °C as an interval in 100 mM potassium phosphate buffer at pH 7.0. As for the pH stability test, pH conditions were settled from 4.5–8.5 with 1.0 as an interval, and protein samples were kept in 100 mM potassium phosphate buffer at each pH condition at 4 °C. Aliquots of the enzyme were taken from 15 min to 6 h, and the retained enzyme activity was measured as described above at pH 7.0 and 30 °C.
Effects of metal ions, chemical protective additives, and salts
Effects of metal ions on enzyme activity of Yll056cp for reduction of each aldehyde were determined in the presence of 0.5 mM Mg2+ (MgCl2·6H2O), 0.5 mM Mn2+ (MnSO4·H2O), 0.5 mM Zn2+ (ZnSO4·7H2O), 0.5 mM Ca2+ (CaCl2), Ni2+ (NiSO4·6H2O), 0.5 mM Fe3+ (FeCl3·6H2O), or 0.25 mM Cu2+ (CuSO4·5H2O) with 10 μl (~1.0 μg) of the purified protein in 50 mM phosphate buffer (pH 7.0). The effects of chemical protective additives, including dithiothreitol (DTT), β-mercaptoethanol, glycerol, and ethylenediaminetetraacetic acid (EDTA), on enzyme activity for reduction of each aldehyde were determined in the presence of each compound at varying concentrations from 1 to 10 mM. To test the effects of salts on enzyme activity, the concentrations of Na+ (NaCl) or K+ (KCl) varied from 100 to 1000 mM.
Sequence analysis
To study the possible mechanism of upregulated transcription of YLL056C under furfural and HMF stress conditions, the promoter region (−1000 to −1 bp) of YLL056C was analyzed. According to known DNA binding motifs deposited in YEASTRACT (Teixeira et al. 2014), DNA binding sites for transcription factors which are involved in stress responses were searched in the promoter region. The amino acid sequence of Yll056cp (GenBank accession no. NP_013044.1), deduced from the YLL056C DNA sequence of the S. cerevisiae S288c, was delivered to the National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/) for protein BLAST analysis. Proteins similar to Yll056cp (coverage >90%, identity >50%) from S. cerevisiae and other species were selected for alignment analysis using the ClustalW method in the MegAlign program 7.1 (DNAStar Inc.). According to the Conserved Domain Database (CCD) (Marchler-Bauer et al. 2013, 2015), the catalytic active and co-factor binding sites and regions were predicted. The aldehyde reductases identified previously from S. cerevisiae, together with the similar proteins selected for alignment analysis with Yll056cp, were used to create a phylogenetic tree of Yll056cp using the neighbor-joining method in the MEGA 6.0 program (Tamura et al. 2013).
Results
Transcriptional response to furfural and HMF
Without furfural or HMF addition in culture as the controls, transcription levels of YLL056C were not significantly changed from 0 to 2 h (Fig. 1a, b). Under 10 mM furfural condition, transcription level of YLL056C did not increase significantly at 2 h (Fig. 1a), compared with the controls. However, its transcription levels at 2 h greatly increased by 37.2-fold (p < 0.01) and 39.8-fold (p < 0.01) at concentrations of 30 and 50 mM, respectively, compared with the control at 0 h (Fig. 1a). The transcription levels of YLL056C in response to HMF were not as high as in response to furfural at the same concentrations of 30 and 50 mM (Fig. 1a, b). Under HMF stress conditions, the transcription levels of YLL056C increased by 4.6-fold (p < 0.01) at concentration of 50 mM, but increased by less than 2.0-fold at concentrations of 10 and 30 mM at 2 h, compared with the control at 0 h (Fig. 1b). Through analysis of the transcriptional regulation elements, we found that there are two potential DNA binding sites for transcription factors Yap1p, Hsf1p, and Pdr1/3p, and four potential DNA binding sites for Stb5p in the promoter region of YLL056C (Fig. 1c). We also found that there is a potential DNA binding site for transcription factor Yrr1p, which shares this binding site with Pdr1/3p (Teixeira et al. 2014).

Stress response of YLL056C to furfural or HMF and putative binding sites for transcription factors. Transcription levels of YLL056C in S. cerevisiae at 2 h after addition of furfural (a) or HMF (b) at final concentrations of 10, 30, and 50 mM were compared with those at 0 and 2 h without furfural or HMF addition as the controls (CK). Mean values are presented in fold change compared to CK at 0 h with vertical error bars representing SD (n = 6). Transcription factor binding sites in the promoter region (−1000 to −1 bp) of YLL056C (c)
Protein localization and expression
A protein localization assay showed that Yll056c-GFP localized to the cytoplasm, but not to the plasma membrane and the nucleus (Fig. 2a, b). According to the forms of the shadow appeared in the yeast cells, it seems that Yll056c-GFP did not probably localize to other subcellular organelles, such as the endoplasmic reticulum, mitochondria, and/or vacuoles, even if it was overexpressed for 24 h under the strong GAL1 promoter control.

