
Abstract
Probiotics are microorganisms that confer beneficial effects on the host; nevertheless, before being allowed for human consumption, their safety must be verified with accurate protocols. In the genomic era, such procedures should take into account the genomic-based approaches. This study aims at assessing the safety traits of Bacillus coagulans GBI-30, 6086 integrating the most updated genomics-based procedures and conventional phenotypic assays. Special attention was paid to putative virulence factors (VF), antibiotic resistance (AR) genes and genes encoding enzymes responsible for harmful metabolites (i.e. biogenic amines, BAs). This probiotic strain was phenotypically resistant to streptomycin and kanamycin, although the genome analysis suggested that the AR-related genes were not easily transferrable to other bacteria, and no other genes with potential safety risks, such as those related to VF or BA production, were retrieved. Furthermore, no unstable elements that could potentially lead to genomic rearrangements were detected. Moreover, a workflow is proposed to allow the proper taxonomic identification of a microbial strain and the accurate evaluation of risk-related gene traits, combining whole genome sequencing analysis with updated bioinformatics tools and standard phenotypic assays. The workflow presented can be generalized as a guideline for the safety investigation of novel probiotic strains to help stakeholders (from scientists to manufacturers and consumers) to meet regulatory requirements and avoid misleading information.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Probiotics are defined as “live microorganisms that, when administered in adequate amounts, confer a health benefit on the host” (Hill et al. 2014). Several microorganisms display probiotic properties, the most common types available being lactic acid bacteria and bifidobacteria. Remarkably, probiotic strains known to date belong to a relatively limited number of species, spanning across very different taxonomic groups (Salvetti et al. 2015); this implies that different properties could be linked to the different taxonomic groups. For the same reason, safety evaluation depends on the nature of the specific microorganism, as different microorganisms could be detrimental to the host via different mechanisms (Sanders et al. 2010).
The safety assessment of (potential) probiotic strains encompasses several aspects, such as their immunological effects, type of administration, dose and duration of consumption, and target population of patients and/or consumers (Sanders et al. 2010); on the microbial side, the absence of the genetic make-up for virulence factor (VF), transmissible antibiotic resistance (AR) and other deleterious characteristics is required by the European Food Safety Authority (EFSA) guidelines (EFSA 2013). The determination of the presence/absence of such genetic traits could be very rapid and cost-effective thanks to the use of available low-cost sequencing techniques (Sanders et al. 2010; Liu et al. 2015; Sun et al. 2015) and could also be seen as a criterion for pre-selection of strains with potential application as probiotics, especially for novel strains which have only limited or no history of safe use. Therefore, next-generation sequencing (NGS) technologies could be readily included in the safety evaluation process and could influence regulatory decisions on the commercial acceptability of the strain (Sanders et al. 2014).
The safety assessments based on the complete genome sequences have been recently performed for Bifidobacterium strains (Bennedsen et al. 2011), for the (potential) probiotic strains Lactobacillus plantarum JDM1 (Zhang et al. 2012) and Bifidobacterium longum JDM301 (Wei et al. 2012), for the bacteriocin-producers Streptococcus salivarius strains NU10 and YU10 (Barbour and Philip 2014), for the surrogate microorganism Enterococcus faecium NRRL B-2354 (Kopit et al. 2014) as well as for novel potential probiotic strains as Butyricicoccus pulicaecorum 25-3T (Steppe et al. 2014) and Lactobacillus helveticus MTCC 5463 (Senan et al. 2015). However, there is still a lack of homogeneity regarding the genetic and phenotypic traits to be assayed, and the proper use of the available bioinformatic tools, which may create confusion among stakeholders involved in this area (scientists, manufacturers, legislative bodies and consumers).
Among the wide number of probiotic products available on the market, a consistent part is represented by spore-forming bacteria, normally members of the genus Bacillus (Hong et al. 2005), which exploit the increased resistance of endospores to environmental stresses with respect to vegetative cells (Sanders et al. 2003). Species most comprehensively studied are Bacillus cereus, Bacillus clausii, Bacillus coagulans, Bacillus licheniformis and Bacillus subtilis (Cutting 2011).
B. coagulans GBI-30, 6086, commercialized under the commercial name GanedenBC30™ (BC30), is a spore-forming lactic acid-producing bacterium with the capacity to resist the harsh conditions typical of the gastrointestinal tract and displays good stability during shelf life (Hyronimus et al. 2000; Maathuis et al. 2010). Several studies demonstrated probiotic properties of this strain such as the aptitude to improve gastrointestinal (GI) quality of life in adults with postprandial intestinal gas-related symptoms (Kalman et al. 2009); the potential to aid in protein, lactose and fructose digestion (Maathuis et al. 2010); the antimicrobial activity in distal regions of the GI tract (Honda et al. 2011) and the capacity to improve some parameters of Clostridium difficile-induced colitis in mice and to limit its recurrence (Fitzpatrick et al. 2011; Fitzpatrick et al. 2012). These data have determined a scientific and commercial relevance of B. coagulans GBI-30, 6086 for human applications, which has been enriched with studies assessing its immunomodulatory properties (Jensen et al. 2010; Benson et al. 2012) and the stimulating effects on other beneficial genres of bacteria as well as organic acid production in the elderly (Nyangale et al. 2014).
The safe history of use of proprietary B. coagulans preparation of GanedenBC30™ has been supported by a toxicological safety assessment (Endres et al. 2009) and by a 1-year chronic oral toxicity study combined with a one-generation reproduction study (Endres et al. 2011). Moreover, the notice of Ganeden Biotech, Inc. to US FDA (Food and Drug Administration) reported unpublished PCR protocols that demonstrated that the strain does not contain genes homologous to those encoding known protein toxins and haemolysin (Ganeden Biotech, Inc. 2011). In the light of these findings, B. coagulans GBI-30, 6086 received the Generally Recognized As Safe (GRAS) status in 2012 from the FDA.
In 2014, we reported the draught genome sequence of B. coagulans GBI-30, 6086 to provide biological information helpful to unveil the genetic basis of its safety and probiosis (Orrù et al. 2014); the aim of the present paper is to critically re-evaluate these genomic data integrating them with phenotypic assays to have a comprehensive view of relevant safety aspects for this strain.
We suggest that this approach could become a structured modus operandi which could be extended to the safety assessment of probiotic bacteria by integrating genomic analyses performed with the modern NGS sequencing platforms and conventional phenotypic tests.
Materials and methods
Bacterial strain and growth conditions
B. coagulans GBI-30, 6086, supplied by Ganeden Biotech, Inc. (Mayfield Heights, OH) was routinely grown in Brain Heart Infusion (BHI) medium (Fluka Analytical, Buchs, Switzerland) at 30 °C in aerobic conditions. Strain GBI-30, 6086 is deposited in the American Type Culture Collection as B. coagulans PTA-6086.
Genome sequencing and taxonomic identification of B. coagulans GBI-30, 6086
The whole genome sequencing of B. coagulans strain GBI-30, 6086 was performed using the Illumina GAIIx sequencer at CRA-Genomic Research Centre. Details about the sequencing, assembly and annotation of the GBI-30, 6086 genome are reported in Orrù et al. (2014).
