
Abstract
Primary bile acids serve important roles in cholesterol metabolism, lipid digestion, host-microbe interactions, and regulatory pathways in the human host. While most bile acids are reabsorbed and recycled via enterohepatic cycling, ∼5% serve as substrates for bacterial biotransformation in the colon. Enzymes involved in various transformations have been characterized from cultured gut bacteria and reveal taxa-specific distribution. More recently, bioinformatic approaches have revealed greater diversity in isoforms of these enzymes, and the microbial species in which they are found. Thus, the functional roles played by the bile acid-transforming gut microbiota and the distribution of resulting secondary bile acids, in the bile acid pool, may be profoundly affected by microbial community structure and function. Bile acids and the composition of the bile acid pool have historically been hypothesized to be associated with several disease states, including recurrent Clostridium difficile infection, inflammatory bowel diseases, metabolic syndrome, and several cancers. Recently, however, emphasis has been placed on how microbial communities in the dysbiotic gut may alter the bile acid pool to potentially cause or mitigate disease onset. This review highlights the current understanding of the interactions between the gut microbial community, bile acid biotransformation, and disease states, and addresses future directions to better understand these complex associations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The composition of the bile acid pool is mediated by bacterial metabolism in the intestinal tract and is intrinsically linked to host physiology. This occurs via a variety of regulatory processes, variation in toxicity among the bile acids, and the microbial ecology of the gut (Ridlon et al. 2014). As such, the function of bile acids as potential mechanisms and treatments for disease states is receiving increasing attention. In this review, we focus on bile acid-mediated regulation associated with bacterial community structure, dysbiosis, and disease states. We provide an overview of the current understanding of the regulation and biotransformation of the bile acid pool by the gut microbial community and address how the complex interactions between bile acids and the gut microbiota influence the onset and treatment of disease states.
The bile acid pool size and composition are inherently linked with microbial community presence, and presumably, composition. Therefore, it seems likely that specific assemblages of microorganisms may result in distinct variations in the bile acid pool. Thus, the current knowledge of the taxa-specific distributions of enzymes involved in bile acid transformation is addressed, followed by a discussion of disease states associated with bile acids. To the extent available, information regarding relationships between the gut microbial community, bile acid transformation, and disease state will be highlighted, along with emerging technologies that allow for a better characterization of the complex interactions between the gut microbiota and the human host associated with bile acid production and metabolism.
Overview of bile acid metabolism
Bile acids are amphipathic biological detergents that primarily function in lipid metabolism but also serve a wide range of regulatory functions throughout the body (Chiang 2009). The primary bile acids, chenodeoxycholic acid (CDCA) and cholic acid (CA), are synthesized from cholesterol in the liver, in a process involving at least 14 enzymes (Russell and Setchells 1992; Chiang 2009). The CDCA molecule has two α-hydroxy groups at the C-3 and C-7 positions, and CA has an additional α-hydroxy group at position C-12 (Hofmann 1999) (Fig. 1). There are two primary pathways by which bile acids are synthesized which have been reviewed in detail (Chiang 2004; Chiang 2009) and are summarized in Fig. 1. The classical bile acid synthesis pathway occurs only in hepatocytes and is controlled by the rate-limiting step converting cholesterol to 7α-hydroxycholesterol (Chiang 2004). Cholic acid is formed through subsequent modification by the enzyme cascades shown in Fig. 1, and CDCA is formed by a similar pathway excluding 12α-hydroxylation by CYP8B1 (Chiang 2004). Alternatively, in a variety of tissues, cholesterol may be oxidized to 27-hydroxycholesterol, which is further modified by the oxysterol hydroxylase CYP7B1 and further converted to primary bile acids (Fig. 1) (Chiang 2004).

Bile acid synthesis pathway in humans. Green arrows reflect the primary process of bile acid synthesis, while blue arrows show an alternative pathway (color figure online)
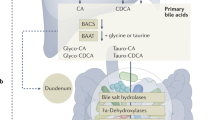
In the liver, primary bile acids are conjugated to either glycine (predominantly in humans) or taurine by the enzymes bile acid-CoA synthase (BACS) and bile acid-amino acid transferase (BAT). Conjugation increases solubility of these hydrophobic (nonpolar) bile acids and concurrently decreases potential cell membrane disruption. Conjugated bile acids subsequently accumulate in the gallbladder (Hofmann 1999; Chiang 2009). Following food intake, the gallbladder releases bile acids into the duodenum, where they contribute to the solubilization of ingested lipids as the bile acids proceed through the small intestine and colon (Hofmann 1999). Bile acids are reabsorbed in the distal ileum, primarily through active transport by the apical sodium-dependent bile salt transporter (ASBT) or the ileal bile acid transporter (IBAT). Typically, this results in a ∼50 to 90% recovery of conjugated bile acids (Hofmann 1999), via enterohepatic circulation, with and an overall bile acid recovery efficiency of ∼95% (Ridlon et al. 2006; Chiang 2009). The cycle of enterohepatic circulation is shown in Fig. 2.

Overview of enterohepatic circulation and microbial processing of bile acids. Gray arrows indicate host processes, and thick black arrows reflect processes performed by the microbiome. Thin, solid arrows reflect the direction of primary acid flow, while dashed arrows reflect secondary bile acids. Open symbols reflect passive transport. BA bile acid, G glycine, T taurine
Bile acids that are not reabsorbed can serve as substrates for microbial metabolism and undergo biotransformation to secondary bile acids (Fig. 3) (Hofmann 1999). The predominant secondary bile acids, formed via 7α-dehydroxylation by gut bacteria, are deoxycholic acid (DCA), formed from CA, and lithocholic acid (LCA), formed from CDCA. Ursodeoxycholic acid (UDCA) has a structure similar to CDCA, and this molecule is also present in small quantities in the total bile acid pool (Hofmann 1999). Once recirculated to the liver, DCA and LCA are both conjugated to glycine or taurine, similar to primary bile acids, with conjugated DCA contributing approximately 20% to the bile acid pool (Hofmann 1999). In contrast, LCA, a highly toxic bile acid, is subsequently sulfated at the C-3 position, after which it is poorly reabsorbed and excreted in the feces (Hofmann 1999).

Chemical structures of secondary bile acids derived from a cholic acid and b chenodeoxycholic acid
Host regulatory pathways of bile acids
Bile acid synthesis is naturally regulated by feedback inhibition from bile acids being recirculated to the liver. What we present here is a brief overview of the regulation of this pathway, focusing on the aspects that are likely to be affected by microbial processes and gut microbial composition, discussed in the following section.
The farnesoid X receptor (FXRα) is the primary regulator of bile acid synthesis in the liver, which inhibits the enzymes that synthesize bile acids from cholesterol precursors (Fig. 1) (Chiang et al. 2000; Yang et al. 2002; Chen and Chiang 2003). The FXRα is activated by hydrophobic (nonpolar) primary and secondary bile acids (i.e., CDCA, CA, DCA, LCA) (Makishima et al. 1999; Hu et al. 2014; Ridlon et al. 2014) and binds to regulatory regions of DNA as a heterodimer with retinoid X receptors (Edwards et al. 2002). The mechanisms of inhibition vary depending on whether FXRα in the liver or intestine is activated (Kim et al. 2007; Chiang 2009), with FXRα working through a small heterodimer partner (SHP) in the liver or fibroblast growth factor 19 (FGF-19; FGF-15 is an ortholog in mice) (Holt et al. 2003; Inagaki et al. 2005; Kim et al. 2007; Chiang 2009). Recently, bile acids have been postulated to regulate proliferation of intestinal cells via FXRα and epidermal growth factor receptor signaling pathways (Dossa et al. 2016). Repression of bile acid synthesis has also been suggested to be coordinately regulated by both hepatic and intestinal FXRα, where intestinal FXRα has a strong effect on CYP7A1, but not CYP8B1. In contrast, hepatic FXRα also inhibits CYP8B1 (Kim et al. 2007).
Bile acid pool size has been recently demonstrated, in mice, to depend on the microbial community present in the intestines (Sayin et al. 2013). It should be noted, however, that mice produce primarily hydrophilic (polar) muricholic acids. Consequently, conclusions generated from mouse models must be separately validated in humans, whose bile acid pool is more hydrophobic (Chiang 2009). When compared to germ-free mice, conventional mice have an overall bile acid pool that is significantly reduced (∼71%). Furthermore, the presence of microbiota was found to have an influence on the site-specific bile acid concentrations in the gallbladder, small intestine, cecum, colon, and feces (Sayin et al. 2013). The authors hypothesized that differences in bile acid concentrations may be due to FXR-mediated reabsorption via the ASBT (Sinha et al. 2008; Sayin et al. 2013), and the disruption of the gut microbiota has been recently shown to increase efficiency of reabsorption (Hu et al. 2014). Similarly, the presence of microbiota resulted in suppression of CYP7A1 and BACS, but not CYP8B1 and BAT, and expression of FXRα, SHP, and FGF-15 were upregulated in the distal ileum, but not in the liver (Sayin et al. 2013). Taken together, results of these studies, done in mice, indicate that the microbiologically mediated formation of hydrophobic secondary bile acids diminishes concentrations of hydrophilic muricholic acid. Thus, removal of the muricholic FXR-antagonists subsequently results in activation of this receptor in the intestine to regulate bile acid homeostasis (Sinha et al. 2008; Sayin et al. 2013; Hu et al. 2014; Ridlon et al. 2014).
In addition to regulation of cholesterol and bile acid homeostasis, bile acids also regulate important metabolic pathways via FXRα and the cell membrane receptor G protein-coupled receptor 5 (TGR5). Altered pathways include those involved in drug, lipoprotein, glucose, and energy metabolism and transport (Nguyen and Bouscarel 2008; Hylemon et al. 2009). A subset of the processes and effects known to be regulated and controlled by bile acids is shown in Table 1, and these processes have been thoroughly reviewed (Nguyen and Bouscarel 2008; Hylemon et al. 2009).
The gut microbial environment
The human colon is colonized by approximately 1011–12 bacteria, with greater than 90% belonging to the phyla Bacteroidetes and Firmicutes (Dethlefsen et al. 2007; The Human Microbiome Consortium 2012). Bacterial diversity in the gut is relatively low, as compared to microbial communities found in water and soil environments, and is comprised of mainly four other phyla, including the Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia (Dethlefsen et al. 2007). Prevalent genera identified thus far include Bacteroides, Eubacterium, Bifidobacterium, Ruminococcus, Peptostreptococcus, Propionibacterium, Clostridium, Lactobacillus, Escherichia, Streptococcus, and the archaeal genus Methanobrevibacter (Dethlefsen et al. 2007). Many of these bacteria, especially some of the clostridia, have been shown to be active in transformation of primary to secondary bile acids (Hofmann 1999; Ridlon et al. 2006).
