
Abstract
The online monitoring of recombinant protein aggregate inclusion bodies during microbial cultivation is an immense challenge. Measurement of scattered and reflected light offers a versatile and non-invasive measurement technique. Therefore, we investigated two methods to detect the formation of inclusion bodies and monitor their production: (1) online 180° scattered light measurement (λ = 625 nm) using a sensor platform during cultivation in shake flask and (2) online measurement of the light reflective interference using a porous Si-based optical biosensor (SiPA). It could be shown that 180° scattered light measurement allows monitoring of alterations in the optical properties of Escherichia coli BL21 cells, associated with the formation of inclusion bodies during cultivation. A reproducible linear correlation between the inclusion body concentration of the non-fluorescent protein human leukemia inhibitory factor (hLIF) carrying a thioredoxin tag and the shift (“Δamp”) in scattered light signal intensity was observed. This was also observed for the glutathione-S-transferase-tagged green fluorescent protein (GFP-GST). Continuous online monitoring of reflective interference spectra reveals a significant increase in the bacterium refractive index during hLIF production in comparison to a non-induced reference that coincide with the formation of inclusion bodies. These online monitoring techniques could be applied for fast and cost-effective screening of different protein expression systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Online detection of recombinant protein inclusion bodies
The monitoring of recombinant protein production is still a bottleneck for effective and sustainable process engineering. A large variety of techniques was developed to determine recombinant protein concentration and especially inclusion bodies (IB) in vivo. An overview is given as follows: (1) fusion of the target protein with green fluorescent protein (GFP) (Hedhammar et al. 2005; Patkar et al. 2002), (2) thioflavin staining (Espargaró et al. 2012), (3) sequence motive specific staining using fluorescein arsenical hairpin (FIAsH) (Ignatova and Gierasch 2004), (4) unspecific DNA/protein staining (Lewis et al. 2004; Medwid et al. 2007), (5) activation of luciferase reporter genes (Kraft et al. 2007), (6) Fourier transform infrared spectroscopy (FTIR) (Ami et al. 2006), (7) flow cytometry stand-alone (Wittrup et al. 1988), and (8) impedance measurement (Upadhyay et al. 2001). Some of these techniques were used in combination with flow cytometry (1, 2, 4) while others used fluorescence spectrometers (3, 5). These current state-of-the-art techniques suffer from several drawbacks, e.g., low robustness, limited universality, expensive reagents, low cell wall permeability of the dye, decrease/loss in target protein yield, and cross-sensitivity. In terms of in vivo online measurement, there are only few examples of commercially available measurement systems which can be adapted to bioreactors, shake flasks, or well plates (Jones et al. 2004; Kensy et al. 2009; Nemecek et al. 2008; Schmidt-Hager et al. 2014; Ude et al. 2014). Especially for shake flasks, which are still widely used to evaluate various, newly constructed expression systems or screening protein production at different conditions, there is no adequate online monitoring system so far to directly measure protein production and IB formation, respectively. While our group has previously demonstrated the concept of monitoring cell growth by 180° scattered light, the challenge of monitoring IB production by optical sensing is still unmet. Light interacting with intracellular homogenous particles of a higher refractive index (RI), compared to the cytoplasm, bends and reflects; in particular, light scattered at angles >40° is sensitive to internal cell structures (Drezek et al. 1999). Thus, in addition to the investigation of intracellular particle size and concentration by the measurement of 180° scattered light (as illustrated in Fig. S2), we introduce a second method to evaluate the optical properties of cells. Herein, cells entrapment within silicon photonic arrays (SiPA) is