The complete 16S rRNA gene sequence of B. coagulans GBI-30, 6086 was retrieved by the genome sequence and searched against the EzTaxon database (http://eztaxon-e.ezbiocloud.net/) (Chun et al. 2007). Then, the 16S rRNA gene sequence of GBI-30, 6086 was aligned with those of B. coagulans DSM 1T, related taxa and other representatives of Bacillus genus using Clustal Omega (Sievers et al. 2011). After the manual editing of the alignment with CLC Main Workbench v. 7.5.1., unknown bases were disregarded and 1142 positions were included in the phylogenetic analysis. A phylogenetic tree was constructed using the number of differences algorithm as substitution model and neighbour-joining (Saitou and Nei 1987) as tree inference method as implemented in MEGA v.6 software package (Tamura et al. 2013).
The availability of the entire genome of B. coagulans GBI-30, 6086 allowed the development of the ribosomal multilocus sequence typing (rMLST) scheme which was based on 49 genes encoding the bacterial ribosome protein subunits (rps genes) as implemented in rMLST database website (http://pubmlst.org/rmlst/).
Ribosomal protein sequences were retrieved from B. coagulans GBI-30, 6086, B. coagulans DSM1T, B. acidiproducens DSM 23148T and B. subtilis 168T and were aligned with Clustal Omega (6128 aminoacids), while Poisson model and neighbour-joining were used to infer the phylogenetic tree.
The statistical reliability of the topology of the phylogenetic trees was evaluated using bootstrapping with 1000 replicates (Felsenstein 1985).
Measurements of antibiotic resistance phenotypes
The minimum inhibitory concentrations (MICs) of several antibiotics were determined following standard protocol ISO 10932:2010 (IDF 223:2010) for 15 antibiotics (ampicillin, chloramphenicol, ciprofloxacin, clindamycin, erythromycin, gentamicin, kanamycin, linezolid, neomycin, rifampicin, streptomycin, tetracycline, trimethoprim, vancomycin and virginiamycin). MICs for kanamycin and streptomycin were re-tested with the same protocol, testing concentrations up to 1500 mg/L.
Measurement of biogenic amine production
B. coagulans GBI-30, 6086 was grown in BHI containing 0.1 % w/v precursors (arginine, histidine, lysine, ornithine, putrescine and tyrosine). Ten-millilitre cultures were centrifuged at 8000g for 10 min at 10 °C, and the supernatants were used for biogenic amine (BA) determination by high-performance liquid chromatography (HPLC) after derivatization with dansyl-chloride (Sigma–Aldrich, Milano, Italy) according to Martuscelli et al. (2000). The BA content was analysed using a PU-2089 Intelligent HPLC quaternary pump, Intelligent UV-VIS multiwavelength detector UV 2070 Plus (Jasco Corporation, Tokyo, Japan) and a manual Rheodyne injector equipped with a 20-μL loop (Rheodyne, Rohnert Park, CA) (Tabanelli et al. 2014). BA production was then quantified according to Tabanelli et al. (2012).
Identification of safety-associated genes
Putative antibiotic resistance genes were identified by querying the Comprehensive Antibiotic Resistance Database (CARD) at http://arpcard.mcmaster.ca/, (McArthur et al. 2013) with the protein sequences derived from the GBI-30, 6086 annotated genes by using a local Protein-protein Basic Local Search Tool (BLASTP). Putative virulence factors were identified by local BLASTP against the Virulence Factor Database (VFDB) (Chen et al. 2012). Only Basic Local Alignment Search Tool (BLAST) results showing more than 30 % identity and 70 % coverage were considered in this study. Putative prophage sequences in the GBI-30, 6086 genome were identified using ProphageFinder (http://bioinformatics.uwp.edu/~phage/DOEResults.php) The clustered regularly interspaced short palindromic regions (CRISPR) were identified using CRISPRFinder (Grissa et al. 2007).
The presence of genes related to biogenic amine production (mainly aminoacids decarboxylases) was searched by BLASTX against the genome of GBI-30, 6086. Seed sequences included the complete operon sequence of tyrosine decarboxylase from B. cereus BAG2X1-1 (JH792376.1) for the production of tyramine, the histidine decarboxylase from a strain of B. coagulans isolated from fermented fish products (AB553281.1) for the production of histamine, the arginine decarboxylase from Bacillus thuringiensis HD1 (CP010005.1), the agmatine deiminase from B. cereus 2_A_57_CT2 (NZ_GL635753.1), the putrescine carbamoyl transferase from Bacillus massilioanorexius AP8 (NZ_CAPG01000089.1), the N-carbamoylputrescine amidase from Bacillus cellulosilyticus DSM 2522 (CP002394.1), the arginine deiminase from B. cereus Al Hakam (CP009651.1) and the ornithine carbamoyltransferase from Bacillus cytotoxicus NVH 391-98 (CP000764.1) involved in the production of putrescine.
The presence of enterotoxin genes was evaluated by searching on the genome the sequences of genes encoding for the haemolysin BL (HBL complex; hblC, hblD, hblA and hblB: AJ007794), the non-haemolytic enterotoxin NHE (NHE complex; nheA, nheB and nheC: Y19005), the enterotoxin T (bceT; D17312), the cytotoxin K (cytK; AJ277962) (Guinebretière et al. 2002) and the cereulide (cesA, cesH, cesP, cesT, cesB, cesC, cesD; DQ360825) (Ehling-Schulz et al. 2006).
Furthermore, the presence of genes involved in the synthesis of lipopeptides, as fengygin (fenA, AF023464; fenB, BACFENB; fenD, CAA09819; fenE, AF023465), surfactins (srfAA, D13262; srfAB, AF233756; sfrAC, CAB12145) (Tapi et al. 2010) and lychenisin (lchAA, lchAB, lchAC; AJ005061) (Yakimov et al. 1998) was also investigated.
Results
Taxonomic identification
The analysis in EzTaxon and the pairwise sequence alignment between the complete 16S rRNA gene sequence of B. coagulans GBI-30, 6086 and B. coagulans DSM 1T showed 99.92 % identity (data not shown), confirming strain GBI-30, 6086 to be allotted to species B. coagulans. This was also supported by the analysis with rMLST, the phylogenetic analyses based on 16S rRNA gene sequence and the concatenation of 49 ribosomal protein sequences [Online Resource 1, Fig.S1 (a) and (b)]. Such identification determined the subsequent analyses, as specific values for Bacillus genus and B. coagulans species in particular have been indicated by EFSA with respect to MIC cut-off value for antibiotic resistance, absence of food poisoning toxins, surfactant activity or enterotoxin activity (EFSA 2014) which have to be confirmed for the safe use of Bacillus strains in animal and human nutrition.
Antibiotic resistance and associated genes
Phenotypic tests were performed, and results were compared to MIC cut-off values for Bacillus species as defined by EFSA. Results obtained showed that strain GBI-30, 6086 was resistant only to kanamycin and streptomycin, MIC values being higher than 1500 mg/L, while MIC cut-off values for Bacillus species are 8 mg/L for both antibiotics according to EFSA guidelines (EFSA 2012) or 64 mg/L for both antibiotics according to a previous European document (European Commission 2003). The strain was susceptible to ampicillin (0.125 mg/L), chloramphenicol (0.25 mg/L), ciprofloxacin (0.03 mg/L), clindamycin (0.125 mg/L), erythromycin (0.125 mg/L), gentamycin (0.031 mg/L), linezolid (0.06 mg/L), neomycin (2 mg/L), rifampicin (0.016 mg/L), tetracycline (0.25 mg/L), trimethoprim (0.063 mg/L), vancomycin (0.063 mg/L) and virginiamycin (0.016 mg/L).