In the colon, nearly all bile acids are biotransformed into secondary bile acids (LCA from CDCA and DCA from CA) by 7α-dehydroxylation and are either reabsorbed via portal circulation or excreted in the stool (Hofmann 1999; Ridlon et al. 2006). The 7α-dehydroxylation reaction has been described as the most quantitatively important process performed by colonic microflora, resulting in the formation of secondary bile acids that are predominantly found in feces (Ridlon et al. 2006). However, current estimates indicate that approximately only 0.0001% of colonic bacteria are capable of performing this reaction, and 16S rRNA sequence analyses performed to date have indicated that these bacteria only belong to the genus Clostridium (Wells et al. 2000; Wells et al. 2003; Ridlon et al. 2006).
The pathway for this reaction is encoded by the bile acid inducible (bai) operon, which has been characterized and is highly conserved in both Cl. scindens and Cl. hylemonae strains, although the efficiency of transformation among these strains differs (Ridlon et al. 2010). The ability to perform 7α-hydroxylation is hypothesized to result in net energy gain and supports a unique niche for these bacteria, possibly by also excluding microbes that are more sensitive to the secondary bile acid end products (Ridlon et al. 2006).
The roles of bile acids in control of microbiota composition in the gut
Bile acids can exert direct influences, both positive and negative, on gut bacteria (Begley et al. 2005a). Several studies have demonstrated that bile acids have an overall negative impact on membrane integrity, likely due to an increase in membrane permeability leading to cell death (Albalak et al. 1996; De Boever et al. 2000). The extent of cell damage is related to the hydrophobicity and structure of bile acids, where more hydrophobic bile acids, and those with two rather than three hydroxy groups are more detrimental to membrane integrity than other bile acids (Hofmann et al. 2001; Begley et al. 2005a). Bile acids also damage DNA and promote increases in enzymes involved in DNA repair (Kandell and Bernstein 1991; Begley et al. 2005a). Moreover, there is further evidence that bile acids may also cause oxidative and pH stress, and may chelate important cellular ions, such as calcium (Bernstein et al. 1999; Begley et al. 2005a). Gram-negative bacteria are currently thought to be more resistant to bile acids than Gram-positive microorganisms, although few studies have characterized bile acid resistance among Gram-negative species (Begley et al. 2005a). Importantly, bile resistance, tolerance, and susceptibility are strain-specific, with large variations in the degree of tolerance within a single bacterial species (Chateau et al. 1994; Záratea et al. 2000).
Bile acids can also have indirect antimicrobial effects mediated by FXRα, the activation of which results in upregulation of genes involved in mucosal defense in the ileum of mice (Inagaki et al. 2006). In a mouse model, biliary duct ligation resulted in overgrowth of aerobic and anaerobic bacteria in the ileum and cecum, but this overgrowth could be blocked by activation of FXRα with the agonist GW4064 (Inagaki et al. 2006). This study further demonstrated that FXRα activation prevented translocation of bacteria across the intestinal mucosal barrier by impeding damage and inflammation that typically results from biliary duct ligation. A subsequent review of the effects of bile acids on mucosal defense suggested that antimicrobial effects are direct when conjugated bile acids are at high concentrations. In contrast, regulation of bacterial densities via signaling occurs when conjugated bile acid concentrations are depleted in the distal ileum (Hofmann and Eckmann 2006).
Several studies have evaluated bacterial community dynamics that are potentially regulated by, or affect the distribution of, bile acids in the small and large intestines (Islam et al. 2011; Ridlon et al. 2013; Kakiyama et al. 2013). Primary bile acids such as taurocholate can provide homing signals to gut bacteria and promote germination of spores, which may be present in dormant and nontoxic forms, and may facilitate recovery of microbiota after dysbiosis induced by antibiotics or toxins (Browne et al. 2016). This mechanism can be exploited by pathogens that have arisen from indigenous gut microbiota, like Clostridium difficile (see below), that germinates into a vegetative, potentially toxigenic form. Primary bile acids may also serve as a control mechanism to prevent outgrowth of pathogenic Gram-negative bacteria in the small intestine (Kakiyama et al. 2013). A recent study in cirrhotic patients with synthetic deficits in production of bile acids found that increases in potentially pathogenic Enterobacteriaceae were positively associated with CDCA concentrations and lower concentrations of secondary bile acids in stool. This likely resulted from a concomitant reduction in typical resident groups of clostridia able to perform 7α-dehydroxylation (i.e., Clostridium cluster XVIa) (Kakiyama et al. 2013).
Separate studies done in rats and mice found that the concentration of CA can regulate gut community structure. Specifically, increased CA concentrations resulted in increases in Firmicutes, especially groups capable of 7α-dehydroxylation, and a decrease of Bacteroidetes (Islam et al. 2011; Ridlon et al. 2013). As suggested by Ridlon et al. (2014), it is still unclear whether the increase in 7α-dehydroxylating bacteria, such as Clostridium cluster XVIa, in response to increases in primary bile acids, reflects (1) an antagonistic effect of bile acids on other community members, (2) increases in production of an antimicrobial compound by these members, or (3) use of these bile acids as an electron acceptor in a fermentative pathway.
Microbial influence on intestinal bile acids
As bile acids move through the small intestine, they are subjected to biotransformation by the resident microbial community. The gut microbiota has been reported to consist primarily of members of the bacterial genera Lactobacillus, Streptococcus, Staphylococcus, and Veillonella, typically at concentrations of 103 to 104 bacteria ml−1 in the duodenum and jejunum (Ridlon et al. 2006). Bacteria in the ileum are mainly comprised of Enterobacteria, Enterococcus, Bacteroides, Clostridium, Lactobacillus, and Veillonella, at much larger concentrations of ∼106 to 108 bacteria ml−1 (Ridlon et al. 2006). Together, these bacteria are responsible for deconjugation of bile acids from glycine or taurine by hydrolases and the oxidation of hydroxy groups (Ridlon et al. 2006). The majority of bile acids (95%) are reabsorbed in the distal ileum and recirculated through the liver (Ridlon et al. 2006; Chiang 2009); however, a small amount (400–800 mg day−1) continue through to the colon where they serve as substrates for a variety of biotransformation reactions by gut bacteria (Ridlon et al. 2006).
Transformation of bile acids
The transformation of bile acids involves several processes including deconjugation to liberate free bile acids, reversible epimerization between α and β orientations, and the oxidation of the 3-, 7-, and 12-hydroxy groups. Hydroxy groups of bile acids, synthesized in the liver, are all in the α orientation resulting in amphipathicity and allowing efficient solubilization of lipid molecules (Ridlon et al. 2006). Deconjugation and epimerization of bile acids result in alteration of the hydrophilicity of bile acids (Armstrong and Carey 1982) and may affect the efficiency of lipid solubilization or affinity of hydroxysteroid dehydrogenases (Ridlon et al. 2006). Furthermore, epimerization of bile acids by gut microbiota, and the accumulation of bile acids with β-oriented hydroxy groups in the bile acid pool, confers a protective effect on the liver against more toxic, hydrophobic, bile acids (Heuman et al. 1991; Hofmann 1995). Therefore, the composition of the microbial community may have significant effects on nutrient absorption and bile acid toxicity, affecting host metabolic responses based on its ability to perform specific bile acid transformations.
The variety of isoforms of the enzymes responsible for these reactions and their distributions among gut microbiota are relatively understudied, and current knowledge is primarily based on biochemical studies in which these enzymes are characterized from single, cultivatable species. However, due to the potentially great variety of isoforms and the potential for limited distribution of these enzymes among only a few microbial species, it is possible that specific community structures have discrete effects on host health via modification of primary and secondary bile acid pools. Furthermore, expression of particular enzymes by certain microbial species may influence community composition by conferring selective advantages to these species in situ.
Results of more recent bioinformatic and metagenomic studies highlight a considerably greater diversity of microorganisms that are capable of performing bile acid transformations, including members of the Archaea, than were previously recognized by biochemical characterization (Jones et al. 2008; Kisiela et al. 2012). Further study of these previously unknown, and likely unculturable, members of the gut microbiota will be important in order to better understand how these groups may influence disease states and community-level processes important to host metabolic processes.
Bile salt hydrolases
The first step in bile acid modification by the microbial community involves the deconjugation of bile acids by bile salt hydrolases (BSHs) (Ridlon et al. 2006). These enzymes share a high degree of structural similarity to penicillin V acylase from Bacillus sphaericus (and other species) and are relatively widely distributed among both Gram-negative and -positive members of the gut microbiota (Ridlon et al. 2006; Kumar et al. 2006; Jones et al. 2008). There are at least nine types of BSHs (Jones et al. 2008), and these differ in size, pH optima, substrate specificity, and genetic organization and regulation (Ridlon et al. 2006). Several studies have suggested that BSHs play a role in human gut colonization, especially by pathogens such as Listeria monocytogenes and Brucella abortus (Begley et al. 2005b; Delpino et al. 2007), and detoxification of bile acids (De Smet et al. 1995). Other studies have suggested that BSHs facilitate scavenging of carbon, nitrogen, and sulfur (Van Eldere et al. 1996; Tanaka et al. 2000).
Bile salt hydrolases have been characterized from species of Bacteroides, Clostridium, Lactobacillus, Bifidobacterium, and Listeria (Ridlon et al. 2006). A more recent functional metagenomic study of approximately 90,000 clones containing DNA fragments from the gut microbiome revealed that 142 clones demonstrated BSH activity and, among the 90 that could be taxonomically assigned, ∼30, 14, and 9% (corresponding to 27, 13, and 8 clones) belonged to the Firmicutes, Bacteroidetes, and Actinobacteria, respectively. Most of the remaining (43%) positive clones could not be assigned (Jones et al. 2008), although updated databases may now allow for classification of many of these. In this study, clones assigned to Firmicutes and Actinobacteria were capable of deconjugating all the glyco- and tauro-conjugated bile acids tested, while Bacteroidetes had specific activity against tauro- but not glyco-conjugated bile acids. Furthermore, one BSH showed a high degree of similarity (56%) to that found in Methanobrevibacter smithii, and the authors demonstrated that this enzyme deconjugated both glyco- and tauro-conjugated bile acids and suggested a role of horizontal gene transfer (transfer of genes between species) in the dissemination of BSHs (Jones et al. 2008).
