
Abstract
In this paper, we present a new counterselection method for deleting fragments from Lactococcus lactis chromosome. The method uses a non-replicating plasmid vector, which integrates into the chromosome and makes the cell sensitive to bacteriocins. The integration vector carries pUC ori functional in Escherichia coli but not in L. lactis, an erythromycin resistance gene for selecting single crossover integrants, and two fragments from L. lactis chromosome for homologous recombinations. In addition, the integration vector is equipped with the Listeria monocytogenes gene mptC encoding the mannose-phosphotransferase system component IIC, the receptor for class IIa bacteriocins. Expression of mptC from the integration vector renders the naturally resistant L. lactis sensitive to class IIa bacteriocins. This sensitivity is then used to select the double crossover colonies on bacteriocin agar. Only the cells which have regained the endogenous bacteriocin resistance through the loss of the mptC plasmid will survive. The colonies carrying the desired deletion can then be distinguished from the wild-type revertants by PCR. By using the class IIa bacteriocins leucocin A, leucocin C or pediocin AcH as the counterselective agents, we deleted 22- and 33-kb chromosomal fragments from the wild-type nisin producing L. lactis strain N8. In conclusion, this counterselection method presented here is a convenient, efficient and inexpensive technique to generate successive deletions in L. lactis chromosome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chromosomal deletions in bacteria are conventionally accomplished by two homologous recombinations using an integration vector with two chromosomal fragments neighbouring the area to be deleted. This approach usually involves two crossover steps. First, two chromosomal fragments flanking the target deletion area are ligated to each other in a vector not replicating in the target host. The construct is then integrated into the chromosome at either of the two homologous regions via single crossover (SCO) recombination. The selection of integrants is typically based on antibiotic resistance. Then, the integrants are screened for a second recombination, resulting in the deletion of the target region, or a reversion to the wild-type genotype. Since the frequency of the second recombination can be low and only part of the double crossover (DCO) colonies carry the deletion, finding the desired strain without selection often requires extensive screening. Therefore, a variety of systems have been developed to either select the DCO or increase its frequency. For instance, temperature-sensitive (TS) pG+Host vectors and Cre-loxP plasmids have been used for Lactococcus lactis genome editing (Biswas et al. 1993; Zhu et al. 2015b). The integration of a pG+Host vector into the chromosome takes place at 37 °C, after which the plasmid is excised by second crossover at 28 °C. TS vectors are good tools for creating single knockout strains. However, culturing L. lactis at 37 °C causes heat-induced mutations, which makes TS vectors less suitable for successive deletions in the same strain (Smith et al. 2012). In the Cre-loxP system, the chromosomal region to be deleted is first replaced by the chloramphenicol resistance gene cat with flanking lox recognition sites for the Cre recombinase (Lambert et al. 2007). Then, via transient expression of the Cre recombinase, the lox-cat-lox cassette is removed. However, there is no actual selection for the gene replacement; instead, the second crossover can be found only by screening for erythromycin sensitivity. In addition, the method is not completely seamless, as it leaves short inactive lox-sites into the chromosome.
Other tools for generating chromosomal deletions are counterselectable vectors, which carry both positive and negative selection markers. The integration of the vector is selected with the positive marker, usually an antibiotic resistance gene, and the loss of the vector through second homologous recombination is selected with the negative marker causing sensitivity to a selective agent. Thus, only those strains, which have undergone the second recombination and lost the integrated vector, will survive. For L. lactis, the vector pCS1966 carrying the orotate transporter gene oroP as a counterselectable marker has been developed (Solem et al. 2008). Expression of oroP renders the cells sensitive to 5′-fluoroorotic acid (FOA), which is then used to select the second recombination. Even though the method seems effective, the FOA selection has some practical inconveniences. Firstly, FOA is rather expensive compound and poorly soluble in aqueous solutions. In addition, for selecting DCO colonies, L. lactis has to be cultured in chemically defined media supplemented with FOA. Moreover, oroP is a common gene among L. lactis strains (Siezen et al. 2011), which causes the risk of unwanted homologous recombinations between the native and the introduced oroP genes.