To elucidate the genetic basis of resistance to aminoglycosides (as kanamycin and streptomycin), the Comprehensive Antibiotic Resistance Database (CARD) was used to search the genome of B. coagulans GBI-30, 6086 for AR-related genes (E < 1e-2, coverage > 70 % and similarity > 30 %). This analysis led to the identification of 109 putative AR genes (Online Resource 2), most of which included transporters (57), genes modulating the antibiotic efflux (9), genes associated with resistance to daptomycin (6), polymyxin (1), streptothricin (1), penicillin (5), vancomycin (13), elfamycin (1), rifampin (2), sulphonamide (1), macrolides (as erythromycin, streptogramin and chloramphenicol) (2), fluoroquinolone (2), aminocoumarin (2) trimethoprim (1), other genes related to a non-specified antibiotic resistance (4) and aminoglycosides (2). The two identified aminoglycoside resistance genes, IE89_07115 and IE89_03650, encode for the ribosomal protein S12 of subunit 30S and an aminoglycoside 3-N-acetyltransferase, respectively.
Regarding IE89_07115, the ribosome alteration is one of the main aminoglycoside resistance mechanisms that can be mediated by 16S rRNA methylases and methyltransferases or intrinsic mechanisms as chromosomal mutations (Van Hoek et al. 2011; Poehlsgaard and Douthwaite 2005). A gene annotated as 16S rRNA methyltransferase (IE89_07580) was retrieved by CARD, but it was shown to be more similar to a gene involved in the resistance to macrolides rather than to aminoglycosides (ermA, e-value: 9e-25; similarity: 31, 33 %; query coverage 78 %). Since no other active rRNA methylases or methyltransferases were detected, we can assume that B. coagulans GBI-30, 6086 underwent events of mutation in IE89_07115, thus, becoming intrinsically resistant. The absence of mobile elements in the surrounding regions of IE89_07115, which is co-localized in the chromosome together with other genes encoding for essential chromosomal genetic information as other ribosomal proteins, suggests the low risk of gene transfer due to the high stability of this region (Courvalin 2006).
As for IE89_03650, this gene is similar (e-value: 3e-41; similarity: 31, 36 %, query coverage 98 %) to the gene encoding for an aminoglycoside 3-N-acetyltransferase from a Micromonospora chalcea isolate (Online Resource 2). The analysis of the flanking regions showed that IE89_03650 is co-localized on the chromosome with a gene encoding for a multidrug transporter MatE (IE89_03645), and this organization is detectable in all available B. coagulans genomes in NCBI (data not shown); no mobile elements as transposases and insertion sequences were found in the flanking regions of the gene, again indicating the very low risk of transfer of IE89_03650 to other bacteria.
The phenotypic and genomic analysis of AR in B. coagulans GBI-30, 6086 showed, for the first time, that this strain is phenotypically resistant to streptomycin and kanamycin. However, since the determinants for this resistance appear to be not easily transferrable to other bacteria, our results support the safety of this strain with respect to antibiotic resistance. Finally, since no other AR phenotypes were observed in GBI-30, 6086, it can be assumed that genes retrieved by in silico analysis were not functional or not expressed at a sufficient level or only partially similar to known resistance genes, but do not represent a harmful trait of this bacterium.
Biogenic amine production and associated genes
HPLC analyses were performed and revealed that the BAs, tyramine, histamine, putrescine, cadaverine and phenyletilamine, and the polyamines, spermine and spermidine, were not produced by B. coagulans GBI-30, 6086 in the conditions used.
Interestingly, on the genomic side, genes for BA production were generally absent, except those encoding for the entire metabolic pathway from arginine to putrescine (arginine decarboxylase, IE89_07650, IE89_01255; agmatinase, IE89_08455) and from putrescine to spermidine (carboxynorspermidine synthase, IE89_08025; carboxynorspermidine decarboxylase, IE89_08020). Although neither putrescine nor spermidine were produced by B. coagulans GBI 30, 6086 in the growth conditions tested, it is interesting to underline that these genes constitute the carboxyspermidine dehydrogenase/carboxyspermidine decarboxylase (CASDH/CASDC) system, which is the dominant polyamine biosynthetic pathway in the human gut microbiota (Hanfrey et al. 2011). As polyamines are important for cell proliferation, growth and development and triamines, such as spermidine, are thought to bind to RNA and influence ribosome function (Lee et al. 2009), further analyses are needed to determine whether those compounds could be produced in gut-like conditions and how this could impact on host physiology. Anyway, since the corresponding BAs were not produced by the strain, it can be assumed that the genes were not functional or not expressed at a sufficient level to produce detectable amounts of BAs.
Putative virulence factors
Putative virulence genes of B. coagulans GBI-30, 6086 were determined by BLAST analysis against the Virulence Factor Database (VFDB), a comprehensive repository of known bacterial virulence factors and other putatively adverse metabolites (Chen et al. 2012). A total of 200 genes putatively related to virulence were identified (E < 1e-2, coverage > 70 % and similarity > 30 %) (Online Resource 3). According to Clusters of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/), most of these genes were defensive or non-classical virulence factors, such as determinants related to transcription, translation, post-translational modifications, ribosomal structure and biogenesis, replication, recombination and repair, cell motility, signal transduction mechanisms, intra- and extracellular transportation, metabolism and transport of lipids, coenzymes, amino acids and carbohydrates, signal transduction mechanisms, cell cycle control, cell division and chromosome partitioning, protein turnover and chaperones, energy production and conversion and membrane biogenesis. In particular, eight genes were classified as related to defence mechanisms and they were annotated as multidrug transporters and resistance proteins (which were also previously detected by CARD), a peroxidase and an alkyl hydroperoxide reductase, notably essential to adapt in response to redox changes (Zuo et al. 2014).
Although the analysis of the GBI-30, 6086 genome against the VFDB revealed the presence of several putative VFs, they could not be considered really harmful. In fact, the majority of them are related to extracellular structures that could also represent essential probiotic traits for the adhesion to the host cells, or for the sporulation mechanism (Online Resource 3), which allow strain GBI-30, 6086 to overcome the harsh conditions in the gut (Cutting 2011; Zhang et al. 2012). Moreover, results are based on BLAST similarity that could detect possible VFs despite the relatively low similarity to known VFs; therefore, the differences in sequence and expression pattern could probably determine the absence of any known detrimental phenotype.
Putatively adverse metabolites
BLASTX analysis showed that B. coagulans GBI-30, 6086 does not carry any known enterotoxin genes. As for lipopeptides, only genes encoding for a long-chain fatty acid-CoA ligase (IE89_02350), an esterase (IE89_12515), a GDSL family lipase (IE89_12715), an ABC transporter ATP-binding protein (IE89_12720) and an ABC transporter permease (IE89_12725) were retrieved, but they were not found to be associated with the production of harmful peptides, thus confirming the toxicological analysis previously performed on this strain (Endres et al. 2009).