Localization of Yll056cp in S. cerevisiae. Localization of Yll056cp was analyzed by measuring the green fluorescence of Yll056c-GFP protein using confocal microscopy. Image of Yll056c-GFP protein in yeast cells (b) taken by using a GFP lens was compared to image of the same cells taken by using a DIC lens as a control (a)
In order to overexpress YLL056C, its DNA sequence was amplified from the S. cerevisiae strain BY4742 and inserted into the NotΙ and XhoΙ sites of the pYES2/NT B to construct the recombinant plasmid pYES2/NT B-YLL056C. A diagnostic PCR test and DNA sequencing analysis demonstrated that the recombinant plasmid pYES2/NT B-YLL056C was successfully constructed (Supplementary material Fig. S1).
The newly constructed recombinant plasmid was transformed into the S. cerevisiae strain INVSc1 for overexpressing YLL056C. The molecular weight of the purified recombinant protein was about 35 kDa according to SDS-PAGE analysis (Supplementary material Fig. S2), which is close to the estimated value of Yll056cp (~32 kDa) plus the additional His-tag residues (~5.4 kDa). These results indicated that YLL056C was successfully overexpressed in S. cerevisiae.
Enzyme activity and kinetic parameters
Enzyme activity assays showed that Yll056cp had an NADH-dependent enzyme activity for reduction of at least seven aldehyde compounds, including furfural (FF), formaldehyde (FA), propionaldhyde (PA), butyraldehyde (BA), isobutyraldhyde (IBA), benzaldehyde (BZA), and glycolaldehyde (GA) (Table 1). This enzyme displayed the highest catalytic activity for reduction of GA (6.5 U/mg), followed by BA (0.78 U/mg), FF (0.47 U/mg), and FA (0.44 U/mg). No NADH-dependent enzyme activity was detected for reduction of HMF, acetaldehyde, and other six aldehyde compounds by Yll056cp (Table 1). When NADPH was used as the co-factor, Yll056cp showed no detectable enzyme activities to reduction of all of the 15 aldehyde compounds tested in this study. We also tested its enzyme activities for reduction of quinones and ketones, and found that this protein had no detectable catalytic activities for reduction of representative quinones of 1,2-naphthoquinone and 9,10-phenanthraquinone and ketones of acetone and pyruvic acid using either NADH or NADPH as the co-factor (Table 1). Kinetic parameters of Yll056cp for reduction of GA, FF, FA, BA, and PA, except for IBA and BZA due to their low specific activities, were characterized using NADH as the co-factor (Table 2). Among the five aldehyde substrates, Yll056cp showed the best maximum velocity (Vmax), catalytic rate constant (Kcat), and catalytic efficiency (Kcat/Km) to GA. Although this enzyme had the good parameters mentioned above to GA, it showed the lowest affinity (Km) to this aldehyde. The highest affinity of this enzyme was observed when FA was used as the substrate (Table 2).
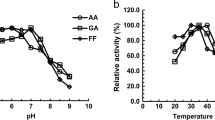
Effects of pH and temperature on enzyme activity
Effects of pH and temperature on enzyme activity of Yll056cp for reduction of GA, FF, FA, PA, and BA were tested in this study. The optimum enzyme activity for reduction of all of the five aldehydes was observed at pH 6.5 (Fig. 3a). Both acidic and alkaline conditions had the most serious effects on enzyme activities of Yll056cp when GA was used as the substrate, and less than 65% of the maximal activities were retained under pH <5.5 and >7.5 conditions. For reduction of BA, this enzyme retained more than 90% of the maximal activities in a broad pH range of 4.5 to 8.0. When FF and FA were used as the substrates, this enzyme showed more preference to acidic than alkaline conditions. The catalytic activities of this enzyme retained more than 80% of the maximal activities at pH 4.5 but dropped quickly as the pH increased in alkaline condition and decreased to less than 29% of the maximal activities at pH 9.0. Similar to GA as the substrate, the functional enzyme conditions of Yll056cp showed a narrow pH range when PA was used as the substrate, and the catalytic activities of this enzyme dropped to less than 80% under pH <5.5 and >7.0 conditions.