Hydroxysteroid dehydrogenases
Hydroxysteroid dehydrogenases (HSDs) are bacterial enzymes that act on the 3-, 7-, and 12-position hydroxy groups of bile acids to catalyze epimerization and oxidation/reduction. This alters the hydrophobicity and toxicity of bile acids and provides potential energy sources for cellular processes (Sherrod and Hylemon 1977; Heuman et al. 1991; Ridlon et al. 2006). Epimerization of hydroxy groups requires the activity of two stereochemically distinct (α or β), position-specific enzymes to form stable oxo-bile acids. The process may be carried out by a single species possessing both enzymes (e.g., Cl. absonum) (Sutherland and Macdonald 1982) or via interspecies interaction (a synergism) in which one species possesses the α-HSD and another the β-HSD (MacDonald et al. 1982). Oxidation and reduction of oxo-bile acids are largely dependent on the redox potential (oxygen state) of the microenvironment in the intestinal tract, such that reduction is favored in the intestinal lumen where redox potential is low (anoxic), and oxidation may be favored closer to the mucosal surface where redox potential is more positive (oxic) (Ridlon et al. 2006).
Bioinformatic analysis has revealed two different 3α-HSDs found in either Cl. scindens or Comomonas testosteroni that have low (22%) amino acid sequence similarity with each other (Kisiela et al. 2012). Homologs to the former 3α-HSD were found not only primarily among the Firmicutes but also in more extremophilic groups (those that survive in extreme environments) including Thermotogae and Aquificae (Kisiela et al. 2012), which is in agreement with prior studies that have characterized these enzymes from several species of abundant, and some less abundant, intestinal bacteria including Cl. hiranosis (Wells and Hylemon 2000), Cl. perfringens (Macdonald et al. 1976), Cl. scindens (Mallonee et al. 1995), Eggerthella lenta (Macdonald et al. 1979), and Peptostreptococcus productus (Edenharder et al. 1989a). Homologs to the latter 3α-HSD in C. testosteroni were identified among several potentially pathogenic groups, primarily within the phylum Actinobacteria (Kisiela et al. 2012). A convergent evolutionary process has been suggested to explain the emergence of these two 3α-HSDs from different ancestors (Kisiela et al. 2012). 3β-HSDs have also been identified within the genera Clostridium (Edenharder et al. 1989b) and Ruminococcus (Akao et al. 1987).
Based on biochemical characterization, 7α- and 7β-HSDs have been found in several of the Bacteroides and Clostridia, and also in E. coli, C. testosteroni, and Ruminococcus spp. (Macdonald et al. 1976; Akao et al. 1987; Yoshimoto et al. 1991; Baron et al. 1991; Coleman et al. 1994; Ferrandi et al. 2012; Ji et al. 2014). Three distinct types of 7α-HSDs were found by bioinformatic analysis that were similar to proteins identified in either (1) E. coli/Br. melitensis (merged model), (2) B. fragilis, or (3) Cl. scindens (Kisiela et al. 2012). Homologs to the Escherichia/Brucella 7α-HSD were found in a variety of species from natural environments as well as gut-associated members of Enterobacteriaceae (i.e., Pseudomonas spp., Cyanothece spp., Comamonas spp., Psychrobacter spp., Acinetobacter spp., Rhodobacter spp., Pseudoalteromonas spp., Brevundimonas spp., Nitrosomonas spp., Fusobacterium spp.), including several potentially pathogenic species. The second 7α-HSD of B. fragilis had homologs not only among other Bacteroides spp. but also among Rhodococcus spp., Strepytomyces spp., Lysinsibacillus spp., and Bacillus spp. The homologs to the Cl. scindens 7α-HSD were restricted to Firmicutes (primarily Clostridium spp.), with some detected in Spirochaetes. The mechanism of dehydroxylation by Cl. scindens is of particular importance due to the predominance of secondary bile acids in feces and the presumed sequestration of these genes within a limited number of Firmicutes (Ridlon et al. 2006). The reaction is catalyzed by enzymes encoded on the bile acid induced bai operon, described in detail elsewhere (Ridlon et al. 2006; Ridlon and Hylemon 2012). Importantly, the process acts only on unconjugated, primary bile acids (Batta et al. 1990) and may provide function in niche restriction via the production of hydrophobic secondary bile acids. This latter supposition is of particular important to the host since the inhibitory effects of secondary bile acids can servehave a protective effect, for example, by inhibiting germination of toxigenic Cl. difficile (described in detail below) (Buffie et al. 2015).
The Firmicutes, especially Clostridium spp., are the predominant group from which 12α/β-HSDs have been identified and characterized both biochemically and bioinformatically (Ridlon et al. 2006; Kisiela et al. 2012). Bioinformatic analyses also revealed homologs to the Clostridium 12α-HSD among the Actinobacteria, Bacteroidetes, and Tenericutes as well as the Methanobacteria within Euryarchaeota (Kisiela et al. 2012). Among the clostridia, 12α- and 12β-HSDs have been characterized from Cl. difficile (Edenharder and Schneider 1985), Cl. leptum (Harris and Hylemon 1978), Cl. paraputrificum (Edenharder and Schneider 1985), Cl. perfringens (Macdonald et al. 1976), and Cl. tertium (Edenharder and Schneider 1985), and are all constitutively expressed, except for the 12β-HSD in Cl. paraputrificum (Edenharder and Pfützner 1988). Similar to other HSDs, the 12α/β-HSDs have been suggested to play a role in host colonization (Macdonald et al. 1976).
Bile-acid-associated diseases states
Altered bile acid pool composition has been associated with several disease states, including recurrent Cl. difficile infection (Wilson 1983), inflammatory bowel diseases (Cima and Pemberton 2001), metabolic syndrome (Mudaliar et al. 2013), and cancers (Nagengast et al. 1995). Recently, the role of the gut microbial community in causing or abrogating disease states has also received considerable attention (Hamilton et al. 2013; Ridaura et al. 2013). Based on current knowledge, it is apparent that both the influence of the bile acid pool on the composition of the bacterial communities, as well as the effect of bacterial metabolism of bile acids, may play important roles in these conditions. As such, the elucidation of these interactions has become a contemporary area of research both to better understand the etiology of these diseases, as well as identify efficacious treatment strategies. In this section, we summarize the current state of research as it relates to the gut microbiome, bile acid metabolism, and associated disease states.
Clostridium difficile infection
Clostridium difficile infection (CDI) is a common nosocomial disease of the colon, generally characterized by diarrhea and abdominal pain. In a typical case of CDI, a patient receives antibiotics, within a healthcare setting, and subsequently ingests spores of Cl. difficile, which can germinate in the newly permissive colonic environment and produce the toxins that cause disease (Kachrimanidou and Malisiovas 2011). The relationship between antibiotics and CDI has been well established in epidemiological and other studies, and antibiotic exposure is far and away the strongest risk factor for CDI (Fashner et al. 2011).
The link between antibiotic usage and risk for CDI is most likely to be the effect of the antibiotic(s) on the native colonic microbiota. In mouse models (Robinson and Young 2010; Buffie et al. 2012; Pérez-Cobas et al. 2013) and humans (Dethlefsen and Relman 2011), antibiotics radically alter the composition of the colonic microbiota as well as its metabolic state. Furthermore, restoration of native microbiota, via fecal microbiota transplantation, results in effective treatment of CDI (Khoruts et al. 2010; Shahinas et al. 2012; Hamilton et al. 2013; van Nood et al. 2013). Originally, it was hypothesized that the increase in space and resources for colonization (allowing niche placement) following antibiotic therapy allowed Cl. difficile to flourish in the colon, providing loss of “colonization resistance” (Brandt and Reddy 2011; Britton and Young 2012). In recent studies, however, it has been suggested that a shift in colonic bile acid composition contributes to a permissive environment in which the bacterium may thrive (Weingarden et al. 2014; Buffie et al. 2015; Weingarden et al. 2016b; Weingarden et al. 2016a). Thus, restoration of gut microbial ecology is due to colonization and chemical signal exchange.
Certain bile acids have long been known to play a role in the growth of Cl. difficile. The primary bile acid cholic acid and its taurine-conjugated derivative, taurocholic acid, have been known for decades to stimulate germination of Cl. difficile spores (Wilson 1983), and sodium taurocholate is a typical reagent used for growing Cl. difficile in vitro (Wilson et al. 1982; Buggy et al. 1985). More recent work has shown that both CA and TCA stimulate the first step of germination of Cl. difficile spores and allow for outgrowth of colonies (Sorg and Sonenshein 2008). In contrast, other bile acids including CDCA, LCA, and UDCA inhibit germination by taurocholate (Sorg and Sonenshein 2009; Sorg and Sonenshein 2010). More significantly, recent studies have shown that there are major shifts in fecal bile acids after antibiotic treatment, characterized by an increase in primary bile acids and a decrease in secondary bile acids, that may promote infection with Cl. difficile (Giel et al. 2010; Theriot et al. 2014). Opposite shifts in fecal bile acids are observed in human patients after fecal microbiota transplantation (Fig. 4) (Weingarden et al. 2014). Thus, the composition of bile acids in the colon may have a significant impact on CDI, with primary bile acids, dominant after antibiotic treatment, promoting the germination of Cl. difficile spores, and secondary bile acids, produced by specific colon bacteria, that are restored after fecal transplantation, inhibiting germination (Weingarden et al. 2016b; Khoruts and Sadowsky 2016; Weingarden et al. 2016a).

Bile acid composition in the colon and Cl. difficile infection. Following antibiotic treatment, the proportion of primary bile acids, including CA, TCA, and CDCA, increases in the colon and feces, along with susceptibility to CDI, possibly by allowing Cl. difficile spores to germinate into vegetative cells (dark blue) (right panel). An FMT can reverse these effects, increasing the proportion of secondary bile acids, such as DCA and LCA, in the feces, and prevent recurrence of CDI, possibly by inhibiting germination of Cl. difficile spores (light blue) (left panel). CA cholic acid, TCA taurocholic acid, CDCA chenodeoxycholic acid, DCA deoxycholic acid, LCA lithocholic acid, FMT fecal microbiota transplantation. Modified from (Weingarden et al. 2014)
Although CDI is primarily a colonic disease, it is thought to be responsible for 10% of diarrhea in patients with a surgically created ileal pouch reservoir after colectomy (Seril and Shen 2014). It has been noted that these patients frequently do not respond to FMT with the same success as patients with intact colons (Hamilton et al. 2012; Borody et al. 2014; Patel et al. 2014). However, we have successfully treated a patient with recurrent CDI of the ileal pouch with oral UDCA, suggesting that even without a normal, intact colonic microbiota, direct replacement of secondary bile acids may be a viable strategy for treatment of CDI pouchitis (Weingarden et al. 2016a). Notably, germination of spores from the strain of Cl. difficile isolated from this patient was found to be inhibited by UDCA, again supporting the idea that bile acid supplements may represent an alternative treatment for CDI in select patients.
Inflammatory bowel disease
Inflammatory bowel disease (IBD) describes a variety of clinical conditions, including Crohn’s disease, ulcerative colitis, and microscopic colitis, which are characterized by chronic inflammation of the gastrointestinal tract (Israel and Kleinman 1994; Maxson et al. 1994). The etiology of each of these conditions is obscure, though it is likely caused by several factors, including host genetics, diet, and gut microbiota (Ek et al. 2014; Huang et al. 2014; Sobczak et al. 2014).