Genes encoding for surfactins, cyclic lipopeptides, which create damages to the host epithelial and sperm cells (From et al. 2007a; From et al. 2007b) produced by all haemolytic Bacillus strains (Salkinoja-Salonen et al. 1999), as well as for other lipopeptides with toxin activity as the fengycin and the lychenisin (EFSA 2011) were not found in B. coagulans GBI 30, 6086.
Focusing on toxins related to gastrointestinal diseases, B. coagulans GBI 30, 6086 does not harbour the genes encoding for the haemolysin BL, the non-haemolytic enterotoxin (Nhe, mostly associated with diarrhoeal outbreaks) (Stenfors-Arnesen et al. 2008), the enterotoxins K and T and the emetic toxin (cereulide) (EFSA 2011).
Stability of the genome of B. coagulans GBI-30, 6086
The genome sequence of B. coagulans strain GBI-30, 6086 was checked manually for the presence of proteins annotated as transposases by the NCBI Prokaryotic Genomes Annotation Pipeline (Orrù et al. 2014). Nine complete transposase-encoding genes were identified, but none of their flanking genes were associated with AR or other putatively adverse genes (see above).
The analysis conducted using ProphageFinder revealed the presence of two prophage-like elements (Table 1), which were found in contigs 58 and 137. The putative prophage region identified on contig 58 lacked genes encoding integrases, while genes encoding for the integrase, the portal protein and the terminase were found in the element of contig 132. However, in the latter, no gene was found for the tail tape measure protein, which is considered one of the phage essential proteins (Canchaya et al. 2003). Furthermore, ProphageFinder did not identify attL and attR sites in both the prophage regions, suggesting that these prophages were defective and non-functional phages.
The genome of B. coagulans GBI-30, 6086 was also analysed for the presence of clustered regularly interspaced short palindromic repeats (CRISPR) (Horvath and Barrangou 2010), and three CRISPR arrays were detected with a total of 41–32-bp repeats (consensus: GTCGCTCCCTACATGGGGGCGTGGATTGAAAT) and 38 spacers of 33 to 36 bp. Two CRISPR arrays were found in two adjacent contigs (contigs 95 and 96), and they could be part of the same CRISPR array. The third CRISPR was found in a contig of 548-bp length (contig 203) with no proteins annotated on it. Five genes putatively encoding for CRISPR associated (cas) proteins were observed in the vicinity of the CRISPR locus found on contig 95. The barrier to entry of foreign DNA elements represented by the presence of a CRISPR system in GBI-30, 6086 may provide an advantage in promoting genome stability.
As a general result, analyses performed in this study, safety of B. subtilis GBI-30, 6086 as a strain that could be used as food additive, are supported by phenotypic and genomic data.
Discussion
In this study, B. coagulans GBI-30, 6086, a strain for which probiotic properties were testified by several studies (Nyangale et al. 2015; Nyangale et al. 2014; Jurenka 2012; Fitzpatrick et al. 2012; Dolin 2009; Hun 2009, Kalman et al. 2009), was analysed with respect to safety aspects. B. coagulans is in the Qualified Presumption of Safety (QPS) list by EFSA as feed additive since 2007 (EFSA 2007) thanks to the certified absence of toxigenic potential. As some species have a long history of safe use in food production while others are less known and may represent a risk for consumers, the Scientific Committee of EFSA developed the QPS based on four pillars: establishing identity, body of knowledge, possible pathogenicity and end-use (Leuschner et al. 2010; EFSA 2007).
The precise and appropriate identification of the bacterial strain thus constitutes the starting point in the assessment of its safety and efficacy, also considering that the principal world regulatory authorities (e.g. FDA and EFSA) applied standards based on taxonomic criteria. Among the techniques developed for taxonomic studies, the analysis of the complete 16S rRNA gene sequences is recommended as the best tool for routine species identification due to its rapidity, reproducibility and multiple data comparisons (Vankerckhoven et al. 2008; van Loveren et al. 2012) although it might have low-resolution power between closely related species (Poretsky et al. 2014). The availability of complete genome sequences offers novel targets that could overcome the 16S rRNA gene shortcoming in accurate species assignment. Bacterial ribosome protein subunits, for instance, are characterized by a variation rate that resolves bacteria into groups at all taxonomic and most typing levels; therefore, their analysis could be combined in a ribosomal multilocus sequence typing (rMLST) approach for the identification of bacteria (Jolley et al. 2012).
In the present study, the taxonomic identification of B. coagulans GBI-30, 6086 was achieved aligning the 16S rRNA gene sequence retrieved by the genome sequence with those of B. coagulans DSM 1T, related taxa and other representatives of Bacillus genus. The 16S rRNA methodology was used in combination with the ribosomal multilocus sequence typing (rMLST) approach using the sequences of 49 genes encoding the bacterial ribosome protein subunits (rps genes).
The presence of up-to-date and internationally recognized databases as EzTaxon, for 16S rRNA gene sequences, and rMLST Database, which include the sequences of the type strains, allows a reliable taxonomic analysis, yielding an accurate and standardized identification.
All bacteria used as feed additive and, more generally, for human consumption must be tested for antibiotic susceptibility and the minimum inhibitory concentration (MIC) determined (Garrigues et al. 2013).
The MIC of the antimicrobials has to be determined in order to assess if the bacterial strain is resistant based on the microbiological cut-off values defined by EFSA. In case the bacterial strain demonstrates high resistance to a specific antimicrobial, AR determinants have to be identified and probability of occurrence of horizontal gene transfer (HGT) must be evaluated (EFSA 2012). AR becomes a safety issue in reason of the risks of spread associated with HGT phenomena. Although Bacillus spp. are widely used as feed additives and probiotics, there is a growing concern over the transfer of AR genes underlined by the fact that many Bacillus strains commercialized as probiotics have been shown to be resistant to chloramphenicol, tetracycline, erythromycin, lincomycin, penicillin, streptomycin (Adimpong et al. 2012) and kanamycin, as reported in this study for B. coagulans GBI 30, 6086.
For Bacillus spp., both EFSA and the Scientific Committee on Animal Nutrition (SCAN) defined the microbiological cut-off values for chloramphenicol, cipro/enrofloxacin, clindamycin, erythromycin, gentamycin, kanamycin, linezolid, quinupristin/dalfopristin, rifampin, streptomycin, tetracycline, trimethoprim and vancomycin. With our surprise, we found for the first time that strain GBI-30, 6086 is sensitive to ampicillin, for which MIC determination is generally not requested for Bacillus species as they are considered as inherently resistant to this antibiotic (European Commission 2003; EFSA 2012).
In order to infer information on the risk of horizontal transfer, analysis of the genetic make-up behind phenotypically detected resistance or susceptibility is crucial. AR gene identification can be achieved thanks to the Comprehensive Antibiotic Resistance Database, a centralized compendium that provides a comprehensive and updated list of AR gene sequences (McArthur et al. 2013). Intrinsic resistance or resistance by mutation of chromosomal genes presents a nearly absent or low risk of horizontal dissemination (Devirgiliis et al. 2011; Van Reenen and Dicks 2011); thus, the microbial strain is considered generally safe. Conversely, the presence of mobile elements in the flanking regions of AR genes (as transposases or insertion elements) could be taken as a clue of a resistance acquired through HGT of the genetic determinant(s); therefore, the strain should not be used as a feed additive (EFSA 2012). In B. coagulans GBI-30, 6086, two AR genes were detected to be putatively involved in aminoglycoside resistance, but the analysis of their flanking regions revealed that these genes are not easily transferable. Moreover, the presence of CRISPR loci, according to Maraffini and Sontheimer 2008, can limit the spread of AR by counteracting multiple route of HGT.