Effects of pH and temperature on enzyme activity of Yll056cp. Effects of pH (a) and temperature (b) on enzyme activity of Yll056cp for reduction of glycolaldehyde (GA), furfural (FF), formaldehyde (FA), propionaldehyde (PA), and butyraldehyde (BA) with NADH as the co-factor were measured in potassium phosphate buffer. Mean values of relative activity are presented with vertical error bars representing standard deviations (n = 3)
The optimum enzyme activity of Yll056cp was observed at either at 30 °C for reduction of FA or at 35 °C for reduction of GA, FF, PA, and BA (Fig. 3b). This enzyme was sensitive to temperature with either one of the five aldehydes as the substrate and showed similar increasing-to-decreasing trends of enzyme activity for the different substrates as the reaction temperature increased. Especially, enzyme activity of this protein dropped very quickly from 35 to 40 °C for reduction of FF, and from 40 to 45 °C for reduction of GA, respectively.
Thermal stability and pH stability
Enzyme activities of Yll056cp remained highly stable (relative activity >90%) for reduction of GA, FF, FA, and PA at room temperature (25 °C) for 6 h (Fig. 4a–c, e) but dropped quickly (relative activity <60%) for reduction of BA at this temperature for 6 h (Fig. 4d). Among the five aldehydes as the substrates, this enzyme also retained the best thermal stability at 30 °C for 6 h and 35 °C for 4 h for reduction of FA. For reduction of all of the five aldehydes, enzyme activities of this protein showed a decreasing trend along with the increase of temperature from 25 to 45 °C and time from 0 to 6 h except for the exceptions mentioned above (Fig. 4). At 45 °C, this enzyme lost its activities completely for reduction of all of these five aldehydes within 6 h.

Thermal stability of Yll056cp. Thermal stability of Yll056cp for reduction of glycolaldehyde (a), furfural (b), formaldehyde (c), butyraldehyde (d), and propionaldehyde (e) were tested by measuring enzyme activities at 25, 30, 35, 40, and 45 °C for 6 h, respectively. Mean values of relative activity are presented with vertical error bars representing standard deviations (n = 3)
Yll056cp showed the best pH stability under near-neutral pH (pH 6.5) conditions among the pH range of 4.5 to 8.5 for reduction of GA, FF, FA, BA, and PA (Fig. 5). Stability of this enzyme varied with pH and substrates. It showed better stability for reduction of GA and FA than reduction of FF, BA, and PA from pH 4.5 to 7.5. Relative activities of this enzyme retained more than 75% at pH 5.5 and 6.5 for reduction of GA, and at pH 4.5, 5.5, 6.5, and 7.5 for reduction of FA for 6 h (Fig. 5a, c). When FF, BA, and PA were used as the substrates, enzyme activities of this protein were quite unstable and apparently decreased at either one of the pH conditions tested in this study, even at the near-neutral pH of 6.5 (Fig. 5b, d, e).

pH stability of Yll056cp. pH stability of Yll056cp for reduction of glycolaldehyde (a), furfural (b), formaldehyde (c), butyraldehyde (d), and propionaldehyde (e) were tested by measuring enzyme activities at pH 4.5, 5.5, 6.5, 7.5, and 8.5 for 6 h, respectively. Mean values of relative activity are presented with vertical error bars representing standard deviations (n = 3)
Effects of metal ions and chemical protective additives on enzyme activity
Of the various metal ions tested in this study, Cu2+ inhibited enzyme activity of Yll056cp most seriously (Table 3). In the presence of 0.25 mM Cu2+, Yll056cp completely lost its enzyme activities for reduction of GA, FF, BA, and PA, and retained trace amount of activity for reduction of FA (relative activity 8.4%). For reduction of GA, this enzyme was only slightly inhibited by 0.5 mM Mg2+, Mn2+, Ca2+, or Fe3+ (relative activity >86%) but greatly inhibited by 0.5 mM Ni2+ or Zn2+ (relative activities of 54.9 and 32.9%, respectively). When FF and FA were used as the substrates, this enzyme was only greatly inhibited by Zn2+ (relative activities of 47.1 and 26.7%, respectively) but significantly activated by Ni2+ (relative activities of 138.7 and 147.4%, respectively), on the contrary. When BA was used as the substrate, Yll056cp was not very sensitive to Mg2+, Mn2+, Ca2+, Ni2+, and Fe3+ (relative activity >71.4%) but greatly inhibited by Zn2+ (relative activity 48.5%). For reduction of PA, this enzyme was greatly inhibited by Mn2+, Zn2+, Ca2+, Ni2+, and Fe3+ (relative activity <54.4%), except for Mg2+ with slightly negative effect (relative activity 99.0%).
