
Abstract
We collected several biofilm samples from Japanese rivers and established a reproducible multi-species biofilm model that can be analyzed in laboratories. Bacterial abundance at the generic level was highly similar between the planktonic and biofilm communities, whereas comparative metatranscriptomic analysis revealed many upregulated and downregulated genes in the biofilm. Many genes involved in iron-sulfur metabolism, stress response, and cell envelope function were upregulated; biofilm formation is mediated by an iron-dependent signaling mechanism and the signal is relayed to stress-responsive and cell envelope function genes. Flagella-related gene expression was regulated depending upon the growth phase, indicating different roles of flagella during the adherence, maturation, and dispersal steps of biofilm formation. Downregulation of DNA repair genes was observed, indicating that spontaneous mutation frequency would be elevated within the biofilm and that the biofilm is a cradle for generating novel genetic traits. Although the significance remains unclear, genes for rRNA methyltransferase, chromosome partitioning, aminoacyl-tRNA synthase, and cysteine, methionine, leucine, thiamine, nucleotide, and fatty acid metabolism were found to be differentially regulated. These results indicate that planktonic and biofilm communities are in different dynamic states. Studies on biofilm and sessile cells, which have received less attention, are important for understanding microbial ecology and for designing tailor-made anti-biofilm drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural environments and multicellular organisms are colonized by various microbes. In many cases, microbial cells form complex communities wherein they communicate (e.g., quorum sensing signals), cooperate (e.g., symbiosis of Clostridium acetobutylicum and Bacillus cereus; Wang et al. 2015), and fight (e.g., production of antimicrobials) each other, instead of existing as isolated cells. Recent studies propose that microbial communities are mostly present in a sessile state and not in a planktonic state (Watnick and Kolter 2000). Since microbes are usually cultured as free planktonic cells in laboratory experiments, the sessile state needs to be given more attention in order to understand microbial activities.
A biofilm is a population of cells growing on a surface and is a type of sessile cell community. Biofilm structures are ubiquitously found on both abiotic and biotic surfaces like kitchen sinks, river stones, and human teeth. Biofilms are known to be responsible for nosocomial infections, contamination of medical devices, chronic bacterial infections, and periodontal diseases (Donlan and Costerton 2002). Microbial cells within biofilm structures are packaged along with extracellular polysaccharides (EPS), nucleic acids, proteins, and other materials, and are tolerant to antimicrobials, heat treatment, and the host immune system (Carvalhais et al. 2015; Stoodley et al. 2002). Therefore, it is difficult to kill microbial cells inside a biofilm and to eliminate the entire biofilm structures. As an example, in the lungs of patients with cystic fibrosis (a genetic disorder), pathogenic Pseudomonas aeruginosa cells form strong biofilms and acquire high tolerance to medical treatments (Magalhães et al. 2016).
Biofilm research so far has mainly focused on biofilm samples consisting of one or a few microbial species, but only a limited number of studies have been performed that concern complex multi-species biofilms. It is therefore important to perform meta 16S ribosomal RNA (rRNA) gene and metatranscriptome analyses of biofilm samples from complex microbiomes or environments. These analyses would be helpful for a detailed understanding of microbial ecology and the development of anti-biofilm drugs.
In the present study, we cultured biofilm samples collected from Japanese rivers and succeeded in establishing a multi-species biofilm model in the laboratory. Culture of this biofilm model resulted in the formation of both planktonic and biofilm cell communities. Although meta 16S rRNA analysis showed little difference between the planktonic and biofilm cell communities, metatranscriptomic analysis revealed various differentially expressed genes.
Materials and methods
Sample collection and biofilm culture
Biofilm samples were cultured in one third strength of Luria broth (1/3 LB; LB broth was purchased from Difco, Sparks, MD, USA) at 25 °C, unless otherwise stated.
Ten river samples were collected from different points of the Tamagawa River, including a rock covered with biofilm from midstream (north latitude 35.610566 and east longitude 139.624680), a stone and its covering slime from midstream (35.610461, 139.625142), a stone and covering slime from downstream (35.541471, 139.752706), brown slime from downstream (35.541471, 139.752706), slime attached to the underside of a rock from downstream (35.541471, 139.752706), slime attached to the underside of a mat from downstream (35.541471, 139.752706), a stone and covering slime from upper stream (35.621639, 139.572920), slime of the top of driftwood from upper stream (35.621642, 139.572930), river water from upstream (35.621700, 139.572478), and river water from downstream (35.541471, 139.752706). Three samples were collected from the Yazawagawa River: river water (35.6037972, 139.6464361), waterfall (35.6037972, 139.6464361), and red clay (35.605759, 139.645380). Since this location is not privately owned or protected, no specific permits were required for the described field studies. A sterilized slide glass was placed in a Petri dish (90 mm diameter), and 10 ml of 1/3 LB along with a drop of the sample (mixture of biofilm and river water) was added. Three-day-old statically cultured cells (both planktonic and biofilm cells) were used to prepare glycerol stocks of these samples with 20 % glycerol and stored at −80 °C.
After a week, inoculation was initiated from glycerol stocks in a similar manner using 50 μl of the glycerol stock solution from each sample. After 3 days, planktonic and biofilm cells were collected separately. (i) The planktonic cells were collected by aspirating 1.5 ml culture broth, (ii) the remaining culture broth was discarded, (iii) the inner part of the dish and the surface of the slide glass were washed twice with 5 ml of 1× PBS [made using 10× PBS(−) purchased from Wako Pure Chemical Industries, Osaka, Japan], (iv) 800 μl of 1× PBS was added, and (v) the biofilm cells were scraped off from the inner part of the dish and the surface of the slide glass using an interdental brush (Yawaraka-Shikan-Burashi; Kobayashi Seiyaku, Tokyo, Japan). Glycerol stocks were prepared again as described above, and these culture and sample collection steps were repeated two more times (thrice in total).
DNA extraction and meta 16S rRNA sequence analysis
Metagenomic DNA from the original biofilm samples and the cultured planktonic and biofilm cells was prepared using a Power Soil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA), according to the manufacturer’s instructions. The V3–V4 regions of the 16S rRNA genes (position 342 to 806 in Escherichia coli numbering) were PCR-amplified using a non-degenerate universal primer set, 342F and 806R (Kato et al. 2015), with EX Taq HS polymerase (Takara Bio, Ohtsu, Japan). All PCR samples were mixed to generate a 21-plex sample library, and the sequences were read by the MiSeq instrument (Illumina, San Diego, CA) in a paired-end-read mode for 300 cycles according to the manufacturer’s protocol.