As such, besides confirming the safety of B. coagulans GBI-30, 6086, those features can open new perspectives regarding the use of this probiotic strain, as multi-(intrinsic) resistant probiotic preparations represent a valuable attractiveness as an adjunct to antibiotic therapy (Hong et al. 2005). In fact, it has been reported that Enterogermina® (Italian product registered 1958 in Italy as a medicinal supplement) contains a mixture of four AR Bacillus strains, each containing a unique spectrum of antibiotic resistance markers introduced by Sanofi Winthrop in order to combine the bacterial therapy with the administration of antibiotics (Green et al. 1999).
Together with the unambiguous identification and the demonstration of a lack of transferable AR determinants, the QPS approach for the safety assessment of microorganisms adopted by EFSA requires the evidence that the candidate strain lacks the capacity of producing toxins or other virulence factors, including BAs. SCAN and EFSA guidance recommends phenotypic assays that prove that the candidate strain is safe (EFSA 2011): in this perspective, Endres and colleagues performed different toxicological assays that indicated the strain GBI-30, 6086 does not have any mutagenic, clastogenic or genotoxic effects (Endres et al. 2009); furthermore, in the present study, a HPLC analysis showed that strain GBI-30, 6086 does not produce any biogenic amine in the tested conditions.
The streamlined screening of any genes on the GBI-30, 6086 genome related to toxins and virulence factor production (including BAs) revealed that GBI-30, 6086 does not harbour any risk-associated genes, thus confirming its safety also from the molecular viewpoint.
Interestingly, the term virulence factor (VF) refers to the elements that allow a microorganism to colonize a host, contributing to the start and development of infection processes. As such, it applies to secreted proteins, cell-surface structures and hydrolytic enzymes that contribute not only to the bacterial pathogenicity but also to adhesion and protection (Wassenaar et al. 2015). Remarkably, many probiosis-related traits could be associated to VFs; therefore, results of in silico analyses have to be carefully evaluated.
In general, the availability of the complete genome sequences allows the wide investigation of genome stability: analysis of transition elements, such as insertion elements (IS), prophages, transposases and CRISPR loci through bioinformatic tools. As for BGI-30, 6086, it harbours non-functional phages and transposases, which are not likely to be involved in risky gene transfer, thus highlighting the stability of its genome.
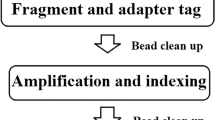
In the current study, a simple, minimum-standard system for the safety assessment of a probiotic bacterial strain which combines both genomic and phenotypic data was followed, and clear and reliable results were obtained. The workflow developed here follows the criteria delineated in the QPS approach: the proper taxonomic analysis based on multiple universal markers (not always considered in the previous safety assessment reports), the investigation of AR,VF and BA genes and genome stability is carried out with updated bioinformatics tools (i.e. CARD compared with the Antibiotic Resistance Database—ARDB, http://ardb.cbcb.umd.edu/), well-defined parameters and in combination with standardized phenotypic procedures (as MIC evaluation and HPLC analysis). Although, in the last few years, several papers have been published regarding the genome-based safety assessments of probiotic microorganisms (Bennedsen et al. 2011; Zhang et al. 2012; Wei et al. 2012; Barbour and Philip 2014; Kopit et al. 2014; Steppe et al. 2014; Senan et al. 2015), the workflow employed in our work and illustrated in Fig. 1 can be recommended as a new and reliable guideline for safety investigation.

Workflow for the safety assessment of probiotics for human use based on both genome and conventional phenotypic analysis. The scheme primarily consists in the proper taxonomic identification (based on 16S rRNA gene sequence and ribosomal proteins), the evaluation of antibiotic resistance, the production of virulence factors and biogenic amines and the analysis of the stability of the genome. Solid line boxes refer to genomic analysis, dotted line boxes refer to conventional phenotypic assays
The DNA sample from the putative probiotics strain is subjected to whole genome sequencing and the sequences are annotated. The functional annotation provides the comprehensive catalogue of the genes present on the genome. This information is subsequently used to proceed with the correct taxonomic identification of the strain using both the 16SrRNA gene sequence and the bacterial ribosome protein subunits sequences (rMLST). The genomic information is then used in association with phenotypic essays to detect the presence of AR, BA and VF genes.
All bacteria used as feed additive must be tested for antibiotic susceptibility and the minimum inhibitory concentration (MIC) determined. AR becomes a safety issue when there is the risk of spread by HGT. When an antibiotic resistance is assessed phenotypically, the genes responsible can be identified by querying the Comprehensive Antibiotic Resistance Database (CARD) with the protein sequences obtained from the functional annotation of the genome. Furthermore, the availability of the complete genome sequences allows the scanning of the genomic context in which the gene responsible for a resistance lies. The presence of transposase or phage elements in the surrounding region should be considered as potentially risky. The BA production is certified by HPLC analysis, and genes can be identified using mainly decarboxylases as seed sequences.
Similar to the AR, the presence of VF is investigated employing the Virulence Factor Database (VFDB); it must be emphasized that VFDB includes already known VFs and may miss important but as yet unidentified ones. However, for well-known and characterized species, this aspect is not predicted to be a crucial limitation. Finally, the safety assessment can be completed with the global analysis of the stability of the genome that includes the investigation of transition elements, such as insertion sequences (IS), prophages, transposases and CRISPR loci with several updated and reliable bioinformatic tools and the analysis of their flanking regions which constitutes a valuable approach to assess if the genome is stable and if the candidate strain is likely to be a donor or a recipient of safety-associated genes (Zhang et al. 2012).
The availability of complete genomic sequences allows the full characterization of the strain and provides the opportunity to decipher the entire genetic complement of a bacterium permitting genomic-based approaches in the evaluation of probiotic safety as the first requirements for marketing authorization and health claim request submission (Miquel et al. 2015). Although many phenotypic methods can be re-placed by the whole genome sequencing technologies, the overall physiology of a strain should also be taken in consideration.
Since probiotics are increasingly gaining ground as a form of preventive medicine and consumers become more conscious about health issues, the knowledge and the results acquired through the genomic analysis of probiotics are expected to ensure the flow of evidence-based, non-misleading, clearly understandable information about probiotics. In order to protect the consumer, approval of health claims for probiotic strains by the regulatory authorities has become very challenging due to the reasonable requirements for probiotic mechanism validation in the target, proper strain characterization and conformity to required product characteristics (Kumar et al. 2015; Hill et al. 2014; Hill and Sanders 2013). As more probiotic strains are used in food, drugs and supplements industry, more attention should be paid to assess the safety of these strains through the latest available technology (Wei et al. 2012). The modus operandi proposed here is intended to be a general template for the safety assessment starting from the genome sequence of a candidate probiotic strain, which can be implemented with genus-specific or species-specific risk-associated issues or genes related to the production of putatively adverse metabolites, depending on the strain used (i.e. toxin genes for Bacillus species) (EFSA 2011).