That gut microbiota may contribute to IBD is an attractive hypothesis, given the intimate relationship between the gut and its commensal microflora. It is well established that the fecal and intestinal mucosal microbiota of human patients with IBD, particularly ileal Crohn’s disease, are highly altered compared to healthy individuals (Swidsinski et al. 2005; Frank et al. 2007; Sokol et al. 2008; Willing et al. 2009). Furthermore, intestinal inflammation in animal models of IBD is highly attenuated in germ-free animals, which lack intestinal microbiota (Sellon et al. 1998; Schultz et al. 1999; Garrett et al. 2010). Patients with underlying IBD also have a lower success rate following FMT for CDI compared to patients without IBD (Khoruts et al. 2016). Despite this relationship, as well as the extensive body of knowledge of intestinal bacterial bile acid metabolism, little work has examined how bile acid metabolism might affect IBD.
Most research on the relationship between bile acids and IBD has focused on how the disease affects both intestinal and serum bile acids. One common complication of IBD is resection of the terminal ileum. Because the terminal ileum is the site of reabsorption of bile acids by the GI tract, the removal of this section of the small intestine can lead to bile acid malabsorption, typically characterized by bile acid-mediated diarrhea or, in large resections of the terminal ileum, a loss of fat digestion (Cima and Pemberton 2001; Domènech et al. 2014; Gothe et al. 2014). Several studies have also noted alterations in the composition of bile acids in the colon, characterized by an increase in conjugated bile acids and decreased secondary bile acids in the feces of patients (Duboc et al. 2013). There are also significant alterations to bile acid concentrations in both portal vein (Holzbach et al. 1980) and systemic serum (Gnewuch et al. 2009) with the disease, even in the absence of small bowel resection.
Although these investigations focused on the effects of IBD on bile acid composition in the colon and blood, recent studies have examined whether bile acids may play a role in the etiology of the disease. In a model of IBD, which uses mice lacking interleukin (IL)-10 and anti-inflammatory cytokine, feeding these mice a diet high in milk fat led to an increase in the concentration of taurocholic acid in the gut. This in turn led to an increase in the abundance of sulfite-producing bacteria and an increase in disease symptoms (Fig. 5) (Devkota et al. 2012). These results suggest that the effects of particular bile acids on IBD is an area worth exploring, even though little work has so far investigated this relationship.

Effects of diet, bile acids, host genetics, and intestinal bacteria on colitis. In genetically susceptible mice (IL-10−/−), a diet rich in milk fat increases taurocholic acid production, which in turn increases the abundance of sulfite-producing bacteria in the colon. Together, these factors increase intestinal inflammation
Another area of potential interest in the relationship between bile acids and IBD is the mediation of inflammation by FXR. In addition to its role in regulating bile acid metabolism by the host, FXR is thought to be involved in the regulation of intestinal innate immunity and permeability (Vavassori et al. 2009). A recent study revealed that treatment with an FXR agonist is protective in mouse models of chemically induced colitis (Gadaleta et al. 2011). These findings suggest that the observed alterations in colonic bile acid composition in IBD not only reflect underlying changes to microbial bile acid metabolism but may also have a key role in mediation of inflammation in these diseases (Ogilvie and Jones 2012). Furthermore, FXR agonists may represent a novel therapeutic option for IBD.
Primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is strongly associated with IBD (Boonstra et al. 2012) and is a chronic cholestatic liver disease characterized by bile duct destruction leading to cirrhosis and liver failure (Hirschfield et al. 2013). PSC may also occur without IBD and, while the etiology is not well understood, is thought to be due to genetic and environmental risk factors (Hirschfield et al. 2013). Incidence of PSC with IBD has been indicated as a risk factor for the development of colorectal cancers (Soetikno et al. 2002), but there is no indication of increased risk in the absence of IBD (Claessen et al. 2009).
Inflammation and obstruction of biliary ducts result in alteration of bile flow and a decrease in resistance to bile acid toxicity (Hohenester et al. 2012; Hirschfield et al. 2013). Several placebo-controlled studies have indicated that administration of UDCA is effective at improving serum liver biochemistry (Chazouillères et al. 1990; Beuers et al. 1992). A more recent study, however, showed that a high dose (∼30 mg kg−1) of UDCA presented significantly higher risk of adverse effects in the group receiving UDCA versus a placebo control (Lindor et al. 2009). Furthermore, the role of the gut microbiota on disease progression was investigated in a study that used a treatment of UDCA alone, or UDCA with metronidazole, and found that while combination therapy improved biochemistry, the use of the antibiotic did not affect overall clinical outcomes (Färkkilä et al. 2004). In addition, high-dose UDCA treatment was suggested to increase the risk of colorectal cancers (Eaton et al. 2011). Currently, the interaction of the gut microbiota and bile acid pool composition as it relates to PSC is enigmatic and will require further study to elucidate how these factors contribute to and may be used to treat this condition. Whether FMT can positively impact PSC is currently unknown, although a clinical trial is currently in progress to address this [https://clinicaltrials.gov/ct2/show/NCT02424175].
Metabolic syndrome
Metabolic syndrome is one of the most common diseases in the USA. According to the Centers for Disease Control and Prevention, the prevalence of one component of metabolic syndrome, obesity, has dramatically increased over the past several decades. It is currently estimated that more than one third (34.9%) of individuals in the USA are obese [http://www.cdc.gov/obesity/data/adult.html]. Metabolic syndrome increases the risk of several diseases, particularly type 2 diabetes and cardiovascular disease (Alberti et al. 2005).
While metabolic syndrome is generally thought to be due to multiple factors, including genetics, the environment, and behavior (Alberti et al. 2005), emerging evidence suggests that the gut microbiota may also contribute to the disease. Early studies demonstrated an increase in the relative proportion of the Firmicutes phylum and a concomitant decrease in Bacteroidetes in genetically obese versus lean mice (Ley et al. 2005). Initially, this trend was thought to be similar in obese versus lean humans (Ley et al. 2006), but these findings have been recently challenged (Walters et al. 2014; Sze and Schloss 2016). Nonetheless, transplantation of fecal or cecal microbiota from several mouse models of obesity into germ-free animals frequently recapitulates the disease phenotype, including weight and fat gain as well as glucose intolerance and triglyceride abnormalities (Turnbaugh et al. 2006; Vijay-Kumar et al. 2010; Cox et al. 2014; Suez et al. 2014). Transplantation of fecal microbiota from obese humans also causes increased weight gain in germ-free mice compared to transplantation with microbiota from lean individuals, suggesting that while differences in the microbiota may exist between lean and obese humans and mice, the effects of the microbiota on phenotype can be recapitulated using human-derived populations (Ridaura et al. 2013). Several mechanisms for these observations have been proposed, including increased caloric extraction from food by the gut microbiota (Turnbaugh et al. 2006) and increased host appetite (Vijay-Kumar et al. 2010; Cox et al. 2014).
Bacterial bile acid metabolism may provide an alternative mechanism underlying the relationship between the gut microbiota and metabolic syndrome. The role of bile acids and their nuclear receptor, FXRα, in metabolic syndrome has been extensively reviewed (Duran-Sandoval et al. 2005; Claudel et al. 2005; Cariou and Staels 2007; Kuipers et al. 2007; Lefebvre et al. 2009; Porez et al. 2012). A therapeutic option for dyslipidemia and other aspects of metabolic syndrome, in fact, is bile acid-binding resins such as cholestyramine, which decrease the concentration of bile acids returned to the liver via enterohepatic cycling and therefore stimulate the conversion of cholesterol to bile acids (Ginsberg and Stalenhoef 2003; Claudel et al. 2005; Turnbaugh et al. 2006; Yamaoka-Tojo et al. 2008). Furthermore, it appears that activation of FXRα can also improve aspects of metabolic syndrome in animal models, including glucose homeostasis (Duran-Sandoval et al. 2005) and triglyceride control (Bilz et al. 2006; Del Bas et al. 2009). A recent Phase II trial in human patients demonstrated increased insulin sensitivity following treatment with an FXRα agonist (Mudaliar et al. 2013). Activation of the bile acid cell membrane receptor TGR5 may also affect metabolic syndrome. TGR5 can stimulate glucagon-like protein 1 production in enteroendocrine cells, leading to improved glucose homeostasis and can activate thyroid hormone in brown adipose tissue and muscle, which increases energy expenditure (Thomas et al. 2008; Zhong 2010; Pols et al. 2011).
Recent work has indicated that bacterial bile acid metabolism can affect FXRα signaling, and therefore metabolic syndrome. Inoculation of mice with E. coli overexpressing a highly active allele of BSH significantly increased FXRα signaling and led to a significant decrease in weight gain and liver triglycerides (Fig. 6) (Joyce et al. 2014).

Bacterial bile acid metabolism can influence metabolic syndrome. Expression of bile salt hydrolase (BSH) by E. coli increases deconjugation of taurocholic acid (TCA) to cholic acid (CA), which increases FXR activation to alter gene expression associated with lipid digestion, adiposignaling, immune-homeostasis, and circadian rhythms, ultimately leading to decreased liver triglycerides and decreased weight gain in the host
The relationship between bacterial bile acid metabolism and metabolic syndrome in humans has also been explored. One study has shown that fecal transplantation with material from lean donors improved insulin sensitivity in individuals with metabolic syndrome (Vrieze et al. 2012). In contrast, administration of oral vancomycin to patients with metabolic syndrome results in substantial changes to the fecal microbiota, along with a significant decrease in fecal secondary bile acids and decreased peripheral insulin sensitivity (Vrieze et al. 2014). Taken together, this work suggests that bacterial bile acid metabolism in the gut can significantly impact metabolic syndrome in the host.
Colorectal cancers
Secondary bile acids, especially DCA, have long been known to accumulate at high levels in the bile acid pool of certain individuals on high fat, “Western” diets (Ridlon et al. 2014). It has been suspected that bacterial metabolism of these bile acids produces carcinogenic or cocarcinogenic compounds (Hill et al. 1975; Nagengast et al. 1995; Bernstein et al. 2005). The mechanisms of action have not been clearly elucidated, but DNA damage and induction of apoptosis are among those most commonly suggested (Bernstein et al. 2005). The majority of studies have investigated the role of bile acids in colon cancer, where patients with cancer had higher levels of fecal bile acids relative to healthy controls or those with other diseases (Hill et al. 1975; Nagengast et al. 1995). In addition, patients with colon cancer also had higher concentrations of 7α-dehydroxylating clostridia in their stool, suggesting a link between these bacteria and the disease state (Hill et al. 1975). On the other hand, epidemiological data did not find an increase in gastrointestinal cancer in patients who had undergone partial ileal resection, a group of patients which may have had increased bile acid load to the colon (Buchwald et al. 1990). A relationship between the gut microbiome and colorectal cancer has been reviewed (Dulal and Keku 2014; Nistal et al. 2015) and Burns and colleagues have identified specific bacterial species that are correlated with colorectal cancer and that the gut microbiota play a pivotal role in cancer development or progression (Burns et al. 2015). As pointed out by Ridlon and coworkers, control of the gut microflora and bile acid metabolism may be key to preventing a large number of human diseases (Ridlon et al. 2016).