Enzyme activities of Yll056cp did not change significantly or were only slightly inhibited by glycerol, β-mercaptoethanol, EDTA, and DTT from concentrations of 1 to 10 mM, when GA, BA, and PA were used as the substrates (Fig. 6a, d, e) (relative activity >70%). At high concentrations (5 and 10 mM), glycerol caused a significant decrease of enzyme activity of Yll056cp for reduction of FF, especially at 10 mM (relative activity ~60%), but this inhibition phenomenon did not occur at low glycerol concentration (1 mM) (Fig. 6b). The other three additives β-mercaptoethanol, EDTA, and DTT did not inhibit enzyme activity of Yll056cp for reduction of FF at any concentration tested in this study. DTT decreased enzyme activity of Yll056cp for reduction of FA at either concentration (from 1 to 10 mM) tested in this study, and β-mercaptoethanol caused this inhibition phenomenon only at high concentrations (5 and 10 mM) but not at low concentration (1 mM) (Fig. 6c). The other two additives, glycerol and EDTA, did not cause this inhibition phenomenon for reduction of FA at any concentration from 1 to 10 mM.

Effects of chemical additives on enzyme activity of Yll056cp. Effects of chemical additives on enzyme activity of Yll056cp for reduction of glycolaldehyde (a), furfural (b), formaldehyde (c), butyraldehyde (d), and propionaldehyde (e) were tested by measuring enzyme activities under different additive concentrations. Mean values of relative activity are presented with vertical error bars representing standard deviations (n = 3)
Effects of salts (NaCl/KCl) on enzyme activity
Effects of salts (Na+ and K+) on enzyme activity varied with aldehyde substrates (Fig. 7). For reduction of GA and PA, enzyme activity of Yll056cp was significantly inhibited by low salt concentration, and the inhibition degree did not increase along with the increase of salt concentration except for KCl at 1000 mM (Fig. 7a, e). When FF and FA were used as the substrates, the activity of this enzyme displayed a decreasing trend along with the increase of salt (NaCl/KCl) concentration, and these two salts had more severe inhibition effects on enzyme activity for reduction of FA than FF (Fig. 7b, c). KCl did not cause significant inhibition on the enzyme activity for reduction of BA at any concentrations (from 100 to 1000 mM) (Fig. 7d). However, the activity for reduction of BA was significantly inhibited by NaCl at 100 mM, but the inhibition degree did not increase along with the increase of NaCl concentration (from 100 to 100 mM).

Effects of salts on enzyme activity of Yll056cp. Effects of salts on enzyme activity of Yll056cp for reduction of glycolaldehyde (a), furfural (b), formaldehyde (c), butyraldehyde (d), and propionaldehyde (e) tested by measuring enzyme activities under different salt concentrations. Mean values of relative activity are presented with vertical error bars representing standard deviations (n = 3)
Protein sequence analysis
Deduced from the DNA sequence of YLL056C, its encoding protein Yll056cp is composed of 298 amino acid residues (Supplementary material Fig. S3a). BLAST analysis found that similar proteins to Yll056cp from S. cerevisiae existed in other Saccharomyces species, including Saccharomyces arboricola, Saccharomyces eubayanus, and Saccharomyces kudriavzevii. Through alignment analysis of their amino acid sequences, we found that these four Yll056cp proteins were comprised of a co-factor binding domain (TGxxGxxG) and a catalytic domain (LSYxxK), respectively, and shared many other conserved amino acid residues (Supplementary material Fig. S3a). Phylogenetic analysis showed that Yll056cp from S. cerevisiae together with Yll056cp proteins from other Saccharomyces species were clustered into an independent group of the atypical SDRs (B), which have a long genetic distance from the zinc-containing MDRs (D) and the non-zinc-containing MDRs (C), but are much closer to the intermediate SDRs (A) (Supplementary material Fig. S3b).