In conclusion, whole genome sequence allows the unveiling of the probiotic potential of a strain and the mechanisms which underline it, thus representing the key to fully meet the health claim requirements in accordance to European or US health and nutrition policies.
The modus operandi followed here is proposed to be a general template for the safety assessment starting from the genome sequence of a candidate probiotic or starter strain, which can be implemented with genus-specific or species-specific risk-associated issues combined with standard phenotypic analysis (EFSA 2011).
The workflow is expected to increase the consistency of future safety assessment, ensuring users (from scientists to manufacturers and consumers) the ability to obtain complete and easily comparable information, to meet regulatory requirements and avoid missing information.
References
Adimpong DB, Sørensen KI, Thorsen L, Stuer-Lauridsen B, Abdelgadir WS, Nielsen DS, Derkx PMF, Jespersen L (2012) Antimicrobial susceptibility of Bacillus strains isolated from primary starters for African traditional bread production and characterization of the bacitracin operon and bacitracin biosynthesis. Appl Environ Microbiol 78:7903–7914. doi:10.1128/AEM.00730-12
Barbour A, Philip K (2014) Variable characteristics of bacteriocin-producing Streptococcus salivarius strains isolated from Malaysian subjects. PLoS One 9:e100541. doi:10.1371/journal.pone.0100541
Bennedsen M, Stuer-Lauridsen B, Danielsen M, Johansen E (2011) Screening for antimicrobial resistance genes and virulence factors via genome sequencing. Appl Environ Microbiol 77:2785–2787. doi:10.1128/AEM.02493-10
Benson KF, Redman KA, Carter SG, Keller D, Farmer S, Endres JR, Jensen GS (2012) Probiotic metabolites from Bacillus coagulans GanedenBC30™ support maturation of antigen-presenting cell in vitro. World J Gastroenterol 18:1875–1883. doi:10.3748/wjg.v18.i16.1875
Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H (2003) Prophage genomics. Microbiol Mol Biol Rev 67:238–276. doi:10.1128/MMBR.67.2.238-276.2003
Chen LH, Xiong ZH, Sun LL, Yang J, Jin Q (2012) VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res 40:D641–D645. doi:10.1093/nar/gkr989
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YM (2007) EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57:2259–2261. doi:10.1099/ijs.0.64915-0
Courvalin P (2006) Antibiotic resistance: the pros and cons of probiotics. Dig Liver Dis 38:S261–S265. doi:10.1016/S1590-8658(07)60006-1
Cutting SM (2011) Bacillus probiotics. Food Microbiol 28:214–220. doi:10.1016/j.fm.2010.03.007
Devirgiliis C, Barile S, Perozzi G (2011) Antibiotic resistance determinant in the interplay between food and gut microbiota. Genes Nutr 6:275–284. doi:10.1007/s12263-011-0226-x
Dolin BJ (2009) Effects of a proprietary Bacillus coagulans preparation on symptoms of diarrhea-predominant irritable bowel syndrome. Methods Find Exp Clin Pharmacol 31:655–659. doi:10.1358/mf.2009.31.10.1441078
EFSA (2007) Opinion of the Scientific Committee. Introduction of a Qualified Presumption of Safety (QPS) approach for assessment of selected microorganisms referred to EFSA. EFSA J 587:1–16. doi:10.2903/j.efsa.2007.587
EFSA (2011) Scientific opinion. Technical guidance on the assessment of the toxinogenic potential of Bacillus species used in animal nutrition. EFSA J 9:2245. doi:10.2903/j.efsa.2011.2445
EFSA (2012) Scientific opinion. Guidance on the assessment of bacterial susceptibility to antimicrobials of human and veterinary importance. EFSA J 10:2740. doi:10.2903/j.efsa.2012.2740
EFSA (2013) Scientific opinion on the maintenance of the list of QPS biological agents intentionally added to food and feed (2013 update). EFSA J 11:3449. doi:10.2903/j.efsa.2013.3449
EFSA (2014) Guidance on the assessment of the toxigenic potential of Bacillus species used in animal nutrition. EFSA J 12:3665. doi:10.2903/j.efsa.2014.3665
Ehling-Schulz M, Guinebretiére M-H, Monthán A, Berge O, Fricker M, Svensson B (2006) Toxin gene profiling of enterotoxic and emetic Bacillus cereus. FEMS Microbiol Lett 260:232–240. doi:10.1111/j.1574-6968.2006.00320.x
Endres JR, Clewell A, Jade KA, Farber T, Hauswirth J, Schauss AG (2009) Safety assessment of a proprietary preparation of a novel probiotic, Bacillus coagulans, as a food ingredient. Food Chem Toxicol 47:1231–1238. doi:10.1016/j.fct.2009.02.018
Endres JR, Qureshi I, Farber T, Hauswirth J, Hirka G, Pasics I, Schauss AG (2011) One-year chronic oral toxicity with combined reproduction toxicity study of a novel probiotic, Bacillus coagulans, as a food ingredient. Food Chem Toxicol 49:1174–1182. doi:10.1016/j.fct.2011.02.012
European Commission (2003) Health & Consumer Protection Directorate-General. Opinion of the scientific committee on animal nutrition on the criteria for assessing the safety of micro-organisms resistant to antibiotics of human clinical and veterinary importance. ec.europa.eu/food/fs/sc/scan/out108_en.pdf
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi:10.2307/2408678
Fitzpatrick LR, Smal JS, Green WH, Karpa KD, Farmer S, Keller D (2012) Bacillus coagulans GBI-30, 6086 limits the recurrence of Clostridium difficile-induced colitis following vancomycin withdrawal in mice. Gut Pathog 4:13. doi:10.1186/1757-4749-4-13
Fitzpatrick LR, Small JS, Green WH, Karpa KD, Keller D (2011) Bacillus coagulans GBI-30 (BC30) improves indices of Clostridium difficile-induced colitis in mice. Gut Pathog 3:16. doi:10.1186/1757-4749-3-16
From C, Hormazabal V, Granum PE (2007a) Food poisoning associated with pumilacidin-producing Bacillus pumilus in rice. Int J Food Microbiol 115:319–324. doi:10.1016/j.ijfoodmicro.2006.11.005
From C, Hormazabal V, Hardy SP, Granum PE (2007b) Cytotoxicity in Bacillus mojavensis is abolished following loss of surfactin synthesis: implications for assessment of toxicity and food poisoning potential. Int J Food Microbiol 117:43–49. doi:10.1016/j.ijfoodmicro.2007.01.013
Ganeden Biotech, Inc. (2011) Notice to US Food and Drug Administration that Bacillus coagulans GBI-30, 6086, a Novel Probiotic, is generally recognized as safe for use in Foods http://www.fda.gov/downloads/Food/IngredientsPackagingLabeling/GRAS/NoticeInventory/UCM277775
Garrigues C, Johansen E, Crittenden R (2013) Pangenomics—an avenue to improved industrial starter cultures and probiotics. Curr Opin Biotechnol 24:187–191. doi:10.1016/j.copbio.2012.08.009