In contrast to the highly hydrophobic DCA, UDCA has been shown in several studies to suppress tumor development, especially in colon cancers (Pardi et al. 2003; Alberts et al. 2005; Centuori and Martinez 2014). However, a more recent analysis found the effects of UDCA to be gender-dependent. While men treated with UDCA showed a reduced risk for developing advanced lesions, some women showed a significantly higher risk (Thompson et al. 2009). These results, as well as the dose-dependent outcomes discussed above (Eaton et al. 2011) and the examination of UDCA in patients with other underlying disorders such as ulcerative colitis and primary sclerosing cholangitis (Pardi et al. 2003), suggest that the mechanism(s) by which DCA and UDCA affect the progression of colorectal cancers is complex and is currently not well understood. Research efforts are currently shifting to examine the role of these bile acids as signaling molecules that regulate pathways involved in the progression of colon cancers to better understand the mechanistic relationships between these molecules and disease progression (Centuori and Martinez 2014). It should be noted, however, that the influence of bile acids on cancer may not be direct. For example, Ridlon et al. (2016) argue that the genotoxic effects of taurocholic acid may be due to bacterial transformation of this bile acid to H2S and cholic acid.
Other cancers
In addition to a probable role in colorectal cancers, secondary bile acids have also been suggested as potential causative agents of cancers in several other body sites (Bernstein et al. 2005). Bile acid exposure resulting from laryngopharyngeal reflux has been suggested to result in carcinoma of the upper aerodigestive tract (Lewin et al. 2003). Similarly, bile acid exposure resulting from gastroesophageal reflux has been hypothesized to eventually lead to esophageal adenocarcinoma (Stamp 2002) and exposure from duodenogastric reflux to gastric stump carcinoma (Kondo 2002). Due to the association of pancreatic cancer with the Western diet, bile acids have also been hypothesized to be associated with pancreatic cancer (Tucker et al. 2004). Similarly, based on the formation of adenocarcinomas near the point of bile acid entry to the small intestine, a role for bile acids in carcinogenesis in the small intestine has also been suggested (Ross et al. 1991).
More recently, alterations of the gut bacterial community associated with obesity and the increased production of DCA have been associated with formation of hepatocellular carcinomas (Yoshimoto et al. 2013). In a mouse model, recirculation of DCA resulted in secretion of pro-inflammatory and pro-tumorigenic factors in the liver, but blocking DCA or reducing the gut microbiota prevented carcinoma development (Yoshimoto et al. 2013). Analysis of the microbiome of mice fed a high fat versus a normal fat diet revealed that the former mice had significantly higher relative abundances of Clostridium clusters XI and XIVa (Yoshimoto et al. 2013) than normal diet mice, which are able to perform 7α-dehydroxylation of primary bile acids. Further analysis revealed that, when fed the high fat diet, Clostridium cluster XI was comprised of a single strain of Cl. sordellii and that there was an increase in the abundance of the baiJ gene which is involved in 7α-dehydroxylation. This strongly suggested that this strain was responsible for the increase in DCA (Yoshimoto et al. 2013).
Conclusions
Bile acids are vital components of cholesterol metabolism, lipid digestion, and other regulatory pathways in the human host. Diet and the gut microbial community interact with the bile acid pool, via biotransformation reactions, that affect the hydrophobicity, toxicity, and regulatory effects of bile acids. Disturbance of the bile acid pool, by disease or temporary antibiotic-induced dysbiosis, may result in a variety of disease states. Consequently, increasing attention is being paid to how microbial community structure, especially that in a dysbiotic state, and microbial processes act on these molecules to cause disturbances. Recent metagenomic and bioinformatic analyses are revealing novel bacterial species and enzymes capable of bile acid transformation, as well as revealing potentially meaningful shifts in community composition associated with these disease states. This knowledge will better inform therapeutic practices to alleviate bile-acid-mediated illness. Future research must focus on elucidating the casual mechanisms by which gut microbiota and bile acids interact, including addressing previously unculturable, unidentified, species that may be critically involved in these processes at the single-species or community level.
References
Akao T, Hattori M, Namba T, Kobashi K (1987) Enzymes involved in the formation of 3 beta, 7 beta-dihydroxy-12-oxo-5 beta-cholanic acid from dehydrocholic acid by Ruminococcus sp. obtained from human intestine. Biochim Biophys Acta 921:275–280
Albalak A, Zeidel ML, Zucker SD, Jackson AA, Donovan JM (1996) Effects of submicellar bile salt concentrations on biological membrane permeability to low molecular weight non-ionic solutes. Biochemistry 35:7936–7945. doi:10.1021/bi960497i
Alberti K, Zimmet P, Shaw J (2005) The metabolic syndrome—a new worldwide definition. Lancet 366:1059–1062. doi:10.1016/S0140-6736(05)67402-8
Alberts DS, Martínez ME, Hess LM, Einspahr JG, Green SB, Bhattacharyya AK, Guillen J, Krutzsch M, Batta AK, Salen G, Fales L, Koonce K, Parish D, Clouser M, Roe D, Lance P (2005) Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J Natl Cancer Inst 97:846–853. doi:10.1093/jnci/dji144
Armstrong MJ, Carey MC (1982) The hydrophobic-hydrophilic balance of bile salts inverse correlation between reverse-phase high performance liquid chromatographic mobilities and micellar cholesterol-solubilizing capacities. J Lipid Res 23:70–80
Baron SF, Franklund CV, Hylemon PB (1991) Cloning, sequencing, and expression of the gene coding for bile acid 7 alpha-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI 12708. J Bacteriol 173:4558–4569
Batta AK, Salen G, Arora R, Shefer S, Batta M, Person A (1990) Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J Biol Chem 265:10925–10928
Begley M, Gahan CGM, Hill C (2005a) The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. doi:10.1016/j.femsre.2004.09.003
Begley M, Sleator RD, Gahan CGM, Hill C (2005b) Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect Immun 73:894–904. doi:10.1128/IAI.73.2.894-904.2005
Bernstein C, Bernstein H, Payne CM, Beard SE, Schneider J (1999) Bile salt activation of stress response promoters in Escherichia coli. Curr Microbiol 39:68–72. doi:10.1007/s002849900420
Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H (2005) Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 589:47–65. doi:10.1016/j.mrrev.2004.08.001
Beuers U, Spengler U, Kruis W, Aydemir ü, Wiebecke B, Heldwein W, Weinzierl M, Pape GR, Sauerbruch T, Paumgartner G (1992) Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology 16:707–714. doi:10.1002/hep.1840160315
Beuers U, Probst I, Soroka C, Boyer JL, Kullak-Ublick GA, Paumgartner G (1999) Modulation of protein kinase C by taurolithocholic acid in isolated rat hepatocytes. Hepatology 29:477–482. doi:10.1002/hep.510290227
Bilz S, Samuel V, Morino K, Savage D, Choi CS, Shulman GI (2006) Activation of the farnesoid X receptor improves lipid metabolism in combined hyperlipidemic hamsters. Curr Vasc Pharmacol 290:E716–E722. doi:10.1152/ajpendo.00355.2005
Boonstra K, van Erpecum KJ, van Nieuwkerk KMJ, Drenth JPH, Poen AC, Witteman BJM, Tuynman HARE, Beuers U, Ponsioen CY (2012) Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm Bowel Dis 18:2270–2276. doi:10.1002/ibd.22938
Borody TJ, Leis S, Pang G, Wettstein AR (2014) Fecal microbiota transplantation in the treatment of recurrent Clostridium difficile infection. In: Rutgeerts P (ed) Fecal microbiota transplantation in the treatment of recurrent Clostridium Difficile infection. UpToDate, Waltham
Bouscarel B, Ceryak S, Gettys TW, Fromm H, Noonan F (1995) Alteration of cAMP-mediated hormonal responsiveness by bile acids in cells of nonhepatic origin. Am J Phys 268:G908–G916
Bouscarel B, Kroll SD, Fromm H (1999) Signal transduction and hepatocellular bile acid transport: cross talk between bile acids and second messengers. Gastroenterology 117:433–452
Brandt LJ, Reddy SS (2011) Fecal microbiota transplantation for recurrent Clostridium difficile infection. J Clin Gastroenterol 45(Suppl):S159–S167. doi:10.1097/MCG.0b013e318222e603
Britton RA, Young VB (2012) Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 20:313–319. doi:10.1016/j.tim.2012.04.001
Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA ,Stares MD, Goulding D, Lawley TD (2016) Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533: 543–546. doi:10.1038/nature17645
Buchwald H, Stoller DK, Campos CT, Matts JP, Varco RL (1990) Partial ileal bypass for hypercholesterolemia. 20- to 26-year follow-up of the first 57 consecutive cases. Ann Surg 212:318–329 31
Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG (2012) Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80:62–73. doi:10.1128/IAI.05496-11
Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MRM, Jenq RR, Taur Y, Sander C, Cross J, Toussaint NC, Xavier JB, Pamer EG (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi:10.1038/nature13828
Buggy BP, Hawkins CC, Fekety R (1985) Effect of adding sodium taurocholate to selective media on the recovery of Clostridium difficile from environmental surfaces. J Clin Microbiol 21:636–637
Burns MB, Lynch J, Starr TK, Knights D, Blekhman R (2015) Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med 7:55. doi:10.1186/s13073-015-0177-8
Cariou B, Staels B (2007) FXR: a promising target for the metabolic syndrome? Trends Pharmacol Sci 28:236–243. doi:10.1016/j.tips.2007.03.002
Centuori SM, Martinez JD (2014) Differential regulation of EGFR-MAPK signaling by deoxycholic acid (DCA) and ursodeoxycholic acid (UDCA) in colon cancer. Dig Dis Sci 59:2367–2380. doi:10.1007/s10620-014-3190-7
Chateau N, Deschamps AM, Sassi AH (1994) Heterogeneity of bile salts resistance in the Lactobacillus isolates of a probiotic consortium. Lett Appl Microbiol 18:42–44. doi:10.1111/j.1472-765X.1994.tb00796.x
Chazouillères O, Poupon R, Capron J-P, Metman E-H, Dhumeaux D, Amouretti M, Couzigou P, Labayle D, Trinchet J-C (1990) Ursodeoxycholic acid for primary sclerosing cholangitis. J Hepatol 11:120–123. doi:10.1016/0168-8278(90)90281-U