Discussion
DNA microarray data indicated that the uncharacterized ORF YLL056C was transcribed at very low level under normal growth conditions but significantly upregulated under furfural and/or HMF stress conditions in S. cerevisiae (Heer et al. 2009; Ma and Liu 2010). Microarray analysis usually gives data with high deviations which has some impact on data reliability. Moreover, this method is not as sensitive as qRT-PCR for transcriptional analysis, and the former has a valid detection limitation to 10 pg of complementary DNA (cDNA), whereas the latter has a better detection limitation to 100 fg of cDNA (Liu and Slininger 2007). Taken these factors into account, transcriptional data of YLL056C in response to furfural and HMF from the DNA microarray should be validated by an independent method. qRT-PCR is considered as the standard method for the validation of gene expression obtained from DNA microarrays (Sinicropi et al. 2007). In this study, we evaluated transcriptional response of YLL056C to furfural and HMF by qRT-PCR and confirmed that it was really significantly upregulated under high furfural or HMF concentrations. Considering YLL056C is not an essential gene with a basal expression under normal growth conditions, its upregulated transcription under furfural and HMF stress conditions indicated that it most probably encodes an aldehyde reductase. Amino acid sequence analysis indicates that the encoded protein by YLL056C is a kind of atypical SDR protein. Enzyme activity assays in this study demonstrated that YLL056C really encodes a protein (Yll056cp) with aldehyde reductase activity. Catalytic conversion of aldehyde to alcohol by aldehyde reductase commonly occurs in the cytoplasm. A protein subcellular localization study by GFP fusion labeling in this research showed that Yll056cp was localized to the cytoplasm, which is consistent with its molecular function. However, a former large-scale subcellular localization study by SWAT-GFP fusion protein and mCherry fusion protein methods showed that Yll056cp was localized to the endoplasmic reticulum and vacuoles, respectively (Yofe et al. 2016). We analyzed the possible reasons causing this localization difference between our study and the former study, and found that there are additional signal peptide sequences in the SWAT-GFP and mCherry fusion proteins containing Yll056cp (Yofe et al. 2016). Signal peptides are short (5–30 amino acids long) amino acid sequences that can guide proteins toward the secretory pathway. The signal peptide sequences in the SWAT-GFP and mCherry fusion proteins containing Yll056cp are the same and both of them are from Kar2p (except for codon alteration in mCherry plasmid) from S. cerevisiae, which is a protein localized to the endoplasmic reticulum in yeast. Therefore, we speculate that, most probably, the signal peptides from Kar2p in the SWAT-GFP and mCherry fusion proteins misled Yll056cp to the endoplasmic reticulum and vacuoles in the former study (Yofe et al. 2016).
YAP1, PDR1, PDR3, RPN4, and HSF1 were identified as key regulatory genes in S. cerevisiae in response to HMF (Ma and Liu 2010). In this study, we searched known binding motifs for these transcription factors and putative binding motifs for Yap1p, Hsf1p, and Pdr1/3p were found in the promoter region of YLL056C. For this reason, we speculate that upregulated transcription of YLL056C should be most probably activated by them, and enhanced expression of YLL056C should accelerate detoxification of toxic furfural to less toxic 2-furanmethanol under furfural stress condition. In this study, we found that Yll056cp has no enzyme activity for reduction of HMF, even if YLL056C was upregulated in response to it. Considering its multiple aldehyde reductase activities, we speculate that Yll056cp likely contributes to the detoxification of endogenous aldehydes derived from damaged proteins as discussed in our previous study (Wang et al. 2016). YLL056C was also found to be upregulated under other kinds of stress conditions, such as salicylic acid, patulin, and heat shock (Iwahashi et al. 2006; Kodo et al. 2013; Wu and Li 2008). Under those stress conditions, serious damages to proteins were reported, which indirectly supported our speculation on the function of Yll056cp for detoxification of endogenous aldehydes in cells. Putative binding motifs for multidrug resistance regulators Yrr1p and Stb5p were also found in the promoter region of YLL056C. Transcriptional regulation of YLL056C by Yrr1p has been demonstrated under toxic compound stress conditions (Lucau-Danila et al. 2003).
Enzymatic tests in this study demonstrated that Yll056cp is a broad-substrate aldehyde reductase, which can reduce not only multiple aliphatic aldehydes (FA, PA, BA, IBA, and GA), but also aromatic aldehyde (BZA) and furan aldehyde (FF). Some aldehydes, such as GA, FF, BA, and BZA, are common toxic inhibitors derived from lignocellulosic hydrolysis during biomass pretreatment (Jayakody et al. 2011; Larsson et al. 1999; Liu 2011), which cannot only inhibit cell growth but also interfere with ethanol production in the lignocellulose-to-ethanol fermentation process. Aldehyde reductase has the function to convert toxic aldehydes to less-toxic or non-toxic corresponding alcohols (Liu 2011). Yll056cp, as an atypical SDR aldehyde enzyme, is shorter than LDR and MDR aldehyde enzymes in length, and is a non-metallo-oxidoreductase, especially (Kavanagh et al. 2008). Ion transport genes are usually downregulated under aldehyde stress conditions (such as furfuran and/or HMF) (Heer et al. 2009; Ma and Liu 2010), which should negatively affect enzyme activities of the metal-containing aldehyde reductases (such as zinc-containing aldehyde reductases Adh6p and Adh7p in S. cerevisiae). Taken its upregulated transcription, enzymatic function, and molecular characteristics together, we speculate that Yll056cp plays an important role under aldehyde stress conditions. Therefore, YLL056C is a good candidate gene for development of tolerant yeast strains by engineering for in situ detoxification of aldehyde inhibitors during lignocellulose-to-ethanol production. To the best of our knowledge, Yll056cp is the first identified atypical SDR aldehyde enzyme in S. cerevisiae. This research provided guidelines for the study of more uncharacterized atypical SDR enzymes from S. cerevisiae and other organisms.