High-quality sequences (of >150 and <450 bases in length) were selected for subsequent processing (i.e., alignment to a standard sense strand and removal of the primer sequences used for sequencing). Only the forward sequences were used for subsequent processing since the reverse sequences were mostly of low quality and merging of each forward and reverse sequence was not possible. Sequence clustering of these high-quality reads was performed using the UCLUST program (Edgar 2010) version 6.0.307 with an identity of ≥97 % and a query and reference coverage of ≥80 %. Chimeric clusters detected by the UCHIME program (Edgar et al. 2011) in the de novo mode and the reference mode [searching the reference Gold Database (DB) (http://drive5.com/uchime/gold.fa)] were removed. Taxonomic assignment of the resulting MiSeq reads was performed using the RDP Classifier program (Wang et al. 2007) version 2.6 with a bootstrap value ≥0.5 against the representative sequences of each 97 % sequence cluster chosen by the UCLUST program. Unless otherwise stated, the programs for read assemblage and analysis in this study were run using default parameters. The 16S rRNA sequence data have been deposited under the BioProject accession number PRJDB4599 in the DDBJ BioProject database (http://trace.ddbj.nig.ac.jp/bioproject/index_e.html).
mRNA extraction and construction of a cDNA library
The culture conditions and sample collection methods were the same as described above. The culture was started with the second glycerol stock of the sample, and 30 Petri dishes were handled in parallel to obtain sufficient amounts of RNA for RNA-seq. Cells were collected separately from the planktonic and the biofilm communities from 12, 24, and 48-h cultures. Total RNA was extracted from the cultured biofilm and planktonic cells using the acid hot phenol method (Aiba et al. 1981). The extracted RNA samples were purified using an RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol for enriching the messenger RNA (mRNA), since most RNAs of <200 nucleotides (including 5.8S rRNA, 5S rRNA, and transfer RNAs (tRNAs), together constituting 15–20 % of total RNA) are selectively excluded. rRNAs were removed from total RNA using a MICROBExpres Bacterial mRNA Enrichment Kit (Ambion, Austin, TX). Complementary DNA (cDNA) libraries were then constructed using a TruSeq Stranded mRNA and Total RNA sample preparation kit (Illumina) and sequenced using MiSeq in the paired-end-read mode for 300 cycles.
RNA-seq analysis
High-quality RNA-seq reads (of >150 and <450 bases in length) were selected for subsequent processing (i.e., alignment to a standard sense strand and removal of primer sequences used for sequencing). RNA-seq reads from each sample were separately assembled using the IDBA-UD program (Peng et al. 2012) version 1.1.0 with the parameters “–mink 20 –maxk 70 –step 5.” Protein coding sequences in the contigs were predicted using the MetaGeneMark program (Zhu et al. 2010) version 2.10 with the parameters “-g 11 -a -d -f G.” Functional annotation of each read was performed by BLASTP search using the parameters “-g T -F F -e 1e-8 -m 8 -b 10 -v 10” against the KEGG protein sequence DB, which was obtained in March 2014. Taxonomic (phylum, class, order, family, and genus levels) and functional [KEGG Orthology (KO)-level] assignment was performed for reads that exhibited the highest BLAST scoring hits with an identity ≥70 % and a BLAST bit-score ≥40. Reads from each sample were mapped to the predicted ORFs using Bowtie2 (Langmead and Salzberg 2012). KO abundance was calculated by dividing the number of reads belonging to the same KO by the median length of proteins annotated to the same KO in the KEGG protein DB. KO abundance was normalized to the number of reads included in each sample. KEGG pathway abundance was calculated as the sum of KO abundances belonging to a single pathway. To compare the abundances of individual KOs and their pathways among different samples, abundances in each RNA-seq sample were normalized to sample read numbers. This method for normalizing functional abundances among samples was applied to all BLAST-based functional assignments in this study. RNA-seq data have been deposited under the BioProject accession number PRJDB4600 in the DDBJ BioProject database (http://trace.ddbj.nig.ac.jp/bioproject/index_e.html).
Determination of biofilm amounts
Samples from glycerol stocks were cultured in a Petri dish (60-mm diameter) containing 2 ml medium. Only planktonic cell communities were collected from the grown samples, and their optical density at 595 nm was measured. To measure the amount of biofilm, the planktonic cells were completely removed, and the remaining biofilm samples were gently washed twice with 2 ml distilled water. The biofilm samples were kept in a fume hood with a blower until the samples dried out (approximately 1 h). Then, 2 ml of 0.1 % crystal violet solution was added to each sample and incubated for 15 min at room temperature. After discarding the crystal violet solution, the biofilm samples were washed twice with 2 ml of distilled water, gently to keep the biofilm intact. The stained biofilm samples were then dried out as previously described, in a fume hood (for approximately 1 h). To each sample, 2 ml of 70 % ethanol was added and the samples were kept for 30 min at room temperature. Finally, 100 μl of the stained ethanol was collected from each sample, and the amounts of the eluted dye were quantified by measuring absorbance at 595 nm.
Results
Establishment of a model biofilm system in a laboratory
Considering that most natural biofilms consist of multiple bacterial species, we believe that the lack of model biofilm systems has hampered intensive research in this area. To establish a biofilm model, 13 river samples were collected, including biofilm structures from both abiotic surfaces and the surrounding river water. These samples were cultured in a Petri dish containing a slide glass (see “Materials and Methods” and Fig. 1a). Three of the samples, SM (a stone and its covering slime from midstream), BD (brown slime from downstream), and WU (river water from upstream), were found to form a clear biofilm on the wall of the dish and on the slide glass after 3 days. Additionally, these three cultures could form a biofilm even when the first cultures were frozen as glycerol stocks and the second inoculation was restarted from these stocks. In the same manner, we confirmed that biofilm formation could be repeated at least two more times.

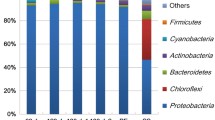
Biofilm model and 16S rRNA gene analysis. a An image of the cultured biofilm BD is shown. b 16S rRNA gene sequences were classified up to the generic level. “O,” “B,” and “P” characters indicate original culture, biofilm community, and planktonic community, respectively. The numerals indicate the number of inoculations repeated
The bacterial species existing in planktonic and biofilm communities of the above samples were analyzed by massive sequencing of 16S rRNA (Fig. 1b). The sequencing results indicate that the generic composition differed depending on the source of the biofilm and that the composition between the planktonic and biofilm communities from the same culture was not clearly different. The generic composition of the original samples (i.e., uncultured samples, Fig. 1b) was also analyzed in a similar manner. We discovered that the variation in the biofilm community was lost upon culture in laboratory conditions. Taken together, we successfully established model biofilm systems, which are reculturable and reproducible in the laboratory. Nevertheless, the fact that species composition might be variable among repeated cultures should be considered.