employed for real-time monitoring of the cell state. SiPA are a class of photonic crystals, characterized by micron-scale periodic pores, that have been recently shown to be compatible for bacterial culture analysis by monitoring small changes in the RI and tracking bacterial cells that are entrapped in the pores (Mirsky et al. 2013; Massad-Ivanir et al. 2014). The term photonic crystal was first used to describe a material comprised of two different RI substances in a sub-wavelength grating structure, consisting of ordered periodic arrays of reflectivity (Yablonovitch 1987). Photonic arrays, or lamellar grating, differ by consisting of ordered periodic topographies larger than the visible wavelength (Fig. S3a). The interference term in photonic arrays is related to a phase delay between the two alternating reflective partners, i.e., light beams reflected from the top and the bottom of the pores (Fig. S3b). When illuminated with a broadband light source, high-order diffraction light modes are coupled, destructively interfering with the zero-order transmitted light. For each particular resonant wavelength, a degree of interference occurs in the reflected light, resulting in an interference spectrum. Further analysis by Fourier transformation termed as interferometric Fourier transform spectroscopy (RIFTS) allows extracting the SiPA intrinsic effective optical thickness (EOT) which is the product of the pore refractive index (n) and the pore depth (L): EOT = 2Ln (Mirsky et al. 2013). Introduction of a biomaterial into the pores induces measurable changes in the EOT that can be monitored and quantified in real-time via RIFTS analysis (Fig. S3c). The advantages of optical monitoring for bacteria cell studies are numerous. Cells’ properties are measured without the use of genetic transfections, dyes, or stains, so a population of cells can be measured repeatedly without disruptions. The detected output signal is highly quantitative, providing repeatable measurements without photobleaching and lacking fluorescence overflow and superior signal-to-noise ratio. Photonic array-based biosensors provide information that is fundamentally different than that obtained by microscopy, as the biosensor responds sensitively to small variations of the cells, within the porous volume. The focus of this study is to investigate whether online 180° scattered light of 625 nm wavelength and online RIFTS measurements could be an appropriate, cost-effective, and fully non-invasive technique to monitor IB formation in vivo.
Materials and methods
Media, buffers, reagents, chemicals, and SDS-PAGE gels
Terrific Broth
24 g L−1 yeast extract (AppliChem, Germany (ApC)), 12 g L−1 tryptone enzymatic digest of casein (Sigma-Aldrich, St. Louis, USA (SA)), 5 g L−1 glycerol (Rotipuran®, Carl-Roth, Germany (CR)), 200 μL L−1 TEGO® Antifoam KS 911 (Evonik-Industries, Essen, Germany (EvI)).
ZYP-31
(F.W. Studier et al., modified): 6.8 g L−1 KH2PO4 (Fluka, St. Louis, USA (FK)), 17.9 g L−1 Na2HPO4 12 H2O (Riedel de Haën, St. Louis, USA (RdH)), 3.3 g L−1 (NH4)2SO4 (CR), 2.0 g L−1 yeast nitrogen base (YNB) salts with amino acids (Y1250 Sigma), 0.06 g L−1 protocatechuic acid (FK), 10 g L−1 glycerol (CR), 200 μL L−1 TEGO® Antifoam KS 911 (EvI).
Phosphate buffered saline
137 mM NaCl, 30 mM Na2HPO4 12 H2O (RdH), 2.7 mM KCl (Merck KGaA, Darmstadt, Germany (Mck)), pH 7.0.
Lysis buffer
50 % (v/v) buffer A, 50 % (v/v) buffer B. A: 50 mM Tris-HCl (ApC), 50 mM NaCl (SA), 0.2 % (w/v) Triton X100 (SA), 50 μL L−1 TEGO® Antifoam KS911 (EvI), 4 mM 4-(2-aminoethyl)-benzenesulfonyl-fluoride-hydrochloride (Pefabloc® SC, (SA)) pH = 7.0.
B: BugBuster® Protein Extraction Reagent (EMD Millipore Corp, Billerica, USA), 0.02083 mg/10 mL lysozyme from chicken egg white (SA, ∼96,000 U mg−1).
Washing buffer
50 mM Tris-HCl (ApC), 10 mM 2-mercaptoethanol (CR), 2 mM EDTA (ApC), 5 % (v/v) glycerol, 0.2 % (w/v) Triton X100 (SA), pH 8.0.
Laemmli buffer
65 mM Tris-HCl (ApC), 2.5 % sodium dodecyl sulfate (SDS) (RdH), 0.005 % (w/v) bromophenol blue (CR), 10 % (v/v) 2-mercaptoethanol (CR), 8.6 % (v/v) glycerol, pH 6.8.
TGS running buffer
Tris-base 25 mM (CR), 86 mM glycine (SA), 3.47 mM SDS (RdH), pH 8.3.