Another counterselection method developed for lactic acid bacteria is based on uracil phosphoribosyltransferase (upp) for Lactobacillus acidophilus, Lactobacillus casei and L. lactis (Goh et al. 2009; Song et al. 2014). The expression of upp from integration vector causes sensitivity to 5-fluorouracil, which can be used to select the DCO colonies. As upp gene is commonly existing and expressed in lactic acid bacteria, the deletion of upp from the host is a prerequisite for its use in counterselection. Another possible inconvenience with the upp system is that regardless of the upp mutation, 5-fluorouracil may still be metabolised and thus be toxic to the mutants (Martinussen and Hammer 1994).
Class IIa (pediocin-like) bacteriocins are Listeria-active peptides, which form a membrane pore leading to cell leakage (Drider et al. 2006). On sensitive cells, the component IIC (MptC) of the mannose-phosphotransferase system (Man-PTS) is responsible for the specific binding of the class IIa bacteriocins (Kjos et al. 2010; Ramnath et al. 2004). L. lactis is naturally resistant to class IIa bacteriocins, as the Lactococcus Man-PTS component IIC (PtnC) and MptC in Listeria are different. However, heterologous expression of listerial mptC gene in L. lactis renders the lactococcal cells sensitive to class IIa bacteriocins, as MptC can form a functional Man-PTS permease complex with the lactococcal component IID (Kjos et al. 2010).
In this study, we describe a counterselection system for genome editing in L. lactis. The integration vector carries erythromycin resistance gene for selecting SCO integrants and the listerial mptC gene for selecting the loss of the vector through the second crossover with class IIa bacteriocin. We demonstrated the efficiency of the bacteriocin counterselection by deleting chromosomal fragments over 30 kb from a wild-type L. lactis strain.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Lactococcus strains were grown in M17 (Oxoid Ltd. Basingstoke, UK) supplemented with 0.5 % (w/v) glucose (M17G) at 30 °C. For L. lactis strains LAC360, LAC409 and LAC428, 0.5 μg nisin/ml was added for plasmid maintenance. Escherichia coli was grown in LB at 37 °C. Listeria monocytogenes WSLC 1033 was grown in brain heart infusion (BHI, Lab M, Lancashire, UK) broth with shaking at 37 °C. Pediocin AcH (Ped) producer Lactobacillus plantarum WHE 92 was grown in MRS (Merck, New Jersey, USA) at 30 °C. For transformant selection, erythromycin (Erm) was used at final concentrations of 250 μg/ml for E. coli and 10 μg/ml for L. lactis. For selecting DCO colonies, leucocin A (LcnA) from L. lactis LAC428 and leucocin C (LecC) from LAC409 were used at the concentrations of 3 and 0.3 μg/ml. Alternatively, LecC and Ped were used as pasteurised cell-free culture supernatants at the concentration of 20 μl/ml.
Nucleic acid techniques
Plasmids from E. coli and L. lactis were isolated with GeneJET Plasmid Miniprep Kit (Thermo Scientific, Waltham, MA, USA). In addition to supplier’s procedure, lysozyme (10 mg/ml) was added to L. lactis cell resuspension solution and incubated at 37 °C for 1 h before lysing the cells.
Fragments for screening, cloning and sequencing were amplified by standard PCR with either Taq DyNAzyme™ II DNA polymerase or Phusion High-Fidelity DNA polymerase (Thermo Scientific) in Eppendorf Mastercycler (Hamburg, Germany). PCR primers used are listed in Table 2. Overlap extension PCR (OE-PCR) was used to fuse the two lactococcal chromosomal fragments for homologous recombinations with the pUC ori from pBluescript (Agilent Technologies, Santa Clara, CA, USA). PCR products were purified with GeneJET PCR Purification Kit, or extracted from agarose gel with GeneJET Gel Extraction Kit (Thermo Scientific). Plasmid constructs and PCR products were sequenced by DNA sequencing service in the Institute of Biotechnology (University of Helsinki, Finland).
T4 DNA ligase, T4 polynucleotide kinase and restriction enzymes were used as recommended by the suppliers (Thermo Scientific; New England Biolabs, Ipswich, MA, USA; Promega, Madison, WI, USA). Plasmids were transferred into E. coli and L. lactis by electroporation with a Bio-Rad Gene Pulser device (Bio-Rad Laboratories, Richmond, CA, USA) essentially as described previously (Holo and Nes 1989; Zabarovsky and Winberg 1990). Two micrograms of each integration vector and 20 ng of transformation control plasmid pLEB579 were used in L. lactis electroporation.