An overview of the RNA-seq analysis of planktonic and biofilm communities
As mentioned, phylogenetic analysis based on 16S rRNA sequences showed little difference between the planktonic and biofilm communities. In order to understand the dynamic but not the static differences among the communities, metatranscriptome analysis by RNA-seq was performed using 12-, 24-, and 48-h cultures (see “Materials and Methods”). Among the above-mentioned three samples, only the sample BD formed sufficient amounts of biofilm to be subjected to RNA-seq, and therefore, only this sample was used further. The growth and biofilm formation kinetics observed during culture are shown in Fig. 2a, b, and we defined 12-, 24-, and 48-h cultures as early, middle, and late stages of biofilm formation, respectively. Growth was found to drift into a period of stagnation around the middle stage of the culture, possibly due to the co-existence of fast and slow growers in this culture.

Growth kinetics and quantitation of biofilm formation. a Growth kinetics of the BD culture are shown. Optical density at 595 nm was measured at three time points. The data are presented as mean ± SD from triplicate experiments. b Quantitation of crystal violet staining of the BD culture is shown. The amounts of crystal violet indicating the amounts of biofilm formed were measured at three time points. The data are presented as mean ± SD from triplicate experiments
From the metatranscriptome data, we first examined the bacterial genera and species that were alive and vital at the specific biofilm formation stages in both communities. To this end, the average expression levels of gyrB, rpoB, and rplC genes were determined (Fig. 3a). These three genes were chosen as a representative of the replication, transcription, and translation machineries and as universal single copy genes (Kato et al. 2015). At the early stage of culture, Aeromonas and Pseudomonas bacteria were dominant, and with time, Pseudomonas bacteria became highly dominant (Fig. 3a). This result is consistent with that shown in Fig. 1b. However, bacteria that were not significantly detected in meta 16S rRNA analysis were also found to be present and vital; these included (i) Acinetobacter at the early and middle stages, (ii) Laribacter and Xanthomonas at the middle and late stages, (iii) Enterobacter at the early stage of the planktonic community, and (iv) Pseudogulbenkiania at the late stage of the biofilm community (Fig. 3a). Although determination of bacterial species was difficult using meta 16S rRNA analysis (Fig. 1b), it was possible with the metatranscriptome data (Fig. 3b). At the species level, at least 20 bacterial species were found to be present and vital, and our culture conditions were feasible for these bacteria. It should be noted that the metatranscriptome data do not reflect the simple presence ratio of bacterial species.

Dynamic differences between planktonic and biofilm communities. a The relative abundance for each genus was calculated from the average of the number of reads assigned to gyrB (KEGG orthology K02470), rpoB (K03043), or rplC (K02906). “B” and “P” characters indicate biofilm and planktonic communities, respectively. b The relative abundance for each species was calculated in the same manner as for panel a
In the next level of metatranscriptome data analysis, the upregulated genes in the biofilm community were identified (Table 1). Over 30 % of the genes were upregulated in the subcategories of “biosynthesis of other secondary metabolites,” “xenobiotics biodegradation and metabolism,” “membrane transport,” and “lipid metabolism” (Table 1, limited to subcategories in which at least 10 genes were detected). These results suggest that changes in cell envelope structure and in material import/export across the cell envelope are important for biofilm formation. In Tables 2, 3, 4, and 5, upregulated genes in the biofilm community at three or two time points are listed, and in Tables S1–S3, the genes upregulated at only one time point are listed. Notably, the genes for cysteine biosynthesis (Fig. 4, see below for details) and iron-sulfur metabolism (iscR, ycdN, iscA, hscB, ftnA, sitA, sitB) were detected to a high extent. Upregulation of a number of stress response genes (bolA, pspC, uspE, pspA, pspB, osmY, cpxP, osmE, treA/treF) is reasonable, because a number of previous studies have reported that environmental stresses induce biofilm formation (O’Toole and Stewart 2005). In relation to the stress response, a functional cell envelope and peptidoglycan biosynthesis is necessary for cell attachment and biofilm maturation (Dong et al. 2011; Loo et al. 2000); therefore, induction of pgm, amiA, yhcB, prc, rfbC, murC, and dacB expression is assumed to be plausible according to the literature (see Tables 2, 3, 4, and 5). The expression of multiple genes for rRNA methyltransferases (rlmB, rsmH), chromosome partitioning (KEGG K03497, mukB), or leucine biosynthesis (leuD, leuC, lrp) was found to be upregulated, but the reason for this upregulation is unclear.

Regulation of cysteine and methionine biosynthesis. The cysteine and methionine biosynthesis pathway is shown. Dark and light red colors indicate genes whose expression is upregulated at least at two time points and one time point, respectively. Dark and light blue colors indicate the downregulated genes at least at two time points and one time point, respectively
The downregulated genes in the biofilm community at all the time points are listed in Table 6, and the downregulated genes at one or two time point(s) are listed in Tables S4–S9. Downregulation of genes involved in the respiratory chain (cyoB, cyoC, nuoI, cyoA, nuoM, cydB/appB), nucleotide biosynthesis (pyrD, purB, purN, preA, uraA, purK, purT, pyrF), fatty acid metabolism (fabF, fadR, fabD, fabH, accA, accD), and DNA repair (mutS, recF, KEGG K03630, dnaE, sulE) was characteristic. Concerning the respiratory chain genes, this result may reflect that the interior of the biofilm is anaerobic. Anaerobic conditions in the biofilm are also suggested by the upregulation of arcB (Table 4), since ArcB represses the genes required for respiratory metabolism and the tricarboxylic acid cycle enzymes. Downregulation of the fatty acid metabolism genes listed above is consistent with upregulation of fadE expression (Table 3) because FadR is a positive and negative transcriptional regulator for fatty acid biosynthesis (fab) and fatty acid degradation (fad) genes, respectively in E. coli (My et al. 2013). We can conclude that fatty acid synthesis decreases during biofilm formation. Repression of DNA repair genes is expected to facilitate replication errors and increase the frequency of spontaneous mutations; since a biofilm acts as the evacuation spot from unfavorable environments, it may be involved in generating new genetic characters and diversity (see “Discussion” section for details). In addition, multiple genes for aminoacyl-tRNA synthase (glnS, glyS, proS, valS; note that serS is exceptionally upregulated) and thiamine biosynthesis (thiG, thiI) were downregulated, but the reason for this downregulation is unclear.
In concluding this section, many upregulated and downregulated genes were identified, and therefore, planktonic and biofilm communities were revealed to be in different dynamic states. The expression of many genes seemed to be consistent with their description in previous studies (references are indicated in Tables 2–6 and S1–S9), whereas some genes were newly identified as biofilm-inducible or biofilm-repressible in this study.
Cysteine and methionine biosynthesis
In Tables 2, 3, 4, 5, and 6 and S1–S9, a number of genes that are involved in cysteine and methionine biosynthesis are listed. The cysteine and methionine biosynthesis pathway is depicted in Fig. 4, and the regulation of individual genes upon biofilm formation is also shown. This figure shows that cysteine and methionine biosynthesis is upregulated and downregulated, respectively. Although the reason for this result is unclear, similar positive and negative effects of these amino acids on biofilm formation have been described earlier (Gnanadhas et al. 2015; Singh et al. 2015; Xu et al. 2014).