15 % polyacrylamide separation gel
37.45 % (v/v) 40 % Rotiphorese® Gel 40 (CR), 0.418 M Tris-base 8.8 (CR), 3.45 mM SDS (RdH), 24.3 % (v/v) H2Odemin, 0.2 % (v/v) TEMED (CR)/2.18 mM APS (CR).
Stacking gel
13.27 % (v/v) 40 % Rotiphorese® Gel 40 (CR), 0.167 M Tris 6.8 (ApC), 1.84 mM SDS (RdH), 66.7 % (v/v) H2Odemin, 0.18 % (v/v) TEMED (CR)/2.0 mM APS (CR).
BSA standard
Bovine albumin (Amresco Inc., Solon, USA), 20 mM Tris-base (CR), pH 8.0.
Colloidal-Coomassie staining solution
0.12 % (w/v) Coomassie G-250 (CR), 10 % (w/v), (NH4)2SO4 (CR), 10 % (v/v) orthophosphoric acid (CR), 20 % methanol (v/v) (VWR International, Darmstadt, Germany (VWR)).
Decolorization agent
10 % (v/v) Ethanol 96 % (CR), 2 % (v/v) orthophosphoric acid (CR).
All solutions were prepared with deionized water produced by Arium® 661 Ultrapure water system (Sartorius Stedim Biotech AG, Göttingen, Germany (SaR)).
SiPA functionalization
N-(3-Trimethoxysilylpropyl)diethylenetriamine (DETAS), wheat germ agglutinin (WGA), ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) (SA), N-hydroxysulfosuccinimide sodium salt (NHS) (SA), morpholinoethanesulfonic acid (MES), succinic anhydride, dimethylsulfoxide (DMSO) (SA), acetonitrile, diisopropylethylamine (DIEA), Lugol’s solution, and N-acetylglucoseamine (NAG) were purchased from (SA, Israel). Acetic acid (BioLab Ltd., Israel). Phosphate buffered saline (PBS) at pH 7.4 (50 mM Na2HPO4, 17 mM NaH2PO4, and 68 mM NaCl). MES buffer (6 mM MES, 4 mM MES sodium salt).
Cultivation methods
Media preparation, cultivation, and induction
The microorganisms, applied media, and cultivation parameters are listed in Table 1. The media were autoclaved at 121 °C for 20 min. Vitamins and tracers were filtered separately with 0.2 μm Luer sterile filters (Minisart®, SaR). Induction was performed with a 0.5 M stock solution of isopropyl β-D-1-thiogalactopyranoside (IPTG) (VWR) to give a final concentration of 2 mM, which was injected via the septum. In the case of antibiotic resistance, the medium was adjusted to the following concentrations: 0.086 mM kanamycin sulfate (CR), 0.14 mM disodium carbenicillin (ApC). Precultures were prepared from 15 % (w/v) glycerol stocks. The main culture was inoculated with a calculated start OD600 of 0.2.
Online shake flask sensor and cultivation equipment
For 180° scattered light measurements, a multisensory platform (SFR OGM FM1, PreSens Precision Sensing GmbH, Regenburg, Germany) was used. This device is able to measure scattered light, dissolved oxygen (DO), and pH simultaneously. The scattered light sensor is basically a setup of a LED (625 nm) and a highly sensitive photodiode measuring at 160–180° angle through the wall of the shake flask. This sensor was observed to be sensitive to morphological, opacity, and color changes of cells. The hardware specification details are described in previous publications (Schmidt-Hager et al. 2014; Ude et al. 2014). Shaking was performed on an orbital shaker with a 25-mm shaking diameter (Certomat® SII, SaR) within an incubator (Certomat® H, B. Braun Biotech International, Berlin, Germany). X-ray sterilized, disposable, 4× baffled shake flasks (Corning® Inc., Corning, USA) equipped with DO- and pH sensor spots (PreSens GmbH, Regensburg, Germany) were used. The batch numbers stated as follows: (1) SFS–HP5–PSt3–500–WB–VEC–v3 ID 13–06–01. (2) SFS–HP5–PSt3–1000–WB–VEC–v3 ID 14–15–02. The detailed calibration data is given in the Online Resource.