Bacteriocins and MICs
LcnA and LecC were obtained from heterologous producers L. lactis LAC428 and LAC409 by precipitating overnight culture supernatants with ammonium sulphate, as described previously (Wan et al. 2013, 2015). The LcnA and LecC concentrations in the crude preparations were estimated to be about 150 and 100 μg/ml, respectively, by comparing band intensities with standard protein samples in SDS-PAGE, as described previously (Wan et al. 2015). L. lactis strain LAC360, which carries the empty cloning vector pLEB690 (Li et al. 2011), was treated in the same way as LAC409 and LAC428 and used as a negative control in minimum inhibitory concentration (MIC) determinations.
To determine the MICs, mptC host L. monocytogenes WSLC 1033, L. lactis N8, the strain MINL18 with replicative mptC plasmid and the strain MINL20 with integrated mptC plasmid were cultured with serial dilutions of LcnA or LecC. Honeycomb microtiter plate wells were filled with 200 μl of appropriate media inoculated with 0.2 % of overnight culture of each indicator strain. Leucocins (LcnA 0, 0.5, 1 μg/ml; LecC 0, 0.01, 0.02, 0.05 μg/ml) and the negative control were added to the wells. The plates were incubated in Bioscreen C (Labsystems, Helsinki, Finland) at 30 °C with constant shaking for Listeria and at 30 °C with 30-s shaking before measurement for L. lactis. The optical density was measured every hour with a wideband filter (420–580 nm). All MIC determinations were performed in triplicate. The MIC was defined as the lowest bacteriocin concentration for which OD value of all three parallel cultures was below 0.1 in 12 h.
Construction of plasmids
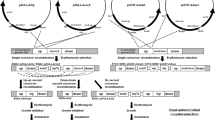
Construction of the replicative mptC plasmid pWUST25 and the integration vectors pWUST27 and pWUST28 is shown in Fig. 1. The mptC from L. monocytogenes WSLC1033 was fused with the nisin-inducible promoter P nisZ from L. lactis N8 to create P nisZ -mptC fragment as follows: Individual fragments of mptC and P nisZ were amplified from L. monocytogenes and L. lactis genome with primers MptC F-MptC R and Pnis F-Pnis R, respectively. The two fragments were then restricted with PciI and NcoI separately, and ligated together. The ligated product was used as the template in PCR with Pnis F-MptC R primers. The P nisZ -mptC fragment was restricted with XhoI and ApaI, ligated with SalI-ApaI digested repAC-ermC fragment amplified from the L. lactis cloning vector pLEB579. The replicon repAC originating from the lactococcal plasmid pSH71 (Gasson 1983) is also functional in E. coli. The ligation mixture was electroporated into E. coli TG1, the right clone was found by PCR screening, and the resulting mptC expression plasmid pWUST25 was isolated and electroporated into L. lactis N8 for testing the MptC function from replicative multicopy plasmid.

Construction of replicative mptC expression plasmid pWUST25 and integrative plasmids pWUST27 and pWUST28 (not in scale). The replicative plasmid was made of a P nisZ -mptC fragment ligated with repAC + ermC fragment. For integration vectors, repAC was replaced by the OE-PCR fusion of pUC ori and two chromosomal fragments (H1 and H2, about 1 kb each) for homologous recombinations. PCR primers used are shown with short arrows. Restriction sites ApaI and SalI/XhoI used for constructing pWUST25 are marked in the end of the primers. TT transcription terminator
For the integrative plasmids, the repAC genes in pWUST25 were replaced with the fusion of pUC ori from pBluescript and two chromosomal fragments (H1, H2, about 1 kb each) from L. lactis N8. The genome sequence of L. lactis N8 was kindly provided by Professor M. Qiao (Nankai University, China). First, the three pieces of H1, H2 and pUC ori were joined by OE-PCR, and the resulting fragment was then ligated blunt-ended with ermC-P nisZ -mptC amplified from pWUST25. The transcription terminator of repAC was included in the ermC-P nisZ -mptC fragment to prevent transcriptional elongation downstream of mptC. The ligation mixtures were electroporated into E. coli TG1, right clones were screened by PCR, and the resulting integration vectors were isolated and electroporated into L. lactis N8. Two integration vectors were constructed: for chromosomal deletions of 22,286 bp from the transposon Tn5481 and a 33,256-bp region containing genes for xylose utilisation. These fragments were chosen, because they are large regions not containing any genes from the core genome of L. lactis (Siezen et al. 2011; M. Qiao, unpublished). Transposon Tn5481 is also known as the nisin-sucrose transposon, but the aimed 22-kb deletion included neither nisin nor sucrose genes. The putative genes included in the two regions to be deleted are listed in Table S1.