Regulation of flagellar genes
Previous studies have shown that the flagella play important roles in biofilm formation in both gram-negative and gram-positive bacteria (Belas 2014). The heat map presented in Fig. 5 shows the expression pattern of flagellar genes. In general, at the early stage of biofilm formation, many genes were upregulated, whereas in the middle stage, the appearance of downregulated genes was remarkable. At the late stage, the expression of genes that were downregulated at the middle stage seemed to be restored. This result is consistent with those of previous studies (Monds and O’Toole 2009; Okuda et al. 2012; Watnick and Kolter 2000). It is generally considered that at the early stage, flagella are necessary to sense and attach to solid surfaces and during biofilm growth, the flagella become obstructive as they destabilize biofilm structure. After biofilm maturation, it is assumed that flagella formation and flagella-associated motility again gains importance for escaping from the biofilm structures and to find new solid surfaces.

Heat map of expression pattern for flagella-related genes. The 29 genes indicated were detected out of 37 genes in the KEGG pathway KO02040 (Flagellar assembly) from the RNA-seq data. The heat map legend at the bottom indicates the color values corresponding to transcription levels, with upregulation shown in red and downregulation in green
Discussion
From the metatranscriptome data (Tables 5, 3, 4, 5, and 6 and S1–S9), we speculated that the iron-dependent signaling mechanism triggers cells to form a biofilm and, as a consequence, genes related to stress response, cell envelope, flagella, and many others are regulated. Iron is reported to be important for Pseudomonas bacteria, both as a positive signaling molecule for biofilm formation and as a structural stabilizer for the biofilm matrix (Balaban 2008). In E. coli, iron-sulfur homeostasis and Fe-S cluster assembly are signals for biofilm formation (Roche et al. 2013; Wu and Outten 2009). These facts raise the question of why iron and sulfur are the chosen signaling molecules for biofilms. In primordial Earth, iron and sulfide were abundant, and the Fe-S cluster is thought to be one of the oldest and most versatile inorganic co-factors (Ayala-Castro et al. 2008; Lill 2009). In addition, Stoody and co-workers proposed the possibility that biofilms were the main form in which microbial cells existed in primordial Earth and that planktonic cells appeared much later during the evolutionary process (Stoodley et al. 2002). Taken together, it is natural that primitive microbes incorporated iron and sulfur in many of their cellular functions including biofilm formation and still utilize them as signaling molecules for transition between the biofilm and planktonic states.
Antimicrobials are widely used globally, but they give bacteria the opportunity to build resistance, allowing the emergence of resistant bacteria. Moreover, cells within biofilms are frequently tolerant to ordinary antimicrobials (Carvalhais et al. 2015; Stoodley et al. 2002). Therefore, the development of anti-biofilm drugs that do not inhibit cell growth is desirable. For some bacteria, iron chelators such as deferoxamine, lactoferrin, and 2,2′-bipyridyl are known to function as anti-biofilm drugs (Banin et al. 2005; Moreau-Marquis et al. 2009; Weinberg 2004). However, iron has a varied effect on bacterial biofilm formation; low iron concentration has a negative effect in Pseudomonas, E. coli, and Vibrio cholerae (Balaban 2008; Wu and Outten 2009), whereas a positive effect of iron is observed in Staphylococcus aureus, Acinetobacter baumannii, and Streptococcus mutants (Balaban 2008). These observations suggest that, when removing undesired biofilms through manipulation of iron concentration, investigating the effect of iron on each biofilm is important. In this context, the metatranscriptome analysis approach described here would be effective for determining the effect of iron. The metatranscriptome data obtained in this study implied that iron had a positive effect on our biofilm model. Our preliminary experiments have shown that addition of 2,2′-bipyridyl to the starting culture indeed reduced the amount of biofilm without reducing bacterial growth (data not shown).
The targets of anti-biofilm drugs are not limited to iron and sulfur metabolism, and all genes regulated in the biofilms can be potential targets. For example, the expression of treA/treF was upregulated in our biofilm (Table 5), and the enzymatic activity of its gene product, α,α-trehalase, is known to be inhibited by casuarine or deoxynojirimycin (Cardona et al. 2009; Forcella et al. 2010). Similar candidates for anti-biofilm drugs are listed in Table S10, and we are now trying to develop novel tailor-made and target-oriented anti-biofilm drugs.
Downregulation of multiple DNA repair genes in the biofilm was observed in this study. Consistent with our result, it has been reported that P. aeruginosa cells within the biofilm community show up to 100-fold increased mutation frequency compared to that in corresponding planktonic cells (Driffield et al. 2008; Luján et al. 2011; Oliver et al. 2000). In addition, bacteria possess a mechanism of stress-induced mutagenesis in which error-prone DNA polymerases are upregulated and error-correcting enzymes are downregulated under stress conditions (Al Mamun et al. 2012; Foster 2007). These facts suggest that biofilm formation is a means for adaptive evolution and generating phenotypic diversity. This idea is further supported by the fact that many biofilm structures contain extracellular DNA, which is a source of genes in horizontal gene transfer (Dominiak et al. 2011; Madsen et al. 2012) and that Streptococcus mutans cells grown in biofilms have a 10- to 600-fold higher rate of natural transformation than planktonic cells (Li et al. 2001).
In addition to cysteine, methionine, and leucine biosynthesis genes (see the “Results” section), glutamate (gdhA, Table 4, upregulated), histidine (hisH, Table 6, downregulated), proline (proB, Table S5, downregulated), and arginine (argE, Table S6, downregulated) biosynthesis genes were identified as biofilm-regulated genes. The positive effects of cysteine and leucine and the negative effects of methionine and arginine on biofilm formation are in good agreement with observations from previous studies (Kolderman et al. 2015; McFarland et al. 2008; Noothalapati Venkata et al. 2011; Singh et al. 2015). The mechanism involved in the effect of amino acids on biofilm formation is unclear, but they are attractive anti-biofilm drugs because of their safety for human health. Intriguingly, high levels of leucine were found to accumulate within microcolonies in E. coli biofilms (Noothalapati Venkata et al. 2011), implying that leucine is a functional component of biofilm structures or a signaling molecule for biofilm formation. Upregulation of lrp (encoding leucine-responsive regulatory protein, Table 4) supports the signaling molecule hypothesis, because Lrp activates transcription from many fimbrial genes that depend on leucine (Noothalapati Venkata et al. 2011).