Chen W, Chiang JYL (2003) Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha). Gene 313:71–82
Chiang JYL (2004) Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol 40:539–551. doi:10.1016/j.jhep.2003.11.006
Chiang JYL (2009) Bile acids: regulation of synthesis. J Lipid Res 50:1955–1966. doi:10.1194/jlr.R900010-JLR200
Chiang JYL, Kimmel R, Weinberger C, Stroup D (2000) Farnesoid X receptor responds to bile acids and represses cholesterol 7α-hydroxylase gene (CYP7A1) transcription. J Biol Chem 275:10918–10924. doi:10.1074/jbc.275.15.10918
Chignard N, Mergey M, Veissière D, Poupon R, Capeau J, Parc R, Paul A, Housset C (2003) Bile salts potentiate adenylyl cyclase activity and cAMP-regulated secretion in human gallbladder epithelium. Am J Physiol - Gastrointest Liver Physiol 284:G205–G212. doi:10.1152/ajpgi.00292.2002
Cima RR, Pemberton JH (2001) Surgical management of inflammatory bowel disease. Curr Treat Options Gastroenterol 4:215–225. doi:10.1007/s11938-001-0034-2
Claessen MMH, Vleggaar FP, Tytgat KMAJ, Siersema PD, van Buuren HR (2009) High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol 50:158–164. doi:10.1016/j.jhep.2008.08.013
Claudel T, Staels B, Kuipers F (2005) The farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol 25:2020–2030. doi:10.1161/01.ATV.0000178994.21828.a7
Coleman JP, Hudson LL, Adams MJ (1994) Characterization and regulation of the NADP-linked 7 alpha-hydroxysteroid dehydrogenase gene from Clostridium sordellii. J Bacteriol 176:4865–4874
Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, Zárate Rodriguez JG, Rogers AB, Robine N, Loke P, Blaser MJ (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158:705–721. doi:10.1016/j.cell.2014.05.052
De Boever P, Wouters R, Verschaeve L, Berckmans P, Schoeters G, Verstraete W (2000) Protective effect of the bile salt hydrolase-active Lactobacillus reuteri against bile salt cytotoxicity. Appl Microbiol Biotechnol 53:709–714. doi:10.1007/s002530000330
De Smet I, Van Hoorde L, Vande Woestyne M, Christiaens H, Verstraete W (1995) Significance of bile salt hydrolytic activities of lactobacilli. J Appl Bacteriol 79:292–301. doi:10.1111/j.1365-2672.1995.tb03140.x
Del Bas JM, Ricketts M-L, Vaqué M, Sala E, Quesada H, Ardevol A, Salvadó MJ, Blay M, Arola L, Moore DD, Pujadas G, Fernandez-Larrea J, Bladé C (2009) Dietary procyanidins enhance transcriptional activity of bile acid-activated FXR in vitro and reduce triglyceridemia in vivo in a FXR-dependent manner. Mol Nutr Food Res 53:805–814. doi:10.1002/mnfr.200800364
Delpino MV, Marchesini MI, Estein SM, Comerci DJ, Cassataro J, Fossati CA, Baldi PC (2007) A bile salt hydrolase of Brucella abortus contributes to the establishment of a successful infection through the oral route in mice. Infect Immun 75:299–305. doi:10.1128/IAI.00952-06
Dethlefsen L, Relman DA (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108(Suppl):4554–4561. doi:10.1073/pnas.1000087107
Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818. doi:10.1038/nature06245
Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487:104–108. doi:10.1038/nature11225
Domènech E, Mañosa M, Lobatón T, Cabré E (2014) Optimizing post-operative Crohn’s disease treatment. Ann Gastroenterol 27:313–319
Dossa AY, Escobar O, Golden J, Frey MR, Ford HR, Gayer CP (2016) Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling
Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, Bridonneau C, Dumetz F, Grill J-P, Masliah J, Beaugerie L, Cosnes J, Chazouillères O, Poupon R, Wolf C, Mallet J-M, Langella P, Trugnan G, Sokol H, Seksik P (2013) Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62:531–539. doi:10.1136/gutjnl-2012-302578
Dulal S, Keku TO (2014) Gut microbiome and colorectal adenomas. Cancer J 20:225–231. doi:10.1097/PPO.0000000000000050
Duran-Sandoval D, Cariou B, Fruchart J-C, Staels B (2005) Potential regulatory role of the farnesoid X receptor in the metabolic syndrome. Biochimie 87:93–98. doi:10.1016/j.biochi.2004.11.018
Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Lindor KD (2011) High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol 106:1638–1645. doi:10.1038/ajg.2011.156
Edenharder R, Pfützner A (1988) Characterization of NADP-dependent 12 beta-hydroxysteroid dehydrogenase from Clostridium paraputrificum. Biochim Biophys Acta 962:362–370
Edenharder R, Schneider J (1985) 12 beta-dehydrogenation of bile acids by Clostridium paraputrificum, C. tertium, and C. difficile and epimerization at carbon-12 of deoxycholic acid by cocultivation with 12 alpha-dehydrogenating Eubacterium lentum. Appl Environ Microbiol 49:964–968
Edenharder R, Pfützner A, Hammann R (1989a) Characterization of NAD-dependent 3α- and 3β-hydroxysteroid dehydrogenase and of NADP-dependent 7β-hydroxysteroid dehydrogenase from Peptostreptococcus productus. Biochim Biophys Acta - Lipids Lipid Metab 1004:230–238. doi:10.1016/0005-2760(89)90272-5
Edenharder R, Pfützner M, Hammann R (1989b) NADP-dependent 3 beta-, 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activities from a lecithinase-lipase-negative Clostridium species 25.11.C. Biochim Biophys Acta 1002:37–44
Edwards PA, Kast HR, Anisfeld AM (2002) BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res 43:1–12
Ek WE, D’Amato M, Halfvarson J (2014) The history of genetics in inflammatory bowel disease. Ann Gastroenterol 27:294–303
Fang Y, Han SI, Mitchell C, Gupta S, Studer E, Grant S, Hylemon PB, Dent P (2004) Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology 40:961–971. doi:10.1002/hep.1840400427
Färkkilä M, Karvonen A-L, Nurmi H, Nuutinen H, Taavitsainen M, Pikkarainen P, Kärkkäinen P (2004) Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial. Hepatology 40:1379–1386. doi:10.1002/hep.20457
Fashner J, Garcia M, Ribble L, Crowell K (2011) Clinical inquiry: what risk factors contribute to C. difficile diarrhea? J Fam Pract 60:545–547
Ferrandi EE, Bertolesi GM, Polentini F, Negri A, Riva S, Monti D (2012) In search of sustainable chemical processes: cloning, recombinant expression, and functional characterization of the 7α- and 7β-hydroxysteroid dehydrogenases from Clostridium absonum. Appl Microbiol Biotechnol 95:1221–1233. doi:10.1007/s00253-011-3798-x
Fischer L, Gukovskaya a S, Penninger JM, Mareninova O a, Friess H, Gukovsky I, Pandol SJ (2007) Phosphatidylinositol 3-kinase facilitates bile acid-induced Ca2+ responses in pancreatic acinar cells. Am J Physiol - Gastrointest Liver Physiol 292:G875–G886. doi:10.1152/ajpgi.00558.2005
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR (2007) Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104:13780–13785. doi:10.1073/pnas.0706625104
Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, Klomp LWJ, Siersema PD, Schipper MEI, Danese S, Penna G, Laverny G, Adorini L, Moschetta A, van Mil SWC (2011) Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60:463–472. doi:10.1136/gut.2010.212159
Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, Gordon JI, Onderdonk AB, Glimcher LH (2010) Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8:292–300. doi:10.1016/j.chom.2010.08.004
Giel JL, Sorg JA, Sonenshein AL, Zhu J (2010) Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS One 5:e8740. doi:10.1371/journal.pone.0008740
Ginsberg HN, Stalenhoef AFH (2003) The metabolic syndrome: targeting dyslipidaemia to reduce coronary risk. Eur J Cardiovasc Prev Rehabil 10:121–128. doi:10.1177/174182670301000207
Gnewuch C, Liebisch G, Langmann T, Dieplinger B, Mueller T, Haltmayer M, Dieplinger H, Zahn A, Stremmel W, Rogler G, Schmitz G (2009) Serum bile acid profiling reflects enterohepatic detoxification state and intestinal barrier function in inflammatory bowel disease. World J Gastroenterol 15:3134–3141
Gothe F, Beigel F, Rust C, Hajji M, Koletzko S, Freudenberg F (2014) Bile acid malabsorption assessed by 7 alpha-hydroxy-4-cholesten-3-one in pediatric inflammatory bowel disease: correlation to clinical and laboratory findings. J Crohns Colitis 8:1072–1078. doi:10.1016/j.crohns.2014.02.027
Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A (2012) Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol 107:761–767. doi:10.1038/ajg.2011.482
Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ (2013) High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4:125–135. doi:10.4161/gmic.23571
Harris JN, Hylemon PB (1978) Partial purification and characterization of NADP-dependent 12α-hydroxysteroid dehydrogenase from Clostridium leptum. Biochim Biophys Acta - Lipids Lipid Metab 528:148–157. doi:10.1016/0005-2760(78)90060-7
Heuman DM, Pandak WM, Hylemon PB, Vlahcevic ZR (1991) Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts:in vitro studies in rat hepatocytes and human erythrocytes. Hepatology 14:920–926. doi:10.1002/hep.1840140527
Hill MJ, Drasar BS, Williams REO, Meade TW, Cox AG, Simpson JEP, Morson BC (1975) Faecal bile acids and clostridia in patients with cancer of the large bowel. Lancet 305:535–539. doi:10.1016/S0140-6736(75)91556-1
Hirschfield GM, Karlsen TH, Lindor KD, Adams DH (2013) Primary sclerosing cholangitis. Lancet 382:1587–1599. doi:10.1016/S0140-6736(13)60096-3
Hofmann AF (1995) Bile acids as drugs: principles, mechanisms of action and formulations. Ital J Gastroenterol 27:106–113
Hofmann AF (1999) The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 159:2647. doi:10.1001/archinte.159.22.2647
Hofmann AF, Eckmann L (2006) How bile acids confer gut mucosal protection against bacteria. Proc Natl Acad Sci U S A 103:4333–4334
Hofmann M, Schumann C, Zimmer G, Henzel K, Locher U, Leuschner U (2001) LUV’s lipid composition modulates diffusion of bile acids. Chem Phys Lipids 110:165–171. doi:10.1016/S0009-3084(01)00131-1
Hohenester S, de B Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RPO, Beuers U (2012) A biliary HCO3 − umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 55:173–183. doi:10.1002/hep.24691
Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA (2003) Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 17:1581–1591. doi:10.1101/gad.1083503
Holzbach RT, Marsh ME, Freedman MR, Fazio VW, Lavery IC, Jagelman DA (1980) Portal vein bile acids in patients with severe inflammatory bowel disease. Gut 21:428–435. doi:10.1136/gut.21.5.428
Hu X, Bonde Y, Eggertsen G, Rudling M (2014) Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J Intern Med 275:27–38. doi:10.1111/joim.12140
Huang H, Vangay P, McKinlay CE, Knights D (2014) Multi-omics analysis of inflammatory bowel disease. Immunol Lett. doi:10.1016/j.imlet.2014.07.014
Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P (2009) Bile acids as regulatory molecules. J Lipid Res 50:1509–1520. doi:10.1194/jlr.R900007-JLR200
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225. doi:10.1016/j.cmet.2005.09.001
Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson J a, Repa JJ, Mangelsdorf DJ, Kliewer S a (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 103:3920–3925. doi:10.1073/pnas.0509592103
Ishizawa M, Matsunawa M, Adachi R, Uno S, Ikeda K, Masuno H, Shimizu M, Iwasaki K, Yamada S, Makishima M (2008) Lithocholic acid derivatives act as selective vitamin D receptor modulators without inducing hypercalcemia. J Lipid Res 49:763–772. doi:10.1194/jlr.M700293-JLR200
Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A (2011) Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141:1773–1781. doi:10.1053/j.gastro.2011.07.046
Israel EJ, Kleinman RE (1994) Inflammatory bowel disease: diagnosis and treatment. Semin Gastrointest Dis 5:95–105
Ji W, Chen Y, Zhang H, Zhang X, Li Z, Yu Y (2014) Cloning, expression and characterization of a putative 7alpha-hydroxysteroid dehydrogenase in Comamonas testosteroni. Microbiol Res 169:148–154. doi:10.1016/j.micres.2013.07.009
Jones BV, Begley M, Hill C, Gahan CGM, Marchesi JR (2008) Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A 105:13580–13585. doi:10.1073/pnas.0804437105
Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, Hill C, Gahan CGM (2014) Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci U S A 111:7421–7426. doi:10.1073/pnas.1323599111
Kachrimanidou M, Malisiovas N (2011) Clostridium difficile infection: a comprehensive review. Crit Rev Microbiol 37:178–187. doi:10.3109/1040841X.2011.556598
Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, White MB, Noble N a, Monteith P, Fuchs M, Thacker LR, Sikaroodi M, Bajaj JS (2013) Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 58:949–955. doi:10.1016/j.jhep.2013.01.003
Kandell RL, Bernstein C (1991) Bile salt/acid induction of DNA damage in bacterial and mammalian cells: implications for colon cancer. Nutr Cancer 16:227–238. doi:10.1080/01635589109514161
Keitel V, Reinehr R, Gatsios P, Rupprecht C, Görg B, Selbach O, Häussinger D, Kubitz R (2007) The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 45:695–704. doi:10.1002/hep.21458
Khoruts A, Sadowsky MJ (2016) Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol 13:508–516. doi:10.1038/nrgastro.2016.98
Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ (2010) Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 44:354–360. doi:10.1097/MCG.0b013e3181c87e02
Khoruts A, Rank KM, Newman KM, Viskocil K, Vaughn BP, Hamilton MJ, Sadowsky MJ (2016) Inflammatory bowel disease affects the outcome of fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin Gastroenterol Hepatol 14:1433–1438. doi:10.1016/j.cgh.2016.02.018
Kim I, Ahn S-H, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ (2007) Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48:2664–2672. doi:10.1194/jlr.M700330-JLR200
Kisiela M, Skarka A, Ebert B, Maser E (2012) Hydroxysteroid dehydrogenases (HSDs) in bacteria: a bioinformatic perspective. J Steroid Biochem Mol Biol 129:31–46. doi:10.1016/j.jsbmb.2011.08.002
Kondo K (2002) Duodenogastric reflux and gastric stump carcinoma. Gastric Cancer 5:16–22. doi:10.1007/s101200200002
Kuipers F, Stroeve JHM, Caron S, Staels B (2007) Bile acids, farnesoid X receptor, atherosclerosis and metabolic control. Curr Opin Lipidol 18:289–297. doi:10.1097/MOL.0b013e3281338d08
Kumar RS, Brannigan JA, Prabhune AA, Pundle AV, Dodson GG, Dodson EJ, Suresh CG (2006) Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum reveals an evolutionary relationship with penicillin V acylase. J Biol Chem 281:32516–32525. doi:10.1074/jbc.M604172200
Lau BW, Colella M, Ruder WC, Ranieri M, Curci S, Hofer AM (2005) Deoxycholic acid activates protein kinase C and phospholipase C via increased Ca2+ entry at plasma membrane. Gastroenterology 128:695–707. doi:10.1053/j.gastro.2004.12.046
Le M, Krilov L, Meng J, Chapin-Kennedy K, Ceryak S, Bouscarel B (2006) Bile acids stimulate PKCalpha autophosphorylation and activation: role in the attenuation of prostaglandin E1-induced cAMP production in human dermal fibroblasts. Am J Physiol - Gastrointest Liver Physiol 291:G275–G287. doi:10.1152/ajpgi.00346.2005
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89:147–191. doi:10.1152/physrev.00010.2008
Lewin JS, Gillenwater AM, Garrett JD, Bishop-Leone JK, Nguyen DD, Callender DL, Ayers GD, Myers JN (2003) Characterization of laryngopharyngeal reflux in patients with premalignant or early carcinomas of the larynx. Cancer 97:1010–1014. doi:10.1002/cncr.11158
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075. doi:10.1073/pnas.0504978102
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023. doi:10.1038/4441022a
Lindor KD, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, Mooney J, Sargeant C, Braaten J, Bernard T, King D, Miceli E, Schmoll J, Hoskin T, Thapa P, Enders F (2009) High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 50:808–814. doi:10.1002/hep.23082
Looby E, Long A, Kelleher D, Volkov Y (2005) Bile acid deoxycholate induces differential subcellular localisation of the PKC isoenzymes beta 1, epsilon and delta in colonic epithelial cells in a sodium butyrate insensitive manner. Int J Cancer 114:887–895. doi:10.1002/ijc.20803
Macdonald IA, Meier EC, Mahony DE, Costain GA (1976) 3α-, 7α- and 12α-hydroxysteroid dehydrogenase activities from Clostridium perfringens. Biochim Biophys Acta - Lipids Lipid Metab 450:142–153. doi:10.1016/0005-2760(76)90086-2
Macdonald IA, Jellett JF, Mahony DE, Holdeman LV (1979) Bile salt 3α- and 12α-hydroxysteroid dehydrogenases from Eubacterium lentum and related organisms. Appl Environ Microbiol 37:992–1000
MacDonald IA, Rochon YP, Hutchison DM, Holdeman LV (1982) Formation of ursodeoxycholic acid from chenodeoxycholic acid by a 7 beta-hydroxysteroid dehydrogenase-elaborating Eubacterium aerofaciens strain cocultured with 7 alpha-hydroxysteroid dehydrogenase-elaborating organisms. Appl Environ Microbiol 44:1187–1195
Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365. doi:10.1126/science.284.5418.1362
Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ (2002) Vitamin D receptor as an intestinal bile acid sensor. Science 296:1313–1316. doi:10.1126/science.1070477
Mallonee DH, Lijewski MA, Hylemon PB (1995) Expression in Escherichia coli and characterization of a bile acid-inducible 3α-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI 12708. Curr Microbiol 30:259–263. doi:10.1007/BF00295498
Maxson CJ, Klein HD, Rubin W (1994) Atypical forms of inflammatory bowel disease. Med Clin North Am 78:1259–1273
Miyake JH, Wang SL, Davis RA (2000) Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7alpha-hydroxylase. J Biol Chem 275:21805–21808. doi:10.1074/jbc.C000275200
Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall H-U, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D (2013) Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145:574–82.e1. doi:10.1053/j.gastro.2013.05.042
Nagengast F, Grubben MJ, van Munster I (1995) Role of bile acids in colorectal carcinogenesis. Eur J Cancer 31:1067–1070. doi:10.1016/0959-8049(95)00216-6
Nguyen A, Bouscarel B (2008) Bile acids and signal transduction: role in glucose homeostasis. Cell Signal 20:2180–2197. doi:10.1016/j.cellsig.2008.06.014
Nistal E, Fernández-Fernández N, Vivas S, Olcoz JL (2015) Factors determining colorectal cancer: the role of the intestinal microbiota. Front Oncol 5:220. doi:10.3389/fonc.2015.00220
Ogilvie LA, Jones BV (2012) Dysbiosis modulates capacity for bile acid modification in the gut microbiomes of patients with inflammatory bowel disease: a mechanism and marker of disease? Gut 61:1642–1643. doi:10.1136/gutjnl-2012-302137
Pardi DS, Loftus EV, Kremers WK, Keach J, Lindor KD (2003) Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 124:889–893. doi:10.1053/gast.2003.50156
Patel LN, Schairer J, Shen B (2014) Fecal transplantation therapy for Clostridium difficile-associated pouchitis. Int J Color Dis 29:263–264. doi:10.1007/s00384-013-1771-0
Pérez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, Otto W, Rojo D, Bargiela R, von Bergen M, Neulinger SC, Däumer C, Heinsen F-A, Latorre A, Barbas C, Seifert J, dos Santos VM, Ott SJ, Ferrer M, Moya A (2013) Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut 62:1591–1601. doi:10.1136/gutjnl-2012-303184
Pols TWH, Noriega LG, Nomura M, Auwerx J, Schoonjans K (2011) The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis 29:37–44. doi:10.1159/000324126
Porez G, Prawitt J, Gross B, Staels B (2012) Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res 53:1723–1737. doi:10.1194/jlr.R024794
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. doi:10.1126/science.1241214
Ridlon JM, Hylemon PB (2012) Identification and characterization of two bile acid coenzyme a transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J Lipid Res 53:66–76. doi:10.1194/jlr.M020313
Ridlon JM, Kang D-J, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. doi:10.1194/jlr.R500013-JLR200
Ridlon JM, Kang D-J, Hylemon PB (2010) Isolation and characterization of a bile acid inducible 7alpha-dehydroxylating operon in Clostridium hylemonae TN271. Anaerobe 16:137–146. doi:10.1016/j.anaerobe.2009.05.004
Ridlon JM, Alves JM, Hylemon PB, Bajaj JS (2013) Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 4:382–387. doi:10.4161/gmic.25723
Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS (2014) Bile acids and the gut microbiome. Curr Opin Gastroenterol 30:332–338. doi:10.1097/MOG.0000000000000057
Ridlon JM, Wolf PG, Gaskins HR (2016) Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 7:201–215
Robinson CJ, Young VB (2010) Antibiotic administration alters the community structure of the gastrointestinal micobiota. Gut Microbes 1:279–284. doi:10.4161/gmic.1.4.12614
Ross RK, Hartnett NM, Bernstein L, Henderson BE (1991) Epidemiology of adenocarcinomas of the small intestine: is bile a small bowel carcinogen? Br J Cancer 63:143–145
Russell DW, Setchells KDR (1992) Bile acid biosynthesis. Biochemistry 31:4737–4749
Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235. doi:10.1016/j.cmet.2013.01.003
Schultz M, Tonkonogy SL, Sellon RK, Veltkamp C, Godfrey VL, Kwon J, Grenther WB, Balish E, Horak I, Sartor RB (1999) IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 276:G1461–G1472
Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB (1998) Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66:5224–5231