Silicon photonic array optical biosensors
Surface functionalization
SiPA microstructures, characterized by a periodic pattern of 4 μm2 pores, were fabricated by photolithography and reactive ion etching processes. Post fabrication, the SiPA chips were thermally oxidized in a tube furnace (Thermolyne, Thermo Scientific, Waltham, USA) at 800 °C for 1 h in ambient air resulting in an oxidized matrix (SiO2). Surface functionalization was constructed in a three-step process. First, the SiO2 was amine silanized with DETAS (2 % v/v) and acetic acid (0.6 % v/v) in 50 % v/v ethanol/distilled water (DW) for 1 h, rinsed three times with 70 % v/v ethanol/DW, and dried under a nitrogen stream. Second, the amine end-groups were reacted with succinic anhydride (1 % v/v) and DIEA (4 % v/v) in acetonitrile for 3 h to the convert surface amines to carboxyls. Next, the crosslinking of amine-reactive NHS (1 % w/v) to surface carboxyls is mediated by carbodiimide chemistry using EDC (2 % w/v) and DMSO (10 % v/v) in MES buffer pH 6 for 1 h. Finally, WGA was dissolved in PBS (1 mg mL−1) and immobilized onto the SiPA by reacting with the NHS activated surface for 1 h.
RIFTS monitoring
The SiPA biosensor was placed in a custom-made flow cell (Fig. 1), and the reflectance spectra of the samples were collected using a charge-coupled device (CCD) USB 4000 spectrometer fitted with a collimating lens coupled to a bifurcated fiber-optic cable. A tungsten light source was directed onto the center of the sample with a spot diameter of 2 mm. All optical parts were purchased from Ocean Optics Inc. (Dunedin, USA) unless mentioned otherwise. Optical data were recorded at a wavelength range of 400–900 nm, with a spectral acquisition time of 30 ms. Illumination of the surface and detection of the reflected light were both performed along an axis coincident with the surface normal. Data were recorded by SpectraSuite software and analyzed by Igor Pro software (Wavemetrics Inc., Lake Oswego, USA) as RIFTS, as previously described (Massad-Ivanir et al. 2014), to retrieve EOT value. The flow cell was heated to 37 °C by infrared lamp Bosotherm 4000 (Boso GmbH & Co. KG, Jungingen, Germany), and temperature was monitored throughout the experiment using infrared thermometer 561 (Fluke corp., Everett, USA). The inlet was fed through a peristaltic pump, and the outlet was directly purged into a waste container. Initially, the flow cell was flushed with 70 % (v/v) ethanol (CR) for 10 min, followed by a constant flow of clean growth media for 30 min, until a stable EOT baseline was obtained. Escherichia coli suspension (108 cell mL−1) was introduced at a flow rate of 50 μL min−1for 20 min, followed by the introduction of a sterile growth medium. For induction experiments, IPTG was added to the growth medium. The SiPA biosensor was optically monitored for 3 h under constant flow conditions (50 μL min−1).

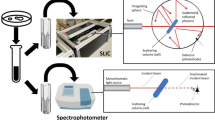
Technical drawing of the experimental setup. The left 1000 mL shake flask was connected to an at-line sampling and OD measuring system. Scattered light was measured online with the SFR OGM FM1 sensor, while the EOT was measured by the SiPA sensor
HRSEM (high resolution scanning electron microscopy) studies
For studying the morphology of bacterial cells, planar SiO2 wafers were functionalized with WGA, as described in section “Surface functionalization.” Cell samples from different suspensions were immediately placed onto the wafer for 30 min, followed by removal of excess media. The adsorbed cells were fixed by gentle flooding with 2.5 % (v/v) glutaraldehyde in phosphate buffer and maintained in preservation solution for 3 days at 4 °C.
For HRSEM analysis of SiPA, 2.5 % (v/v) glutaraldehyde in phosphate buffer was injected into the flow cell at the end of the optical experiment. After incubation of 15 min, the SiPA samples were collected into sealed vials and kept in their preservation solution, for 3 days, at 4 °C. Samples were prepared for HRSEM observation by serial dehydration with increasing ethanol fraction up to 100 % ethanol and finally vacuum dried. The samples were carbon coated by graphite sputtering and studied by a high-resolution scanning electron microscope (Carl Zeiss Ultra Plus HRSEM).