EPS is a key component for maintaining biofilm architecture, possibly acting as an intercellular cement. Quorum sensing positively regulates EPS production and facilitates the development of mixed species populations (Madsen et al. 2012). LsrB (Table 5, upregulated) is a subunit of the transporter complex for the autoinducer-2 molecule, which is produced by many bacterial species and is one of the quorum-sensing signaling molecules (Shao et al. 2007). In addition, PssA (Table 4, upregulated) catalyzes the first step of EPS synthesis (Chen et al. 2014), and a pssA mutation in Rhizobium leguminosarum results in complete inability to form biofilms (Russo et al. 2006). Collectively, we conclude that EPS plays an important role in our multi-species biofilm community. However, the EPS produced by some bacteria is also known to interfere with biofilm formation by other bacteria, and some bacteria produce enzymes to degrade quorum-sensing molecules possibly to interfere with the inclusion of unfavorable bacteria in the biofilm (Madsen et al. 2012). Since the generic compositions of planktonic and biofilm communities were almost indistinguishable in this study (Fig. 1b), it seemed that there were no such interference mechanisms in this case.
Phosphotransacetylase (Pta, Table 6, downregulated at all the time points) and acetate kinase (AckA, Table S3, upregulated only at 48 h) are enzymes that convert acetyl-CoA to acetyl-phosphate and acetyl-phosphate to acetate, respectively. The role of these proteins on biofilm formation has been discussed in many bacteria, but both positive and negative effects have been reported. A consensus view is that acetyl-phosphate functions as a signaling molecule through two-component response regulators (possibly, as a phosphate donor) and its intracellular level influences biofilm formation (Kim et al. 2015; Wolfe et al. 2003). In E. coli, the acetyl phosphate level affects the expression of about 100 genes including those related to flagella synthesis, type 1 pili, capsule, and stress effectors (Wolfe et al. 2003). In our biofilm community, the intracellular acetyl-phosphate level is expected to be lower than that in the planktonic community. Many genes for two-component response regulators are listed in Tables 2, 3, 4, and 5 and in S1–S9, and thus, the expression of these genes may be regulated by acetyl-phosphate. As discussed above, iron, leucine, and autoinducer-2 may also act as signaling molecules for biofilm formation. Studies on the interplay between these signaling molecules are the next important issue.
For upregulated and downregulated genes, it is necessary to determine expression dynamics accurately using quantitative real-time RT-PCR. However, quantification of these genes according to KO is difficult, because multiple orthologous genes are assigned to one KO and a universal primer set to amplify all the orthologous genes is not designed. One solution is quantifying each orthologous gene separately and combining the results. In addition, quantifying the amount of proteins expressed from the upregulated and downregulated genes is important in the future.
The multi-species biofilm has been poorly studied so far, because it is too complex to understand overall activities and functions. However, recent advances in next-generation sequencer have enabled us to study the multi-species biofilm as a system without investigating individual microbial cells. There is, however, still one hurdle; examples of multi-species biofilm in nature, marine sediments, chronic wounds, and dental plaques are well known (Si et al. 2015; Yang et al. 2011), but none of these biofilms can be maintained easily, safely, and over a long duration. Therefore, we believe that a study on reproducing multi-species biofilms in the laboratory is important to quantitatively and repeatedly compare planktonic and biofilm samples. The biofilm model used in this study may differ from the original biofilm because of its culture in rich media (Fig. 1b). It is important to establish model biofilms wherein microbial populations are maintained in a nature-identical manner for deeper understanding of biofilms and for designing tailor-made anti-biofilm drugs.
References
Aiba H, Adhya S, de Crombrugghe B (1981) Evidence for two functional gal promoters in intact Escherichia coli cells. J Biol Chem 256:11905–11910.
Al Mamun AAM, Lombardo MJ, Shee C, Lisewski AM, Gonzalez C, Lin D, Nehring RB, Saint-Ruf C, Gibson JL, Frisch RL, Lichtarge O, Hastings PJ, Rosenberg SM (2012) Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338:1344–1348. doi:10.1126/science.1226683
Allegrucci M, Hu FZ, Shen K, Hayes J, Ehrlich GD, Post JC, Sauer K (2006) Phenotypic characterization of Streptococcus pneumoniae biofilm development. J Bacteriol 188:2325–2335. doi:10.1128/JB.188.7.2325–2335.2006
Amini S, Goodarzi H, Tavazoie S (2009) Genetic dissection of an exogenously induced biofilm in laboratory and clinical isolates of E. coli. PLoS Pathog 5:e1000432. doi:10.1371/journal.ppat.1000432
Ayala-Castro C, Saini A, Outten FW (2008) Fe-S cluster assembly pathways in bacteria. Microbiol Mol Biol Rev 72:110–125. doi:10.1128/MMBR.00034-07
Balaban N (2008) Control of biofilm infections by signal manipulation. Springer, USA
Banin E, Vasil ML, Greenberg EP (2005) Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A 102:11076–11081. doi:10.1073/pnas.0504266102
Barnhart MM, Lynem J, Chapman MR (2006) GlcNAc-6P levels modulate the expression of Curli fibers by Escherichia coli. J Bacteriol 188:5212–5219. doi:10.1128/JB.00234-06
Belas R (2014) Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol 22:517–527
Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, Haagensen JAJ, Molin S, Prensier G, Arbeille B, Ghigo JM (2004) Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol 51:659–674. doi:10.1046/j.1365-2958.2003.03865.x
Cardona F, Parmeggiani C, Faggi E, Bonaccini C, Gratteri P, Sim L, Gloster TM, Roberts S, Davies GJ, Rose DR, Goti A (2009) Total syntheses of casuarine and its 6-O-α-glucoside: complementary inhibition towards glycoside hydrolases of the GH31 and GH37 families. Chemistry 15:1627–1636. doi:10.1002/chem.200801578