Generation of deletion mutants
After electroporation into L. lactis N8, transformants with integrated plasmid were selected with erythromycin, and the correct SCO integration was confirmed by PCR and bacteriocin sensitivity on M17G + Erm + LecC plate. For the second crossover, integrants were grown o/n in M17G + Erm broth, followed by five transfers (1 % inoculum) in M17G broth without Erm (about 35 generations). The cultures were serially diluted to 10−4, from which 0.1 ml were spread onto M17G agar plates supplemented with LcnA, LecC, or Ped. For comparison, the o/n M17G + Erm broth culture was diluted to 10−2, and spread similarly onto M17G + bacteriocin agar plates. The plates were incubated overnight, and the obtained colonies were confirmed not to carry integration vectors by Erm sensitivity on M17G + Erm agar plate. The bacteriocin resistant, Erm-sensitive DCO colonies were screened by PCR with corresponding H1 F − H2 R primers, and the correct chromosomal deletions were confirmed by sequencing. Growth of the deletion strains in M17 broth supplemented with 0.5 % glucose or 1 % xylose were examined using Bioscreen C (Labsystems) with 1 % inoculums and measuring the optical density every hour with a wideband filter (420–580 nm).
For generating double deletion mutant, the Δ22-kb L. lactis strain MINL25 was used as the host in transformation to uptake the integration vector for the 33-kb deletion. Consequent SCO and DCO strains were obtained and screened by the same above-mentioned method.
Results
Expression of mptC in L. lactis N8
Previously, it has been demonstrated that expression of listerial mptC in a nonsensitive L. lactis strain without Man-PTS resulted in sensitivity to class IIa bacteriocins (Kjos et al. 2010). Since L. lactis N8 carries the ptn genes encoding Man-PTS, the first aim here was to test whether the expression of listerial mptC makes the wild-type L. lactis N8 sensitive to class IIa bacteriocins. As listerial component IIC can form a functional Man-PTS permease complex with lactococcal component IID, expressing only the IIC component from Listeria could be sufficient to make L. lactis N8 sensitive to class IIa bacteriocins. The gene mptC from Listeria was cloned into a replicative plasmid under the control of a nisin promoter, and the construct was introduced into L. lactis N8, resulting in the mptC expression strain MINL18. Sensitivity to bacteriocins was first tested by spot-on-lawn method on plates, and compared with Listeria and the wild-type L. lactis N8. L. monocytogenes WSLC 1033 and the strain MINL18 showed high sensitivity to class IIa bacteriocins leucocins A and C and pediocin AcH, as large halos were obtained on indicator plates, whereas the host strain N8 was completely resistant (Fig. 2). On both Listeria and MINL18 plates, small colonies grew inside the bacteriocin halos, indicating that L. lactis can develop bacteriocin resistance similarly as Listeria. Even though the inhibition zones were bigger on Listeria plate, the MICs of both L. monocytogenes WSLC 1033 and L. lactis MINL18 were determined to be 1 μg/ml for LcnA and 0.02 μg/ml for LecC. In conclusion, the expression of listerial mptC made the wild-type L. lactis sensitive to class IIa bacteriocins.

Sensitivity to class IIa bacteriocins LcnA, LecC and pediocin of a wild-type Lactococcus lactis N8, b Listeria monocytogenes WSLC 1033 and c L. lactis N8 derivative MINL18 carrying replicative mptC expression plasmid. Leucocins A and C were used as crude preparations, pediocin as cell-free culture supernatant. Three microliters of leucocins and 10 μl of pediocin were spotted on indicator lawns. A LcnA, C LecC, P pediocin
Construction of the integration vectors
The integration vectors for chromosomal deletions were constructed by replacing the repAC from the replicative mptC plasmid pWUST25 with pUC ori and two fragments from L. lactis N8 chromosome for homologous recombinations (Fig. 1). The strategy to obtain chromosomal deletion through two homologous recombinations is presented in Fig. 3.