Optical density measurement and sampling
The OD600 was measured with an at-line sampling and dilution system connected to a 3–4 °C cooling tray and a VIS spectrometer (Libra S11, Biochrom Ltd., Cambridge, UK) (Fig. 1, Fig. S1, Table S1). This device was used because manual sampling represents a significant disturbance to the process and a continuous measurement. For this purpose, customized multiport flask caps were used (Fig. S4). More information regarding this system is given in the Online Resource. Two-milliliter samples were collected, and OD600 was measured in a 15-min time interval. To flood the SiPA biosensor with growing cells and for IPTG induction, the measurement was stopped (i.e., data gaps). Samples were frozen at −20 °C.
Online scattered light shift analysis
Prior to the analysis, the scattered light raw data were median filtered with a running window of n = 45. The window was aligned backwards since data prediction was not considered for regular online measurement so far. The difference between growth curves of an induced and non-induced culture (“Δamp”) was defined as the difference of the median filtered scattered light raw data and a linear elongation of the growth phase before induction (Fig. S5). It was assumed that the trend of the growth after induction is also linear because this behavior was observed in the control cultivation without induction. Since there are small variations of the growth rate between different cultivations, the specific linear elongation (linear function) was preferred to the usage of one control cultivation as methodology. The linear function was determined by two points; one point within the early growth phase (yellow dot), the second localized shortly before induction (yellow square). The linear function was shifted to run through the first point after induction (green diamond) due to the small disturbance caused by the stop of the shaking. This disturbance leads to a marginal lower signal which would cause a negative Δamp at the initial stage of the production phase. Δamp was subsequently calculated by subtraction of the filtered scattered light signal and the linear function. Δamp was correlated with the IB concentration (section “Protein quantification”).
Flow cytometry
Samples of the culture were taken with their OD600 adjusted to approx. 0.02 with PBS to provide 200–1000 events per second. To distinguish cells from debris, cells were stained with 10 μL mL−1 SYBR® Green I (10,000×, Molecular Probes®, Thermo Fisher Scientific, Waltham, USA) solution for 20 min at 37 °C in a thermomixer at 600 rpm (Eppendorf AG, Hamburg, Germany (Epd)). The cells stained were washed and resuspended in PBS. Samples were analyzed by Epics® XL-MCL™ (Beckman Coulter Inc., Brea, USA) using an air-cooled 488-nm argon ion laser. Cell population was discriminated by the SYBR Green signal, which was collected by a 525/40 band pass filter. A total of 20,000 events were analyzed per sample. Forward scatter (FSC) and side scatter (SSC) were calculated with the software WinMDI 2.9 (J. Trotter, The Scripps Research Institute, La Jolla, USA). Measurement parameters (signal, volts, gain, signal amplification): FS, 1000, 10, 40; SS, 1000, 100, 400; FL1, 767, 1. Discriminators FL1: 20.
Protein quantification
Cell disruption and quantitative SDS-PAGE
Cells were normalized to OD600 = 3 according to Eq. 1. The calculated sample volume was transferred into a 1.5-mL reaction tube and centrifuged at 21,000×g for 5 min. The pellet was washed once with PBS, and 500 μL of lysis buffer was added to the pellet which was resuspended subsequently. The samples were treated by an ultrasonic probe (Labsonic® M, SaR) performing 3 cycles (100 % at 0.6 Hz) for 1.5 min undergoing a 20-min pause between the cycles. The samples were liquid cooled to 4 °C. Afterwards, the samples were incubated in a thermomixer (Epd) at 27 °C for 30 min while shaking at 1000 rpm. The samples were cooled on ice for 20 min and centrifuged at 21,000×g for 7 min. The supernatant was used for analysis of the soluble protein fraction. The pellet was washed once with 1 mL washing buffer. All working steps were performed at 4 °C. The pellet was resuspended in 200 μL of Laemmli buffer and incubated in a thermomixer (1000 rpm) at 95 °C for 10 min. Supernatants were diluted (1:2) with Laemmli buffer and incubated at 95 °C just as the samples. Different concentrations of bovine serum albumin (BSA) were used for calibration (stated on the gels). The BSA standards were treated like the supernatants. Detailed information concerning loading and scanning are given in the Online Resource.