Carvalhais V, Cerca N, Vilanova M, Vitorino R (2015) Proteomic profile of dormancy within Staphylococcus epidermidis biofilms using iTRAQ and label-free strategies. Appl Microbiol Biotechnol 99:2751–2762. doi:10.1007/s00253-015-6434-3
Chao Y, Marks LR, Pettigrew MM, Hakansson AP (2015) Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol 4:194. doi:10.3389/fcimb.2014.00194
Chauhan PS, Nautiyal CS (2010) The purB gene controls rhizosphere colonization by Pantoea agglomerans. Lett Appl Microbiol 50:205–210. doi:10.1111/j.1472-765X.2009.02779.x
Chen LH, Köseoğlu VK, Güvener ZT, Myers-Morales T, Reed JM, D’Orazio SEF, Miller KW, Gomelsky M (2014) Cyclic di-GMP-dependent signaling pathways in the pathogenic Firmicute Listeria monocytogenes. PLoS Pathog 10:e1004301. doi:10.1371/journal.ppat.1004301
Ding Y, Peng N, Du Y, Ji L, Cao B (2014) Disruption of putrescine biosynthesis in Shewanella oneidensis enhances biofilm cohesiveness and performance in Cr(VI) immobilization. Appl Environ Microbiol 80:1498–1506. doi:10.1128/AEM.03461-13
Domenech M, Garciá E, Moscoso M (2011) In vitro destruction of Streptococcus pneumoniae biofilms with bacterial and phage peptidoglycan hydrolases. Antimicrob Agents Chemother 55:4144–4148. doi:10.1128/AAC.00492-11
Domenech M, Ramos-Sevillano E, García E, Moscoso M, Yuste J (2013) Biofilm formation avoids complement immunity and phagocytosis of Streptococcus pneumoniae. Infect Immun 81:2606–2615. doi:10.1128/IAI.00491-13
Dominiak DM, Nielsen JL, Nielsen PH (2011) Extracellular DNA is abundant and important for microcolony strength in mixed microbial biofilms. Environ Microbiol 13:710–721. doi:10.1111/j.1462-2920.2010.02375.x
Domka J, Lee J, Bansal T, Wood TK (2007) Temporal gene-expression in Escherichia coli K-12 biofilms. Environ Microbiol 9:332–346. doi:10.1111/j.1462-2920.2006.01143.x
Dong J, Signo KSL, Vanderlinde EM, Yost CK, Dahms TES (2011) Atomic force microscopy of a ctpA mutant in Rhizobium leguminosarum reveals surface defects linking CtpA function to biofilm formation. Microbiology 157:3049–3058. doi:10.1099/mic.0.051045-0
Donlan RM, Costerton JW (2002) Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol 15:167–193. doi:10.1128/CMR.15.2.167
Dressaire C, Moreira RN, Barahona S, Alves de Matos AP, Arraiano CM (2015) BolA is a transcriptional switch that turns off motility and turns on biofilm development. MBio 6:e02352–e02314. doi:10.1128/mBio.02352-14
Driffield K, Miller K, Bostock JM, O’Neill AJ, Chopra I (2008) Increased mutability of Pseudomonas aeruginosa in biofilms. J Antimicrob Chemother 61:1053–1056. doi:10.1093/jac/dkn044
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Felek S, Muszyński A, Carlson RW, Tsang TM, Hinnebusch BJ, Krukonis ES (2010) Phosphoglucomutase of Yersinia pestis is required for autoaggregation and polymyxin B resistance. Infect Immun 78:1163–1175. doi:10.1128/IAI.00997-09
Forcella M, Cardona F, Goti A, Parmeggiani C, Cipolla L, Gregori M, Schirone R, Fusi P, Parenti P (2010) A membrane-bound trehalase from Chironomus riparius larvae: purification and sensitivity to inhibition. Glycobiology 20:1186–1195. doi:10.1093/glycob/cwq087
Foster PL (2007) Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 42:373–397. doi:10.1080/10409230701648494
Frese SA, Mackenzie DA, Peterson DA, Schmaltz R, Fangman T, Zhou Y, Zhang C, Benson AK, Cody LA, Mulholland F, Juge N, Walter J (2013) Molecular characterization of host-specific biofilm formation in a vertebrate gut symbiont. PLoS Genet 9:e1004057. doi:10.1371/journal.pgen.1004057
Ge X, Kitten T, Chen Z, Lee SP, Munro CL, Xu P (2008) Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect Immun 76:2551–2559. doi:10.1128/IAI.00338-08
Giaouris E, Samoilis G, Chorianopoulos N, Ercolini D, Nychas GJ (2013) Differential protein expression patterns between planktonic and biofilm cells of Salmonella enterica serovar Enteritidis PT4 on stainless steel surface. Int J Food Microbiol 162:105–113. doi:10.1016/j.ijfoodmicro.2012.12.023
Gnanadhas DP, Elango M, Datey A, Chakravortty D (2015) Chronic lung infection by Pseudomonas aeruginosa biofilm is cured by L-methionine in combination with antibiotic therapy. Sci Rep 5:16043. doi:10.1038/srep16043
Hamilton S, Bongaerts RJM, Mulholland F, Cochrane B, Porter J, Lucchini S, Lappin-Scott HM, Hinton JCD (2009) The transcriptional programme of Salmonella enterica serovar Typhimurium reveals a key role for tryptophan metabolism in biofilms. BMC Genomics 10:599. doi:10.1186/1471-2164-10-599
Heilmann C, Hussain M, Peters G, Götz F (1997) Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol 24:1013–1024. doi:10.1046/j.1365-2958.1997.4101774.x
Ito A, May T, Kawata K, Okabe S (2008) Significance of rpoS during maturation of Escherichia coli biofilms. Biotechnol Bioeng 99:1462–1471. doi:10.1002/bit.21695
Kato H, Mori H, Maruyama F, Toyoda A, Oshima K, Endo R, Fuchu G, Miyakoshi M, Dozono A, Ohtsubo Y, Nagata Y, Hattori M, Fujiyama A, Kurokawa K, Tsuda M (2015) Time-series metagenomic analysis reveals robustness of soil microbiome against chemical disturbance. DNA Res 22:dsv023. doi:10.1093/dnares/dsv023
Kim JN, Ahn SJ, Burne RA (2015) Genetics and physiology of acetate metabolism by the Pta-Ack pathway of Streptococcus mutans. Appl Environ Microbiol 81:5015–5025. doi:10.1128/AEM.01160-15
Knobloch JK, Nedelmann M, Kiel K, Bartscht K, Horstkotte MA, Dobinsky S, Rohde H, Mack D (2003) Establishment of an arbitrary PCR for rapid identification of Tn917 insertion sites in Staphylococcus epidermidis: characterization of biofilm-negative and nonmucoid mutants. Appl Environ Microbiol 69:5812–5818. doi:10.1128/AEM.69.10.5812
Kolderman E, Bettampadi D, Samarian D, Dowd SE, Foxman B, Jakubovics NS, Rickard AH (2015) L-Arginine destabilizes oral multi-species biofilm communities developed in human saliva. PLoS ONE. doi:10.1371/journal.pone.0121835