Scheme of using mptC-based selection/counterselection integration vector for constructing gene knockout in L. lactis (not in scale). a Plasmid carrying two homologous regions (H1, H2) flanking the region to be deleted in L. lactis N8 genome is integrated into the chromosome at either of the homologous regions, and the integrant is selected by erythromycin resistance. The resulting integrant becomes sensitive to class IIa bacteriocins. b A second recombination event at H1 or H2 leads to the excision of the integrated plasmid. Only the cells which have lost the integration vector can survive in the presence of class IIa bacteriocin. The second recombination at the same homologous region (H1) as in the integration event reverts the integrant to the wild type. If the second recombination takes place at H2, the event results in a chromosomal deletion between H1 and H2. Chr chromosomal DNA; diagonal stripes the region to be deleted
Two integration vectors were constructed, aiming to delete a 22-kb region in the transposon Tn5481, and a 33-kb region containing xylose utilisation genes from L. lactis N8 chromosome.
Single crossover
The constructed integration vectors and the transformation control plasmid pLEB579 were eletroporated into L. lactis N8, and the transformants were selected with erythromycin. Number of colonies obtained from 2 μg of electroporated integration vectors aiming at 22- and 33-kb deletions was 5 and 7, respectively, while transformation frequency was about 106 colonies per microgram of pLEB579. Correct integrants were verified by PCR, and their sensitivity to class IIa bacteriocins was tested on M17G + Erm agar by spot-on-lawn method. All three bacteriocins, leucocin A, leucocin C and pediocin AcH, caused large inhibition zones, showing that L. lactis with integrated mptC renders the cells sensitive to the bacteriocins (Fig. 4). The MIC for L. lactis integrant was the same as those of the replicative multicopy mptC plasmid (1 μg/ml for LcnA, 0.02 μg/ml for LecC).

Class IIa bacteriocin sensitivity of Lactococcus lactis N8 carrying integrated mptC-vectors. Indicators: a MINL25 for 22-kb deletion. b MINL26 for 33-kb deletion. LcnA and LecC were used as crude preparations, pediocin as cell-free culture supernatant. Three microliters of leucocins and 10 μl of pediocin were spotted on indicator lawns. A LcnA, C LecC, P pediocin
Selecting the DCO colonies and finding the deletions
To find the clones, which has lost the integration plasmid through second recombination, two strategies were tested: first, the integrants were grown in M17G broth without Erm for about 35 generations. Alternatively, the integrants were cultured in M17G with Erm overnight. Both cultures were then serially diluted and plated onto bacteriocin agar. Twenty microliters per milliliter of LecC or Ped producer culture supernatant was shown to be sufficient to select DCO colonies, virtually without background or bacteriocin resistant mutants. From Erm-free pre-culture, about 1 % of the plated cells grew on bacteriocin plates, whereas from antibiotic pre-culture, about 0.01 % of the cells were resistant to bacteriocins. Nearly all (about 99 %) of the obtained colonies were confirmed to be Erm-sensitive, showing the excision of the integration vector through the second recombination. These colonies either contained the desired chromosomal deletion, or had reverted to the wild-type, as presented in Fig. 3.
The deletion strains were distinguished from the wild-type revertants by PCR and confirmed by sequencing. The proportion of the 22- and 33-kb deletions was about 10 and 40 %. The growth of the deletion strains on glucose and xylose was then examined. Compared to the wild-type, the deletions did not significantly affect the strains’ growth on glucose (Fig. 5). As expected, the 33-kb deletion strain did not grow on xylose, showing that the deletion altered the strain’s phenotype.

Growth of L. lactis N8 (WT) and the deletion strains Δ22- and Δ33-kb on M17 supplemented with glucose or xylose. The optical density was measured with a wideband filter (420–580 nm) in Bioscreen C. Error bars standard deviation of the mean from three parallel cultures. The optical density of the growth in M17 without sugar is subtracted from the values in the graph
Double deletion
To test if the presented counterselection method can be used to generate multiple deletions in the same strain, 2 μg of the integration vector pWUST28 aiming at deleting the 33-kb xylose region was introduced into the Δ22-kb strain MINL25 yielding four SCO colonies. The same selection-counterselection procedure was followed, and the deletion of 33-kb region was confirmed by PCR, sequencing and xylose negative phenotype. About 8 % of the colonies on bacteriocin selection plates contained the correct double deletion. In conclusion, the method described in this paper can be used to create successive chromosomal deletions in L. lactis strain.