Green DH, Wakeley PR, Page A, Barnes A, Baccigalupi L, Ricca E, Cutting SM (1999) Characterization of two Bacillus probiotics. Appl Environ Microbiol 65:4288–4291
Grissa I, Vergnaud G, Pourcel C (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi:10.1093/nar/gkm360
Guinebretière M-H, Broussolle V, Nguyen-The C (2002) Enterotoxigenic profiles of food-poisoning and food-borne Bacillus cereus strains. J Clin Microbiol 40:3053–3056. doi:10.1128/JCM.40.8.3053-3056.2002
Hanfrey CC, Pearson BM, Hazeldine S, Lee J, Gaskin DJ, Woster PM, Phillips MA, Michael AJ (2011) Alternative spermidine biosynthetic route is critical for growth of Campylobacter jejuni and is the dominant polyamine pathway in the human gut microbiota. J Biol Chem 286:43301–43312. doi:10.1074/jbc.M111.307835
Hill C, Sanders ME (2013) Rethinking probiotics. Gut Microbes 4:269–270. doi:10.4161/gmic.25143
Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, Calder PC, Sanders ME (2014) Expert consensus document. International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 8:506–514. doi:10.1038/nrgastro.2014.66
Honda H, Gibson GR, Farmer S, Keller D, McCartney AL (2011) Use of continuous culture fermentation system to investigate the effect of GanedenBC30 (Bacillus coagulans GBI-30, 6086) supplementation on pathogen survival in the human gut microbiota. Anaerobe 17:36–42. doi:10.1016/j.anaerobe.2010.12.006
Hong HA, Duc le H, Cutting SM (2005) The use of bacterial spore formers as probiotics. FEMS Microbiol Rev 29:813–835. doi:10.1016/j.femsre.2004.12.001
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. doi:10.1126/science.1179555
Hun L (2009) Bacillus coagulans significantly improved abdominal pain and bloating in patients with IBS. Postgrad Med 121:119–124. doi:10.3810/pgm.2009.03.1984
Hyronimus B, Le Marrec C, Sassi AH, Deschamps A (2000) Acid and bile tolerance of spore-forming lactic acid bacteria. Int J Food Microbiol 61:193–197. doi:10.1016/S0168-1605(00)00366-4
Jensen GS, Benson KF, Carter SG, Endres JR (2010) GanedenBC30™ cell wall and metabolites: anti-inflammatory and immune modulating effects in vitro. BMC Immunol 11:15. doi:10.1186/1471-2172-11-15
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MC (2012) Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158:1005–1015. doi:10.1099/mic.0.055459-0
Jurenka JS (2012) Bacillus coagulans. Altern Med Rev 17:76–81
Kalman DS, Schwartz HI, Alvarez P, Feldman S, Pezzullo JC, Krieger DR (2009) A prospective, randomized, double-blind, placebo-controlled parallel-group dual site trial to evaluate the effects of a Bacillus coagulans-based product on functional intestinal gas symptoms. BMC Gastroenterol 9:85. doi:10.1186/1471-230X-9-85
Kopit LM, Kim EB, Siezen RJ, Harris LJ, Marco ML (2014) Safety of the surrogate microorganism Enterococcus faecium NRRL B-2354 for use in thermal process validation. Appl Environ Microbiol 80:1899–1909. doi:10.1128/AEM.03859-13
Kumar H, Salminen S, Verhagen H, Rowland I, Heimbach J, Bañares S, Young T, Nomot T, Lalonde M (2015) Novel probiotics and prebiotics: road to the market. Curr Opin Biotechnol 32:99–103. doi:10.1016/j.copbio.2014.11.021
Lee J, Sperandio V, Frantz DE, Longgood J, Camilli A, Phillips MA, Michael AJ (2009) An alternative polyamine biosynthetic pathway is widespread in bacteria and essential for biofilm formation in Vibrio cholerae. J Biol Chem 284:9899–9907. doi:10.1074/jbc.M900110200
Leuschner RGK, Robinson TP, Hugas M, Cocconcelli P, Richard-Forget F, Klein G, Licht TR, Nguyen-The C, Querol A, Richardson M, Suarez JE, Thrane U, Valk JM, von Wright A (2010) Qualified Presumption of Safety (QPS) is a generic risk assessment approach applied by the European Food Safety Authority (EFSA). Trends Food Sci Technol 21:425–435. doi:10.1016/j.tifs.2010.07.003
Liu S, Zhao L, Ren F, Sun E, Zhang M, Guo H (2015) Complete genome sequence of Bifidobacterium adolescentis BBMN23, a probiotic strain from healthy centenarian. J Biotechnol 198:44–45. doi:10.1016/j.jbiotec.2015.02.003
Maathuis AJH, Keller D, Farmer S (2010) Survival and metabolic activity of the GanedenBC30 strain of Bacillus coagulans in a dynamic in vitro model of the stomach and small intestine. Benef Microbes 1:31–36. doi:10.3920/BM2009.0009
Maraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. doi:10.1126/science.1165771
Martuscelli M, Crudele MA, Gardini F, Suzzi G (2000) Biogenic amine formation and oxidation by Staphylococcus xylosus strains from artisanal fermented sausages. Lett Appl Microbiol 31:228–232. doi:10.1046/j.1365-2672.2000.00796.x
McArthur AG, Waglechner N, NizamF YA, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O’Brien JS, Pawlowski AC, Piddock LJ, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. doi:10.1128/AAC.00419-13
Miquel S, Beaumont M, Martín R, Langella P, Braesco V, Thomas M (2015) A proposed framework for an appropriate evaluation scheme for microorganisms as novel foods with a health claim in Europe. Microb Cell Factories 14:48. doi:10.1186/s12934-015-0229-1
Nyangale EP, Farmer S, Cash HA, Keller D, Chernoff D, Gibson GR (2015) Bacillus coagulans GBI-30, 6086 modulates Faecalibacterium prausnitzii in older men and women. J Nutr 145:1446–1452. doi:10.3945/jn.114.199802
Nyangale EP, Farmer S, Keller D, Chernoff D, Gibson RG (2014) Effect of prebiotics on the fecal microbiota of elderly volunteers after dietary supplementation of Bacillus coagulans GBI-30, 6086. Anaerobe 30:75–81. doi:10.1016/j.anaerobe.2014.09.002
Orrù L, Salvetti E, Cattivelli L, Lamontanara A, Michelotti V, Capozzi V, Spano G, Keller D, Cash H, Martina A, Torriani S, Felis GE (2014) Draft genome sequence of Bacillus coagulans GBI-30, 6086, a widely used spore-forming probiotic strain. Genome Announc 2:e01080–e01014. doi:10.1128/genomeA.01080-14
Poehlsgaard J, Douthwaite S (2005) The bacterial ribosome as a target for antibiotics. Nat Rev Microbiol 3:870–881. doi:10.1038/nrmicro1265
Poretsky R, Rodriguez-R LM, Luo C, Tsementzi D, Konstantinidis KT (2014) Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS One 9:e93827. doi:10.1371/journal.pone.0093827
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Salkinoja-Salonen MS, Vuorio R, Andersson MA, Kamfer P, Andersson MC, Honkanen-Buzalski T, Scoging AC (1999) Toxigenic strains of Bacillus licheniformis related to food poisoning. Appl Environ Microbiol 65:4637–4645