Kuboniwa M, Tribble GD, James CE, Kilic AO, Tao L, Herzberg MC, Shizukuishi S, Lamont RJ (2006) Streptococcus gordonii utilizes several distinct gene functions to recruit Porphyromonas gingivalis into a mixed community. Mol Microbiol 60:121–139. doi:10.1111/j.1365-2958.2006.05099.x
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lazarevic V, Soldo B, Médico N, Pooley H, Bron S, Karamata D (2005) Bacillus subtilis alpha-phosphoglucomutase is required for normal cell morphology and biofilm formation. Appl Environ Microbiol 71:39–45. doi:10.1128/AEM.71.1.39
Li G, Hamamoto K, Kitakawa M (2012) Inner membrane protein YhcB interacts with RodZ involved in cell shape maintenance in Escherichia coli. ISRN Mol Biol 2012:1–8. doi:10.5402/2012/304021
Li YH, Lau PCY, Lee JH, Ellen RP, Cvitkovitch DG (2001) Natural genetic transformation of Streptococcus mutans growing in biofilms. J Bacteriol 183:897–908. doi:10.1128/JB.183.3.897-908.2001
Lill R (2009) Function and biogenesis of iron-sulphur proteins. Nature 460:831–838. doi:10.1038/nature08301
Longo PL, Ota-Tsuzuki C, Nunes ACR, Fernandes BL, Mintz K, Fives-Taylor P, Mayer MPA (2009) Aggregatibacter actinomycetemcomitans arcB influences hydrophobic properties, biofilm formation and adhesion to hydroxyapatite. Brazilian J Microbiol 40:550–562
Loo CY, Corliss DA, Ganeshkumar N (2000) Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol 182:1374–1382. doi:10.1128/JB.182.5.1374-1382.2000
Luján AM, Maciá MD, Yang L, Molin S, Oliver A, Smania AM (2011) Evolution and adaptation in Pseudomonas aeruginosa biofilms driven by mismatch repair system-deficient mutators. PLoS One 6:e27842. doi:10.1371/journal.pone.0027842
Ma Q, Wood TK (2009) OmpA influences Escherichia coli biofilm formation by repressing cellulose production through the CpxRA two-component system. Environ Microbiol 11:2735–2746. doi:10.1111/j.1462-2920.2009.02000.x
Madsen JS, Burmølle M, Hansen LH, Sørensen SJ (2012) The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol 65:183–195. doi:10.1111/j.1574-695X.2012.00960.x
Magalhães AP, Azevedo NF, Pereira MO, Lopes SP (2016) The cystic fibrosis microbiome in an ecological perspective and its impact in antibiotic therapy. Appl Microbiol Biotechnol 100:1163–1181. doi:10.1007/s00253-015-7177-x
McFarland KA, Lucchini S, Hinton JCD, Dorman CJ (2008) The leucine-responsive regulatory protein, Lrp, activates transcription of the fim operon in Salmonella enterica serovar typhimurium via the fimZ regulatory gene. J Bacteriol 190:602–612. doi:10.1128/JB.01388-07
Monds RD, O’Toole GA (2009) The developmental model of microbial biofilms: ten years of a paradigm up for review. Trends Microbiol 17:73–87
Moreau-Marquis S, O’Toole GA, Stanton BA (2009) Tobramycin and FDA-approved iron chelators eliminate Pseudomonas aeruginosa biofilms on cystic fibrosis cells. Am J Respir Cell Mol Biol 41:305–313. doi:10.1165/rcmb.2008-0299OC
My L, Rekoske B, Lemke JJ, Viala JP, Gourse RL, Bouveret E (2013) Transcription of the Escherichia coli fatty acid synthesis operon fabHDG is directly activated by FadR and inhibited by ppGpp. J Bacteriol 195:3784–3795. doi:10.1128/JB.00384-13
Nachin L, Nannmark U, Nyström T (2005) Differential roles of the universal stress proteins of Escherichia coli in oxidative stress resistance, adhesion, and motility. J Bacteriol 187:6265–6272. doi:10.1128/JB.187.18.6265-6272.2005
Niba ETE, Naka Y, Nagase M, Mori H, Kitakawa M (2007) A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Res 14:237–246. doi:10.1093/dnares/dsm024
Noothalapati Venkata HN, Nomura N, Shigeto S (2011) Leucine pools in Escherichia coli biofilm discovered by Raman imaging. J Raman Spectrosc 42:1913–1915. doi:10.1002/jrs.2952
Novak EA, Shao H, Daep CA, Demuth DR (2010) Autoinducer-2 and QseC control biofilm formation and in vivo virulence of Aggregatibacter actinomycetemcomitans. Infect Immun 78:2919–2926. doi:10.1128/IAI.01376-09
O’Toole GA, Stewart PS (2005) Biofilms strike back. Nat Biotechnol 23:1378–1379. doi:10.1038/nbt1105-1378
Ojha A, Hatfull GF (2007) The role of iron in Mycobacterium smegmatis biofilm formation: the exochelin siderophore is essential in limiting iron conditions for biofilm formation but not for planktonic growth. Mol Microbiol 66:468–483. doi:10.1111/j.1365-2958.2007.05935.x
Okuda S, Tsuchiya Y, Kiriyama C, Itoh M, Morisaki H (2012) Virtual metagenome reconstruction from 16S rRNA gene sequences. Nat Commun 3:1203. doi:10.1038/ncomms2203
Oliver A, Cantón R, Campo P, Baquero F, Blázquez J (2000) High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254. doi:10.1126/science.288.5469.1251
Patel CN, Wortham BW, Lines JL, Fetherston JD, Perry RD, Oliveira M a. (2006) Polyamines are essential for the formation of plague biofilm. J Bacteriol 188:2355–2363. doi:10.1128/JB.188.7.2355-2363.2006
Peng Y, Leung HC, Yiu SM, Chin FY (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi:10.1093/bioinformatics/bts174
Pettigrew MM, Marks LR, Kong Y, Gent JF, Roche-Hakansson H, Hakansson AP (2014) Dynamic changes in the Streptococcus pneumoniae transcriptome during transition from biofilm formation to invasive disease upon influenza A virus infection. Infect Immun 82:4607–4619. doi:10.1128/IAI.02225-14
Pobre V, Arraiano CM (2015) Next generation sequencing analysis reveals that the ribonucleases RNase II, RNase R and PNPase affect bacterial motility and biofilm formation in E. coli. BMC Genomics 16:72. doi:10.1186/s12864-015-1237-6
Post D, Held JM, Ketterer MR, Phillips NJ, Sahu A, Apicella MA, Gibson BW (2014) Comparative analyses of proteins from Haemophilus influenzae biofilm and planktonic populations using metabolic labeling and mass spectrometry. BMC Microbiol 14:2322. doi:10.1186/s12866-014-0329-9
Pysz MA, Conners SB, Montero CI, Shockley KR, Johnson MR, Ward DE, Kelly RM (2004) Transcriptional analysis of biofilm formation processes in the anaerobic, hyperthermophilic bacterium Thermotoga maritima. Appl Environ Microbiol 70:6098–6112. doi:10.1128/AEM.70.10.6098-6112.2004