Discussion
In this study, we present a new counterselection method for generating markerless chromosomal deletions in wild-type nisin producing L. lactis. The method is based on bacteriocin sensitivity by expressing the L. monocytogenes gene mptC encoding Man-PTS component IIC, the receptor for class IIa bacteriocins. This is the first time when bacteriocin sensitivity marker is used for counterselection. First step in the method is to integrate mptC plasmid vector into the chromosome by homologous recombination. As a result, the integrant becomes resistant to erythromycin and sensitive to class IIa bacteriocins. Second step is to plate the integrant onto bacteriocin agar to select the cells which have lost the integrated vector through second homologous recombination. Thus, the bacteriocin selection completely eliminates the screening of DCO colonies using replica plates with and without antibiotic. The position of the second crossover determines if the DCO colony contains the desired chromosomal deletion or if it has reverted to wild-type. These two genome types can be differentiated for example by PCR. It is noticeable that the ratio between the deletion and wild-type may not be 1 to 1, but it can vary considerably depending on the genes to be deleted. In this study, the proportion of the colonies carrying the deletion varied from about 8 to 40 %. Considering the large sizes of the deletions (22 and 33 kb), the frequencies are reasonably high, possibly because these regions did not contain any L. lactis core genes and their deletion did not affect the growth in M17G. In addition to the two deletions presented in this paper, we have used the method for other non-essential knockouts, of which the biggest was about 35 kb (data not shown). Also in these cases, the deletion frequencies have varied from about 10 to 40 %.
In the conventional gene knockout procedure by two crossover events, the integrant is pre-cultured in non-selective media for many generations before plating and searching of second crossover colonies. Here, we showed that this pre-culturing step in bacteriocin counterselection was unnecessary, as second crossover colonies could be obtained by plating the integrant directly from the selective media onto bacteriocin plate. The second crossover in the cells could have happened already in the antibiotic broth culture before plating. Because Erm is a bacteriostatic antibiotic, these Erm-sensitive cells may have survived overnight with the antibiotic. About 0.01 % of cells from Erm containing culture were bacteriocin resistant, thus giving the second crossover frequency 10−4.
The integrants showed high sensitivity to the bacteriocins. According to the MIC determinations, the bacteriocin sensitivity was in the same range as in the mptC host L. monocytogenes WSLC 1033. We have noticed that for high bacteriocin sensitivity and efficient DCO selection, strong expression of mptC is needed (unpublished results). The expression of mptC in the integration vector was controlled by the strong nisin-inducible promoter P nisZ (Li et al. 2011). However, nisin promoter is a suitable choice only for strains carrying the nisin-specific two-component signal transduction system (Kuipers et al. 1995). To apply the method in non-nisin L. lactis strains, other promoter should be used. Many strong promoters have been characterised for L. lactis, some of which could replace the P nisZ used here. For constitutive gene expression, the phosphopentomutase promoter P8 has been shown to be even stronger than the nisin promoter (Zhu et al. 2015a). However, high expression of mptC may cause stability problems in E. coli. In that case, inducible expression system, such as the agmatine-controlled expression system ACE (Linares et al. 2015) or the zinc-regulated expression system Zirex (Mu et al. 2013) could be applied. Yet, the suitability of a promoter should be tested for each L. lactis strain.
Currently, there is an increasing interest of using the CRISPR/Cas9 system for genomic deletions in bacteria, including lactic acid bacteria (Oh and van Pijkeren 2014; van Pijkeren and Britton 2014). However, so far, there are no reports about its use in Lactococcus genome editing. Instead, chromosomal deletions in L. lactis have often been generated by using pG+Host thermosensitive vectors (Biswas et al. 1993). These vectors are useful tools for single knockouts, but since culturing L. lactis at 37 °C induces spontaneous heat resistant mutations, consecutive uses of the thermosensitive vector in the same strain seems impossible (Smith et al. 2012). The bacteriocin counterselection presented here did not have such a limitation, as a new mptC integration vector could be introduced into the Δ22-kb strain, and the loss of the vector could be selected with bacteriocin resulting in the strain with two chromosomal deletions.