Enzyme assays in vitro indicated that Yll056cp strictly uses NADH as the co-factor for reduction of aldehyde compounds. NADH and/or NADPH can be used as the co-factor(s) to catalyze the conversion of an aldehyde to its corresponding alcohol by different reductases (Liu 2011). NADH is important on ethanol fermentation and NADPH is required for the synthesis of fatty acids, amino acids, and nucleotides for cell growth in S. cerevisiae (Van Dijken and Scheffers 1986). Preference of Yll056cp to NADH rather than NADPH as the co-factor for reduction of aldehydes in vivo avoids competition for NADPH with cell growth in theory. Therefore, Yll056cp is a good candidate gene for in situ detoxification of aldehydes derived from lignocellulosic biomass. Reduction of aldehyde compounds by NADH-dependent aldehyde reductases in vivo will deplete NADH in cells, which will decrease the reduction rate of aldehydes, especially at high aldehyde concentrations. Enhanced transcription of genes for regeneration of NADH in the glycolysis pathway in evolutionarily engineered yeast is considered to be contributed to acquired tolerance to aldehyde inhibitors (Liu et al. 2009a).
Like other aldehyde reductases (Li et al. 2015; Liu and Moon 2009; Liu et al. 2008; Ma et al. 2013; Moon and Liu 2015; Wang et al. 2016), Yll056cp identified in this study did not show enzyme activities for reduction of all of the 15 aldehydes but showed enzyme activities for reduction 7 of them (Table 1). The molecular structure of Yll056cp from S. cerevisiae or its closely related protein can help to elucidate its catalytic mechanism. However, only one atypical SDR enzyme from a Gram-negative bacterium Vibrio vulnificus has been studied at structural and biochemical level till now (Buysschaert et al. 2013). Unfortunately, this atypical SDRvv is quite different from Yll056cp in both the co-factor binding motif and the catalytic active site motif. Therefore, it cannot be used to predict the functional structure, amino acid residues, and motifs of Yll056cp, but have to be structurally studied on Yll056cp itself in the future. In this study, we found that enzyme characteristics of Yll056cp are not always consistent but mostly variable with substrate, pH, temperature, metal ions, chemical protective additives, and salts. Whether the enzyme activities of Yll056cp can be affected by other factors, such as anions, is not determined in this study but needs to be studied in the future. Enzyme activity and characteristics are mainly related to molecular weight, structure, group polarity from the substrate aspect, and the molecular structure from the enzyme aspect. The exact relationships within substrate, enzyme, and reaction conditions should be elucidated along with the structural insights into Yll056cp from S. cerevisiae.
References
Buysschaert G, Verstraete K, Savvides SN, Vergauwen B (2013) Structural and biochemical characterization of an atypical short-chain dehydrogenase/reductase reveals an unusual cofactor preference. FEBS J 280(5):1358–1370. doi:10.1111/febs.12128
Drew D, Newstead S, Sonoda Y, Kim H, von Heijne G, Iwata S (2008) GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat Protoc 3(5):784–798. doi:10.1038/nprot.2008.44
Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418(6896):387–391. doi:10.1038/nature00935
Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11(4):355–360. doi:10.1002/yea.320110408
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274(5287): 546, 563–567. doi:10.1126/science.274.5287.546
Heer D, Heine D, Sauer U (2009) Resistance of Saccharomyces cerevisiae to high concentrations of furfural is based on NADPH-dependent reduction by at least two oxireductases. Appl Environ Microbiol 75(24):7631–7638. doi:10.1128/AEM.01649-09
Iwahashi Y, Hosoda H, Park JH, Lee JH, Suzuki Y, Kitagawa E, Murata SM, Jwa NS, Gu MB, Iwahashi H (2006) Mechanisms of patulin toxicity under conditions that inhibit yeast growth. J Agric Food Chem 54(5):1936–1942. doi:10.1021/jf052264g
Jayakody LN, Hayashi N, Kitagaki H (2011) Identification of glycolaldehyde as the key inhibitor of bioethanol fermentation by yeast and genome-wide analysis of its toxicity. Biotechnol Lett 33(2):285–292. doi:10.1007/s10529-010-0437-z