Ren D, Bedzyk LA, Thomas SM, Ye RW, Wood TK (2004) Gene expression in Escherichia coli biofilms. Appl Microbiol Biotechnol 64:515–524. doi:10.1007/s00253-003-1517-y
Ren D, Zuo R, González BAF, Bedzyk LA, Eldridge GR, Pasmore ME, Wood TK (2005) Differential gene expression for investigation of Escherichia coli biofilm inhibition by plant extract ursolic acid. Appl Environ Microbiol 71:4022–4034. doi:10.1128/AEM.71.7.4022-4034.2005
Roche B, Aussel L, Ezraty B, Mandin P, Py B, Barras F (2013) Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim Biophys Acta 1827:455–469. doi:10.1016/j.bbabio.2013.05.001
Russo DM, Williams A, Edwards A, Posadas DM, Finnie C, Dankert M, Downie JA, Zorreguieta A (2006) Proteins exported via the PrsD-PrsE type I secretion system and the acidic exopolysaccharide are involved in biofilm formation by Rhizobium leguminosarum. J Bacteriol 188:4474–4486. doi:10.1128/JB.00246-06
Sha J, Rosenzweig JA, Kozlova EV, Wang S, Erova TE, Kirtley ML, van Lier CJ, Chopra AK (2013) Evaluation of the roles played by Hcp and VgrG type 6 secretion system effectors in Aeromonas hydrophila SSU pathogenesis. Microbiology 159:1120–1135. doi:10.1099/mic.0.063495-0
Shao H, Lamont RJ, Demuth DR (2007) Autoinducer 2 is required for biofilm growth of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect Immun 75:4211–4218. doi:10.1128/IAI.00402-07
Shemesh M, Tam A, Aharoni R, Steinberg D (2010) Genetic adaptation of Streptococcus mutans during biofilm formation on different types of surfaces. BMC Microbiol 10:51. doi:10.1186/1471-2180-10-51
Si X, Quan X, Wu Y (2015) A small-molecule norspermidine and norspermidine-hosting polyelectrolyte coatings inhibit biofilm formation by multi-species wastewater culture. Appl Microbiol Biotechnol 99:10861–10870. doi:10.1007/s00253-015-6943-0
Singh P, Brooks JF, Ray VA, Mandel MJ, Visick KL (2015) CysK plays a role in biofilm formation and colonization by Vibrio fischeri. Appl Environ Microbiol 81:5223–5234. doi:10.1128/AEM.00157-15
Stoodley P, Sauer K, Davies DG, Costerton JW (2002) Biofilms as complex differentiated communities. Annu Rev Microbiol 56:187–209. doi:10.1146/annurev.micro.56.012302.160705
Tenorio E, Saeki T, Fujita K, Kitakawa M, Baba T, Mori H, Isono K (2003) Systematic characterization of Escherichia coli genes/ORFs affecting biofilm formation. FEMS Microbiol Lett 225:107–114. doi:10.1016/S0378-1097(03)00507-X
Tremblay YDN, Deslandes V, Jacques M (2013) Actinobacillus pleuropneumoniae genes expression in biofilms cultured under static conditions and in a drip-flow apparatus. BMC Genomics 14:364. doi:10.1186/1471-2164-14-364
Ueda A, Attila C, Whiteley M, Wood TK (2009) Uracil influences quorum sensing and biofilm formation in Pseudomonas aeruginosa and fluorouracil is an antagonist. Microb Biotechnol 2:62–74. doi:10.1111/j.1751-7915.2008.00060.x
Vieira HLA, Freire P, Arraiano CM (2004) Effect of Escherichia coli morphogene bolA on biofilms. Appl Environ Microbiol 70:5682–5684. doi:10.1128/AEM.70.9.5682-5684.2004
Waite RD, Paccanaro A, Papakonstantinopoulou A, Hurst JM, Saqi M, Littler E, Curtis MA (2006) Clustering of Pseudomonas aeruginosa transcriptomes from planktonic cultures, developing and mature biofilms reveals distinct expression profiles. BMC Genomics 7:162. doi:10.1186/1471-2164-7-162
Wang G, Wu P, Liu Y, Mi S, Mai S, Gu C, Wang G, Liu H, Zhang J, Børresen BT, Mellemsæther E, Kotlar HK (2015) Isolation and characterisation of non-anaerobic butanol-producing symbiotic system TSH06. Appl Microbiol Biotechnol 99:8803–8813. doi:10.1007/s00253-015-6864-y
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Watnick P, Kolter R (2000) Biofilm, city of microbes. J Bacteriol 182:2675–2679. doi:10.1128/JB.182.10.2675-2679.2000
Weinberg ED (2004) Suppression of bacterial biofilm formation by iron limitation. Med Hypotheses 63:863–865
Wolfe AJ, Chang DE, Walker JD, Seitz-Partridge JE, Vidaurri MD, Lange CF, Prüß BM, Henk MC, Larkin JC, Conway T (2003) Evidence that acetyl phosphate functions as a global signal during biofilm development. Mol Microbiol 48:977–988. doi:10.1046/j.1365-2958.2003.03457.x
Wu Y, Outten FW (2009) IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J Bacteriol 191:1248–1257. doi:10.1128/JB.01086-08
Xu D, Li Y, Gu T (2014) D-Methionine as a biofilm dispersal signaling molecule enhanced tetrakis hydroxymethyl phosphonium sulfate mitigation of Desulfovibrio vulgaris biofilm and biocorrosion pitting. Mater Corros 65:837–845. doi:10.1002/maco.201206894
Yang L, Liu Y, Wu H, Hóiby N, Molin S, Song Z (2011) Current understanding of multi-species biofilms. Int J Oral Sci 3:74–81. doi:10.4248/IJOS11027
Yu X, Liang X, Liu K, Dong W, Wang J, Zhou M (2015) The thiG gene is required for full virulence of Xanthomonas oryzae pv. oryzae by preventing cell aggregation. PLoS One 10:e0134237. doi:10.1371/journal.pone.0134237
Zhang L, Hinz AJ, Nadeau JP, Mah TF (2011) Pseudomonas aeruginosa tssC1 links type VI secretion and biofilm-specific antibiotic resistance. J Bacteriol 193:5510–5513. doi:10.1128/JB.00268-11
Zhang XS, García-Contreras R, Wood TK (2007) YcfR (BhsA) influences Escherichia coli biofilm formation through stress response and surface hydrophobicity. J Bacteriol 189:3051–3062. doi:10.1128/JB.01832-06
Zhu W, Lomsadze A, Borodovsky M (2010) Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 12:e132. doi:10.1093/nar/gkq275
Acknowledgments
We are grateful to Ms. Tomoko Narisawa for the technical assistance. This work was supported in part by KAKENHI (26450110). Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals.
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
ESM 1
(PDF 714 kb)
The supplementary materials accompanying this article are found at the publisher’s website.
Rights and permissions
About this article

Cite this article
Nakamura, Y., Yamamoto, N., Kino, Y. et al. Establishment of a multi-species biofilm model and metatranscriptomic analysis of biofilm and planktonic cell communities. Appl Microbiol Biotechnol 100, 7263–7279 (2016). https://doi.org/10.1007/s00253-016-7532-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7532-6