In addition to the thermosensitive vectors, two counterselectable markers have been published for genome editing of lactic acid bacteria: the gene oroP encoding orotate transporter in L. lactis (Solem et al. 2008), and the gene upp encoding uracil phosphoribosyltransferase in L. acidophilus, L. casei and L. lactis (Goh et al. 2009; Song et al. 2014). Expression of the genes caused sensitivity to 5-fluoroorotate or 5-fluorouracil, which were then used to select the loss of the integration vector through the second recombination event. The bacteriocin selection presented in this paper has several practical advantages over those two counterselection methods. First of all, selection of the DCO colonies with bacteriocin does not require chemically defined or semi-defined media, but simply commercial M17 agar supplemented with glucose and class IIa bacteriocin. Moreover, the chromosomal deletions could be made in a wild-type L. lactis without pre-treatments, e.g. deletion of endogenous Man-PTS. In fact, the Man-PTS permease component IID (PtnD) from the L. lactis host is probably needed for a functional bacteriocin receptor. As shown by Kjos et al. (2010), PtnD can form an active permease with listerial MptC. Like most L. lactis strains (Siezen et al. 2011), also N8 carries ptnABCD genes for Man-PTS (M. Qiao, unpublished). Hence, presumably because of the native ptnD gene, the introduction of mptC alone was enough to make L. lactis sensitive to bacteriocins.
For selection, purification of bacteriocins was not needed, but they could be applied as crude precipitates or pasteurised cell-free supernatants from bacteriocin producer cultures. Only 20 μl/ml of leucocin C or pediocin containing supernatant could select the loss of the integrated plasmid. When tested on M17G + Erm agar, the majority of the bacteriocin resistant colonies were Erm-sensitive, confirming the excision of the integration vector. Only about 1 % of the colonies obtained on bacteriocin agar turned out to be resistant to Erm, indicating a development of spontaneous resistance to bacteriocins. As the recombination frequency here was shown to be approximately 10−4, the frequency of developing resistance was thus 10−6, similar as what has been reported with L. monocytogenes (Gravesen et al. 2002).
In conclusion, the bacteriocin counterselection presented here is a convenient method to generate chromosomal deletions in L. lactis. The method was efficient, simple, inexpensive, and it could be used successively in the same wild-type strain.
References
Beasley SS, Takala TM, Reunanen J, Apajalahti J, Saris PEJ (2004) Characterization and electrotransformation of Lactobacillus crispatus isolated from chicken crop and intestine. Poult Sci 83(1):45–48
Biswas I, Gruss A, Ehrlich SD, Maguin E (1993) High-efficiency gene inactivation and replacement system for gram-positive bacteria. J Bacteriol 175(11):3628–3635
Drider D, Fimland G, Héchard Y, McMullen LM, Prévost H (2006) The continuing story of class IIa bacteriocins. Microbiol Mol Biol Rev 70(2):564–582
Ennahar S, Aoude-Werner D, Sorokine O, Van Dorsselaer A, Bringel F, Hubert JC, Hasselmann C (1996) Production of pediocin AcH by Lactobacillus plantarum WHE 92 isolated from cheese. Appl Environ Microbiol 62(12):4381–4387
Gasson MJ (1983) Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol 154(1):1–9
Goh YJ, Azcárate-Peril MA, O’Flaherty S, Durmaz E, Valence F, Jardin J, Lortal S, Klaenhammer TR (2009) Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol 75(10):3093–3105
Gravesen A, Jydegaard Axelsen AM, Mendes da Silva J, Hansen TB, Knøchel S (2002) Frequency of bacteriocin resistance development and associated fitness costs in Listeria monocytogenes. Appl Environ Microbiol 68(2):756–764
Holo H, Nes IF (1989) High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol 55(12):3119–3123
Kjos M, Salehian Z, Nes IF, Diep DB (2010) An extracellular loop of the mannose phosphotransferase system component IIC is responsible for specific targeting by class IIa bacteriocins. J Bacteriol 192(22):5906–5913