Salvetti E, Torriani S, Felis GE (2015) A survey on established and novel probiotic strains for probiotic applications. In: Foerst P, Santivarangkna C (ed). Advances in probiotic technology. CRC Press, pp. 26–44
Sanders ME, Akkermans LMA, Haller D, Hammerman C, Heimbach J, Hormannsperger G, Huys G, Levy DD, Lutgendorff F, Mack D, Phothirath P, Solano-Aguilar G, Vaughan E (2010) Safety assessment of probiotics for human use. Gut Microbes 1:164–185. doi:10.4161/gmic.1.3.12127
Sanders ME, Klaenhammer TR, Ouwehand AC, Pot B, Johansen HJT, Marco ML, Tennilä RRP, Franz C, Pagé N, Pridmore RD, Leyer G, Salminen S, Charbonneau D, Call E, Lenoir-Wijnkoop I (2014) Effects of genetic, processing, or product formulation changes on efficacy and safety of probiotics. Ann N Y Acad Sci 1309:1–18. doi:10.1111/nyas.12363
Sanders ME, Morelli L, Tompkins TA (2003) Sporeformers as human probiotics: Bacillus, Sporolactobacillus and Brevibacillus. Compr Rev Food Sci Food Saf 2:101–110. doi:10.1111/j.1541-4337.2003.tb00017.x
Senan S, Prajapati JB, Joshi CG (2015) Feasibility of genome-wide screening for biosafety assessment of probiotics: a case study of Lactobacillus helveticus MTCC 5463. Probiotics Antimicro Proteins 7:249–258. doi:10.1007/s12602-015-9199-1
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using ClustalOmega. Mol Syst Biol 7:539. doi:10.1038/msb.2011.75
Stenfors-Arnesen LP, Fagerlund A, Granum PE (2008) From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev 32:579–606. doi:10.1111/j.1574-6976.2008.00112.x
Steppe M, Nieuwerburgh FV, Vercauteren G, Boyen F, Eeckhaut DD, Haesebrouck DR, van Immerseel F (2014) Safety assessment of the butyrate-producing Butyricicoccus pullicaecorum strain 25-3T, a potential probiotic for patients with inflammatory bowel disease, based on oral toxicity tests and whole genome sequencing. Food Chem Toxicol 72:129–137. doi:10.1016/j.fct.2014.06.024
Sun E, Zhao L, Ren F, Liu S, Zhang M, Guo H (2015) Complete genome sequence of Bifidobacterium animalis subsp. lactis A6, a probiotic strain with high acid resistance ability. J Biotechnol 200:8–9. doi:10.1016/j.jbiotec.2015.02.016
Tabanelli G, Montanari C, Bargossi E, Lanciotti R, Gatto V, Felis G, Torriani S, Gardini F (2014) Control of tyramine and histamine accumulation by lactic acid bacteria using bacteriocin forming lactococci. Int J Food Microbiol 190:14–23. doi:10.1016/j.ijfoodmicro.2014.08.023
Tabanelli G, Torriani S, Rossi F, Rizzotti L, Gardini F (2012) Effect of chemico-physical parameters on the histidine decarboxylase (HdcA) enzymatic activity in Streptococcus thermophilus PRI60. J Food Sci 77:M231–M237. doi:10.1111/j.1750-3841.2012.02628.x
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Tapi A, Chollet-Imbert M, Scherens B, Jacques P (2010) New approach for the detection of non-ribosomal peptide synthetase genes in Bacillus strains by polymerase chain reaction. Appl Microbiol Biotechnol 85:1521–1531. doi:10.1007/s00253-009-2176-4
Van Hoek AHAM, Mevius D, Guerra B, Mullany P, Roberts AP, Aarts HJM (2011) Acquired antibiotic resistance genes: an overview. Front Microbiol 2:203. doi:10.3389/fmicb.2011.00203
Van Loveren H, Sanz Y, Salminen S (2012) Health claims in Europe: probiotics and prebiotics as case examples. Annu Rev Food Sci Technol 3:247–261. doi:10.1146/annurev-food-022811-101206
Van Reenen CA, Dicks LM (2011) Horizontal gene transfer amongst probiotic lactic acid bacteria and other intestinal microbiota: what are the possibilities? A review. Arch Microbiol 193:157–168. doi:10.1007/s00203-010-0668-3
Vankerckhoven V, Huys G, Vancanneyt M, Vael C, Klare I, Romond M-B, Entenza J, Moreillon P, Wind R, Knol J, Wiertz E, Pot B, Vaughan EE, Kahlmeter G, Goossens H (2008) Biosafety assessment of probiotics used for human consumption: recommendations from the EU-PROSAFE project. Trends Food Sci Technol 19:102–114. doi:10.1016/j.tifs.2007.07.013
Wassenaar TM, Zschüttig A, Beimfohr C, Geske T, Auerbach C, Cook H, Zimmermann K, Gunzer F (2015) Virulence genes in a probiotic E. coli product with a recorded long history of safe use. Eur J Microbiol Immunol (Bp) 5:81–93. doi:10.1556/EUJMI-D-14-00039
Wei Y-X, Zhang Z-Y, Liu C, Malakar PK, Guo X-K (2012) Safety assessment of Bifidobacterium longum JDM301 based on complete genome sequences. World J Gastroenterol 18:479–488. doi:10.3748/wjg.v18.i5.479
Yakimov MM, Kröger A, Slepack TN, Giuliano L, Timmis KN, Golyshin PN (1998) A putative lichenysin A synthetase operon in Bacillus licheniformis: initial characterization. Biochim Biophys Acta 1399:141–153. doi:10.1016/S0167-4781(98)00096-7
Zhang Z-Y, Liu C, Zhu Y-Z, Wei Y-X, Tian F, Zhao G-P, Guo X-K (2012) Safety assessment of Lactobacillus plantarum JDM1 based on the complete genome. Int J Food Microbiol 153:166–170. doi:10.1016/j.ijfoodmicro.2011.11.003
Zuo F, Yu R, Khaskheli GB, Ma H, Chen L, Zeng Z, Mao A, Chen S (2014) Homologous overexpression of alkyl hydroperoxide reductase subunit C (ahpC) protects Bifidobacterium longum strain NCC2705 from oxidative stress. Res Microbiol 165:581–589. doi:10.1016/j.resmic.2014.05.040
Acknowledgments
The authors would like to thank Fausto Gardini (University of Bologna) for the help with HPLC analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by the Italian Ministry for Development in the framework of the project “Pass-World–pasta e salute nel mondo—Industria 2015” (MI01_00138). GS is supported by Ministry of Education, University and Research (PON02_00186_2937475) in the framework of the project named “Protocolli innovativi per lo sviluppo di alimenti funzionali” (Pro. Ali. Fun.). Vittorio Capozzi was supported by a grant of the Apulian Region in the framework of ‘FutureInResearch’ programme (practice code 9OJ4W81).
Conflict of interest
The authors declare that they have no conflict of interest, with the exception of HC and DK who are paid employees of Ganeden Biotech Inc.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Elisa Salvetti and Luigi Orrù made equal contributions to this article.
Electronic supplementary material
ESM 1
(PDF 880 kb)
Rights and permissions
About this article

Cite this article
Salvetti, E., Orrù, L., Capozzi, V. et al. Integrate genome-based assessment of safety for probiotic strains: Bacillus coagulans GBI-30, 6086 as a case study. Appl Microbiol Biotechnol 100, 4595–4605 (2016). https://doi.org/10.1007/s00253-016-7416-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7416-9