Jörnvall H, Persson M, Jeffery J (1981) Alcohol and polyol dehydrogenases are both divided into two protein types, and structural properties cross-relate the different enzyme activities within each type. Proc Natl Acad Sci 78(7):4226–4230
Jörnvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D (1995) Short-chain dehydrogenases/reductases (SDR). Biochemistry 34:6003–6013. doi:10.1021/bi00018a001
Jörnvall H, Höög JO, Persson B (1999) SDR and MDR: completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett 445(2–3):261–264. doi:10.1016/S0014-5793(99)00130-1
Jörnvall H, Landreh M, Östberg LJ (2015) Alcohol dehydrogenase, SDR and MDR structural stages, present update and altered era. Chem Biol Interact 234:75–79. doi:10.1016/j.cbi.2014.10.017
Kallberg Y, Persson B (2006) Prediction of coenzyme specificity in dehydrogenases/reductases: a hidden Markov model-based method and its application on complete genomes. FEBS J 273(6):1177–1184. doi:10.1111/j.1742-4658.2006.05153.x
Kallberg Y, Oppermann U, Jörnvall H, Persson B (2002) Short-chain dehydrogenases/reductases (SDRs): coenzyme-based functional assignments in completed genomes. Eur J Biochem 269(18):4409–4417. doi:10.1046/j.1432-1033.2002.03130.x
Kavanagh KL, Jörnvall H, Persson B, Oppermann U (2008) The SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci 65:3895–3906. doi:10.1007/s00018-008-8588-y
Kodo N, Matsuda T, Doi S, Munakata H (2013) Salicylic acid resistance is conferred by a novel YRR1 mutation in Saccharomyces cerevisiae. Biochem Biophys Res Commun 434(1):42–47. doi:10.1016/j.bbrc.2013.03.069
Larsson S, Palmqvist E, Hahn-Hägerdal B, Tengborg C, Stenberg K, Zacchi G, Nilvebrant NO (1999) The generation of inhibitors during dilute acid hydrolysis of softwood. Enzym Microb Technol 24:151–159. doi:10.1016/S0141-0229(98)00101-X
Li X, Yang R, Ma M, Wang X, Tang J, Zhao X, Zhang X (2015) A novel aldehyde reductase encoded by YML131W from Saccharomyces cerevisiae confers tolerance to furfural derived from lignocellulosic biomass conversion. Bioenerg Res 8(1):119–129. doi:10.1007/s12155-014-9506-9
Liu ZL (2011) Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl Microbiol Biotechnol 90:809–825. doi:10.1007/s00253-011-3167-9
Liu ZL, Moon J (2009) A novel NADPH-dependent aldehyde reductase gene from Saccharomyces cerevisiae NRRL Y-12632 involved in the detoxification of aldehyde inhibitors derived from lignocellulosic biomass conversion. Gene 446(1):1–10. doi:10.1016/j.gene.2009.06.018
Liu ZL, Slininger PJ (2007) Universal external RNA controls for microbial gene expression analysis using microarray and qRT-PCR. J Microbiol Methods 68(3):486–496. doi:10.1016/j.mimet.2006.10.014
Liu ZL, Moon J, Andersh BJ, Slininger PJ, Weber S (2008) Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxymethylfurfural by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 81(4):743–753. doi:10.1007/s00253-008-1702-0
Liu ZL, Ma M, Song M (2009a) Evolutionarily engineered ethanologenic yeast detoxifies lignocellulosic biomass conversion inhibitors by reprogrammed pathways. Mol Gen Genomics 282(3):233–244. doi:10.1007/s00438-009-0461-7
Liu ZL, Palmquist DE, Ma M, Liu J, Alexander NJ (2009b) Application of a master equation for quantitative mRNA analysis using qRT-PCR. J Biotechnol 143(1):10–16. doi:10.1016/j.jbiotec.2009.06.006
Lucau-Danila A, Delaveau T, Lelandais G, Devaux F, Jacq C (2003) Competitive promoter occupancy by two yeast paralogous transcription factors controlling the multidrug resistance phenomenon. J Biol Chem 278(52):52641–52650. doi:10.1074/jbc.M309580200
Ma M, Liu ZL (2010) Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4 and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genomics 11:660. doi:10.1186/1471-2164-11-660
Ma M, Wang X, Zhang X, Zhao X (2013) Alcohol dehydrogenases from Scheffersomyces stipitis involved in the detoxification of aldehyde inhibitors derived from lignocellulosic biomass conversion. Appl Microbiol Biotechnol 97(18):8411–8425. doi:10.1007/s00253-013-5110-8
Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41:D348–D352. doi:10.1093/nar/gks1243
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43:D222–D226. doi:10.1093/nar/gku1221
Moon J, Liu ZL (2015) Direct enzyme assay evidence confirms aldehyde reductase function of Ydr541cp and Ygl039wp from Saccharomyces cerevisiae. Yeast 32(4):399–407. doi:10.1002/yea.3067