Kuipers OP, Beerthuyzen MM, de Ruyter PGGA, Luesink EJ, de Vos WM (1995) Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J Biol Chem 270(45):27299–27304
Lambert JM, Bongers RS, Kleerebezem M (2007) Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol 73(4):1126–1135
Li R, Takala TM, Qiao M, Xu H, Saris PEJ (2011) Nisin-selectable food-grade secretion vector for Lactococcus lactis. Biotechnol Lett 33(4):797–803
Linares DM, Alvarez-Sieiro P, del Rio B, Ladero V, Redruello B, Martin MC, Fernandez M, Alvarez MA (2015) Implementation of the agmatine-controlled expression system for inducible gene expression in Lactococcus lactis. Microb Cell Factories 14:208
Martinussen J, Hammer K (1994) Cloning and characterization of upp, a gene encoding uracil phosphoribosyltransferase from Lactococcus lactis. J Bacteriol 176(21):6457–6463
Mu D, Montalban-Lopez M, Masuda Y, Kuipers OP (2013) Zirex: a novel zinc-regulated expression system for Lactococcus lactis. Appl Environ Microbiol 79(14):4503–4508
Oh J-H, van Pijkeren J-P (2014) CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42(17):e131
Ramnath M, Arous S, Gravesen A, Hastings JW, Hechard Y (2004) Expression of mptC of Listeria monocytogenes induces sensitivity to class IIa bacteriocins in Lactococcus lactis. Microbiology 150(Pt 8):2663–2668
Sambrook J, Russell D (2001) Molecular cloning. A laboratory manual. 3era. Ed. 1: 1.32–1.34. Cold Spring Harbour Lab. Press, New York
Siezen RJ, Bayjanov JR, Felis GE, van der Sijde MR, Starrenburg M, Molenaar D, Wels M, van Hijum SAFT, van Hylckama Vlieg JE (2011) Genome-scale diversity and niche adaptation analysis of Lactococcus lactis by comparative genome hybridization using multi-strain arrays. Microb Biotechnol 4(3):383–402
Smith WM, Pham TH, Lei L, Dou J, Soomro AH, Beatson SA, Dykes GA, Turner MS (2012) Heat resistance and salt hypersensitivity in Lactococcus lactis due to spontaneous mutation of llmg_1816 (gdpP) induced by high-temperature growth. Appl Environ Microbiol 78(21):7753–7759
Solem C, Defoor E, Ruhdal Jensen P, Martinussen J (2008) Plasmid pCS1966, a new selection/counterselection tool for lactic acid bacterium strain construction based on the oroP gene, encoding an orotate transporter from Lactococcus lactis. Appl Environ Microbiol 74(15):4772–4775
Song L, Cui H, Tang L, Qiao X, Liu M, Jiang Y, Cui W, Li Y (2014) Construction of upp deletion mutant strains of Lactobacillus casei and Lactococcus lactis based on counterselective system using temperature-sensitive plasmid. J Microbiol Methods 102:37–44
van Pijkeren JP, Britton RA (2014) Precision genome engineering in lactic acid bacteria. Microb Cell Factories 13(Suppl 1):S10
Wan X, Li R, Saris PEJ, Takala TM (2013) Genetic characterisation and heterologous expression of leucocin C, a class IIa bacteriocin from Leuconostoc carnosum 4010. Appl Microbiol Biotechnol 97(8):3509–3518
Wan X, Saris PEJ, Takala TM (2015) Genetic characterization and expression of leucocin B, a class IId bacteriocin from Leuconostoc carnosum 4010. Res Microbiol 166(6):494–503
Zabarovsky ER, Winberg G (1990) High efficiency electroporation of ligated DNA into bacteria. Nucleic Acids Res 18(19):5912
Zhu D, Liu F, Xu H, Bai Y, Zhang X, Saris PEJ, Qiao M (2015a) Isolation of strong constitutive promoters from Lactococcus lactis subsp. lactis N8. FEMS Microbiol Lett 362(16):fnv107
Zhu D, Zhao K, Xu H, Zhang X, Bai Y, Saris PEJ, Qiao M (2015b) Construction of thyA deficient Lactococcus lactis using the Cre-loxP recombination system. Ann Microbiol 65(3):1659–1665
Acknowledgments
This work was supported by Academy of Finland (project no. 268922).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Table S1
(PDF 306 kb)
Rights and permissions
About this article

Cite this article
Wan, X., Usvalampi, A.M., Saris, P.E.J. et al. A counterselection method for Lactococcus lactis genome editing based on class IIa bacteriocin sensitivity. Appl Microbiol Biotechnol 100, 9661–9669 (2016). https://doi.org/10.1007/s00253-016-7828-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7828-6