Seril DN, Shen B (2014) Clostridium difficile infection in patients with ileal pouches. Am J Gastroenterol 109:941–947. doi:10.1038/ajg.2014.22
Shah SA, Looby E, Volkov Y, Long A, Kelleher D (2005) Ursodeoxycholic acid inhibits translocation of protein kinase C in human colonic cancer cell lines. Eur J Cancer 41:2160–2169. doi:10.1016/j.ejca.2005.06.015
Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, Weese S, Wong A, Low DE, Pillai DR (2012) Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. MBio 3:e00338–e00312. doi:10.1128/mBio.00338-12
Sherrod JA, Hylemon PB (1977) Partial purification and characterization of NAD-dependent 7α-hydroxysteroid dehydrogenase from Bacteroides thetaiotaomicron. Biochim Biophys Acta - Lipids Lipid Metab 486:351–358. doi:10.1016/0005-2760(77)90031-5
Sinha J, Chen F, Miloh T, Burns RC, Yu Z, Shneider BL (2008) Beta-klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes. Am J Physiol Gastrointest Liver Physiol 295:G996–G1003. doi:10.1152/ajpgi.90343.2008
Sobczak M, Fabisiak A, Murawska N, Wesołowska E, Wierzbicka P, Wlazłowski M, Wójcikowska M, Zatorski H, Zwolińska M, Fichna J (2014) Current overview of extrinsic and intrinsic factors in etiology and progression of inflammatory bowel diseases. Pharmacol Reports 66:766–775. doi:10.1016/j.pharep.2014.04.005
Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO (2002) Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointest Endosc 56:48–54. doi:10.1067/mge.2002.125367
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J, Blugeon S, Bridonneau C, Furet J-P, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105:16731–16736. doi:10.1073/pnas.0804812105
Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM (2002) Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc Natl Acad Sci U S A 99:13801–13806. doi:10.1073/pnas.212494599
Sorg JA, Sonenshein AL (2008) Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190:2505–2512. doi:10.1128/JB.01765-07
Sorg JA, Sonenshein AL (2009) Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J Bacteriol 191:1115–1117. doi:10.1128/JB.01260-08
Sorg JA, Sonenshein AL (2010) Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol 192:4983–4990. doi:10.1128/JB.00610-10
Stamp DH (2002) Three hypotheses linking bile to carcinogenesis in the gastrointestinal tract: certain bile salts have properties that may be used to complement chemotherapy. Med Hypotheses 59:398–405. doi:10.1016/S0306-9877(02)00125-1
Staudinger JL, Goodwin B, Jones S a, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer S a (2001) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 98:3369–3374. doi:10.1073/pnas.051551698
Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, Israeli D, Zmora N, Gilad S, Weinberger A, Kuperman Y, Harmelin A, Kolodkin-Gal I, Shapiro H, Halpern Z, Segal E, Elinav E (2014) Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514:181–186. doi:10.1038/nature13793
Sutherland J, Macdonald I (1982) The metabolism of primary, 7-oxo, and 7 beta-hydroxy bile acids by Clostridium absonum. J Lipid Res 23:726–732
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43:3380–3389. doi:10.1128/JCM.43.7.3380-3389.2005
Sze M, Schloss PD (2016) Looking for a signal in the noise: revisiting obesity and the microbiome
Tanaka H, Hashiba H, Kok J, Mierau I (2000) Bile salt hydrolase of Bifidobacterium longum -biochemical and genetic characterization. Appl Environ Microbiol 66:2502–2512. doi:10.1128/AEM.66.6.2502-2512.2000
The Human Microbiome Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi:10.1038/nature11234
Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB (2014) Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. doi:10.1038/ncomms4114
Thomas C, Auwerx J, Schoonjans K (2008) Bile acids and the membrane bile acid receptor TGR5--connecting nutrition and metabolism. Thyroid 18:167–174. doi:10.1089/thy.2007.0255
Thompson PA, Wertheim BC, Roe DJ, Ashbeck EL, Jacobs ET, Lance P, Martínez ME, Alberts DS (2009) Gender modifies the effect of ursodeoxycholic acid in a randomized controlled trial in colorectal adenoma patients. Cancer Prev Res 2:1023–1030. doi:10.1158/1940-6207.CAPR-09-0234
Tucker ON, Dannenberg AJ, Yang EK, Fahey TJ (2004) Bile acids induce cyclooxygenase-2 expression in human pancreatic cancer cell lines. Carcinogenesis 25:419–423. doi:10.1093/carcin/bgh010
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. doi:10.1038/nature05414
Van Eldere J, Celis P, De Pauw G, Lesaffre E, Eyssen H (1996) Tauroconjugation of cholic acid stimulates 7 alpha-dehydroxylation by fecal bacteria. Appl Environ Microbiol 62:656–661
van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JFWM, Tijssen JGP, Speelman P, Dijkgraaf MGW, Keller JJ (2013) Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368:407–415. doi:10.1056/NEJMoa1205037
Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S (2009) The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183:6251–6261. doi:10.4049/jimmunol.0803978
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science 328:228–231. doi:10.1126/science.1179721
Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A (2002) Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540:49–55. doi:10.1113/jphysiol.2002.017525
Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JFWM, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JET, Bloks VW, Groen AK, Heilig HGHJ, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JBL, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143:913–6.e7. doi:10.1053/j.gastro.2012.06.031
Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS, van Nood E, Holleman F, Knaapen M, Romijn JA, Soeters MR, Blaak EE, Dallinga-Thie GM, Reijnders D, Ackermans MT, Serlie MJ, Knop FK, Holst JJ, van der Ley C, Kema IP, Zoetendal EG, de Vos WM, Hoekstra JBL, Stroes ES, Groen AK, Nieuwdorp M (2014) Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol 60:824–831. doi:10.1016/j.jhep.2013.11.034
Walters WA, Xu Z, Knight R (2014) Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett 588:4223–4233. doi:10.1016/j.febslet.2014.09.039
Weingarden AR, Chen C, Bobr A, Yao D, Lu Y, Nelson VM, Sadowsky MJ, Khoruts A (2014) Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol - Gastrointest Liver Physiol 306:G310–G319. doi:10.1152/ajpgi.00282.2013
Weingarden AR, Chen C, Zhang N, Graiziger CT, Dosa PI, Steer CJ, Shaughnessy MK, Johnson JR, Sadowsky MJ, Khoruts A (2016a) Ursodeoxycholic acid inhibits Clostridium difficile spore germination and vegetative growth, and prevents the recurrence of ileal pouchitis associated with the infection. J Clin Gastroenterol 50:624–630. doi:10.1097/MCG.0000000000000427
Weingarden AR, Dosa PI, DeWinter E, Steer CJ, Shaughnessy MK, Johnson JR, Khoruts A, Sadowsky MJ (2016b) Changes in colonic bile acid composition following fecal microbiota transplantation are sufficient to control Clostridium difficile germination and growth. PLoS One 11:e0147210. doi:10.1371/journal.pone.0147210
Wells JE, Hylemon PB (2000) Identification and characterization of a bile acid 7alpha-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7alpha-dehydroxylating strain isolated from human feces. Appl Environ Microbiol 66:1107–1113
Wells JE, Berr F, Thomas LA, Dowling RH, Hylemon PB (2000) Isolation and characterization of cholic acid 7alpha-dehydroxylating fecal bacteria from cholesterol gallstone patients. J Hepatol 32:4–10
Wells JE, Williams KB, Whitehead TR, Heuman DM, Hylemon PB (2003) Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7α-dehydroxylating bacteria in human feces. Clin Chim Acta 331:127–134. doi:10.1016/S0009-8981(03)00115-3
Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, Tysk C, Jansson JK (2009) Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis 15:653–660. doi:10.1002/ibd.20783
Wilson KH (1983) Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J Clin Microbiol 18:1017–1019
Wilson KH, Kennedy MJ, Fekety FR (1982) Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J Clin Microbiol 15:443–446
Xie W, Radominska-pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM (2001) An essential role for nuclear receptors SXR ͞ PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A 98:3375–3380
Yamaoka-Tojo M, Tojo T, Izumi T (2008) Beyond cholesterol lowering: pleiotropic effects of bile acid binding resins against cardiovascular disease risk factors in patients with metabolic syndrome. Curr Vasc Pharmacol 6:271–281. doi:10.2174/157016108785909698
Yang Y, Zhang M, Eggertsen G, Chiang JYL (2002) On the mechanism of bile acid inhibition of rat sterol 12alpha-hydroxylase gene (CYP8B1) transcription: roles of alpha-fetoprotein transcription factor and hepatocyte nuclear factor 4alpha. Biochim Biophys Acta 1583:63–73
Yoshimoto T, Higashi H, Kanatani A, Lin XS, Nagai H, Oyama H, Kurazono K, Tsuru D (1991) Cloning and sequencing of the 7 alpha-hydroxysteroid dehydrogenase gene from Escherichia coli HB101 and characterization of the expressed enzyme. J Bacteriol 173:2173–2179
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499:97–101. doi:10.1038/nature12347
Záratea G, Gonzaleza S, Chaia AP, Oliver G (2000) Effect of bile on the β -galactosidase activity of dairy propionibacteria. Lait 80:267–276
Zhong M (2010) TGR5 as a therapeutic target for treating obesity. Curr Top Med Chem 10:386–396. doi:10.2174/156802610790980576
Acknowledgements
This work was supported by the NIH grant 1R21-AI114722-01 (MJS and AK), Minnesota’s Discovery, Research and InnoVation Economy grant from the University of Minnesota (MJS and AK), and the University of Minnesota Clinical and Translational Science Institute UL1TR000114 grant via the National Center for Advancing Translational Sciences of the NIH (ARW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interests.
Ethical approval
Any studies with humans or animals performed by any of the authors were approved by the University of Minnesota Institutional Review Board (IRB) and Institutional Animal Care and Use Committee (IACUC). As is known by the authors, referenced studies adhere to applicable international, national, and/or institutional guidelines for the care and use of animals.
Additional information
Alexander Khoruts and Michael J. Sadowsky these authors contributed equally to this work.
Rights and permissions
About this article

Cite this article
Staley, C., Weingarden, A.R., Khoruts, A. et al. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol 101, 47–64 (2017). https://doi.org/10.1007/s00253-016-8006-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-8006-6