Newstead S, Kim H, von Heijne G, Iwata S, Drew D (2007) High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci 104(35):13936–13941. doi:10.1073/pnas.0704546104
Nordling E, Jörnvall H, Persson B (2002) Medium-chain dehydrogenases/reductases (MDR): family characterizations including genome comparisons and active site modeling. Eur J Biochem 269(17):4267–4276. doi:10.1046/j.1432-1033.2002.03114.x
Peralba JM, Cederlund E, Crosas B, Moreno A, Julià P, Martinez SE, Persson B, Farrés J, Parés X, Jörnvall H (1999) Structural and enzymatic properties of a gastric NADP(H)-dependent and retinal-active alcohol dehydrogenase. J Biol Chem 274(37):26021–26026. doi:10.1074/jbc.274.37.26021
Persson B, Jeffery J, Jörnvall H (1991) Different segment similarities in long-chain dehydrogenases. Biochem Biophys Res Commun 177(1):218–223. doi:10.1016/0006-291X(91)91970-N
Persson B, Hedlund J, Jörnvall H (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the MDR superfamily. Cell Mol Life Sci 65(24):3879–3894. doi:10.1007/s00018-008-8587-z
Schwartz MF, Jörnvall H (1976) Structural analyses of mutant and wild-type alcohol dehydrogenases from Drosophila melanogaster. Eur J Biochem 68:159–168. doi:10.1111/j.1432-1033.1976.tb10774.x
Sinicropi D, Cronin M, Liu M-L (2007) Gene expression profiling utilizing microarray technology and RT-PCR. In: Ferrari M, Ozkan M, Heller M (eds) BioMEMS and biomedical nanotechnology, Volume II: Micro/Nano technologies for genomics and proteomics. Springer-Verlag, Heidelberg, pp 23–46
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. doi:10.1093/molbev/mst197
Teixeira MC, Monteiro PT, Guerreiro JF, Gonçalves JP, Mira NP, dos Santos SC, Cabrito TR, Palma M, Costa C, Francisco AP, Madeira SC, Oliveira AL, Freitas AT, Sá-Correia I (2014) The YEASTRACT database: an upgraded information system for the analysis of gene and genomic transcription regulation in Saccharomyces cerevisiae. Nucleic Acids Res 42:D161–D166. doi:10.1093/nar/gkt1015
Thatcher DR, Sawyer L (1980) Secondary-structure prediction from the sequence of Drosophila melanogaster (fruitfly) alcohol dehydrogenase. Biochem J 187(3):884–886. doi:10.1042/bj1870884
Van Dijken JP, Scheffers WA (1986) Redox balances in the metabolism of sugars by yeasts. FEMS Microb Rev 32(3–4):199–224. doi:10.1016/0378-1097(86)90291-0
Wang X, Ma M, Liu ZL, Xiang Q, Li X, Liu N, Zhang X (2016) GRE2 from Scheffersomyces stipitis as an aldehyde reductase contributes tolerance to aldehyde inhibitors derived from lignocellulosic biomass. Appl Microbiol Biotechnol 100(15):6671–6682. doi:10.1007/s00253-016-7445-4
Wu WS, Li WH (2008) Identifying gene regulatory modules of heat shock response in yeast. BMC Genomics 9:439. doi:10.1186/1471-2164-9-439
Yofe I, Weill U, Meurer M, Chuartzman S, Zalckvar E, Goldman O, Ben-Dor S, Schütze C, Wiedemann N, Knop M, Khmelinskii A, Schuldiner M (2016) One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat Methods 13(4):371–378. doi:10.1038/nmeth.3795
Zhao X, Tang J, Wang X, Yang R, Zhang X, Gu Y, Li X, Ma M (2015) YNL134C from Saccharomyces cerevisiae encodes a novel protein with aldehyde reductase activity for detoxification of furfural derived from lignocellulosic biomass. Yeast 32(5):409–422. doi:10.1002/yea.3068
Acknowledgments
We thank Z. Lewis Liu, Bioenergy Research Unit, NCAUR-ARS, U.S. Department of Agriculture (Peoria, IL, USA) for providing the calibrated mRNA control mix as the reference mRNA for the qRT-PCR analyses. This work was supported by the National Natural Science Foundation of China (No. 31570086) and the Talent Introduction Fund of Sichuan Agricultural University (No. 01426100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals by any of the authors.
Additional information
Han-Yu Wang and Di-Fan Xiao contributed equally to this work.
Electronic supplementary material
ESM 1
(PDF 391 kb)
Rights and permissions
About this article

Cite this article
Wang, HY., Xiao, DF., Zhou, C. et al. YLL056C from Saccharomyces cerevisiae encodes a novel protein with aldehyde reductase activity. Appl Microbiol Biotechnol 101, 4507–4520 (2017). https://doi.org/10.1007/s00253-017-8209-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8209-5