*V = volume
Densitometry and protein concentration
Densitometry was performed with ImageJ (National Institutes of Health, Bethesda, USA) as shown in Figure S6. For a reproducible area determination, a constant rectangular size was used rigorously (200 × 105 px). Calibration and samples were analyzed in a separate lane sequence. For preparative error estimation (Eq. 6), the calibration was loaded on each gel and selected time points (every 2nd sample after induction) of the samples to be analyzed were loaded again on a second gel. The second gel corresponded technically to the same PAGE run. In addition, densitometrical area determination was carried out twice for software error estimation (Eq. 7) delivering a data set as follows: area1gel1, area2gel1, area1gel2, area2gel2. BSA calibrations were fitted using second grade polynomial or Bleasdale-Nelder (BN) function (Eq. 2) (Bleasdale and Nelder 1960) within the software Origin® 8.5.1 (OriginLab Corp., Northampton, UK). Mean values of BSA band areas on two different gels were used for calibration data fitting. Mean values of each sample data set (see above) were used for sample concentration calculation. Sample concentrations were calculated according to Eq. 3 (for polynomial) and Eq. 4 (for BN), respectively. Normalization, lysis volume, and dilution with Laemmli buffer were considered in the calculation of the protein concentration per volume culture broth (Eq. 5). The received concentrations were subsequently correlated with Δamp values. Error bars in IB concentration plots were determined by calculating the standard deviation of area1gel1, area2gel1, area1gel2, area2gel2 including preparative (Eq. 6) and software error (Eq. 7). With this value, a protein concentration was calculated the same way as with the samples. Error calculation results are given in Tab. S2.
* c = concentration.
Results
Online 180° scattered light analysis and flow cytometry analysis for inclusion body detection
Two different strains of E. coli BL21 (DE3) producing GFP-GST and human leukemia inhibitory factor (hLIF), respectively, were tested regarding online scattered light during inclusion body formation. The latter was investigated in more detail by additional methods, i.e., offline FSC/SCC, HRSEM, and EOT measurement in defined medium. The online scattered light measurement during cultivation of E. coli BL21 (DE3) producing GFP-GST in TB medium exhibited a characteristic bend approx. 10 min after induction with IPTG. In comparison with a non-induced control, it was apparent that an additional scattered light signal (Δamp) was caused in consequence of the induction (Fig. 2). The accumulation of inclusion bodies could be proved by growth normalized SDS-PAGE of the insoluble fraction (Fig. S7), while the plot of Δamp and IB concentration showed a linear correlation up to 100 mg L−1 (Fig. S8). A temporary bend which also occurred in the control cultivation 6.75 h after inoculation could be referred to a stop in medium acidification and was not associated to the induction (Fig. S13). The same effect was observed during production of hLIF in ZYP-31, which was successfully reproduced in three E. coli BL21 cultivations (ID 150708-150710). Cultivation 150708 and a control (ID 150714) were presented by relevant measurements in Fig. 3 a, b. The course of the scattered light bend showed to be linear within the first 1.5 h of protein production while heading towards saturation after 2 h. The maximum Δamp values of all three cultivations were comparable and ranged between 27,500 and 30,000 rel. AU. Towards the end of the protein production phase, Δamp was observed to decrease 8.8 h after inoculation. Very close to that event, an increase of DO from 0 to 43 % (ID 150708-150709) and 0 % to 25 % (ID 150710) was measured online (Fig. S9). The measurement of Δamp was verified with an established measurement method, i.e., SSC and FSC measurement. While SSC was increasing up to two-folds, FSC did not change significantly in the context of induction (Fig. 3 a, b). SDS-PAGE gels confirmed that a majority of the insoluble protein fraction was composed of IB, while no detectable concentration of soluble hLIF was found in the supernatant during cultivation at 37 °C (Fig. 4). A total of 160–170 mg L−1 of insoluble hLIF was reached in the cultivation broth 2.5 h after induction (Fig. 3 a). A linear correlation between the increases of Δamp and IB concentration could be revealed up to 160 mg L−1 (Fig. 5 a), whereas the correlation of SSC and IB was observed to be less significant in terms of a linearity within the investigated calibration range (Fig. 5 b). Regarding lower IB concentrations, a coefficient of determination of r = 0.99 was determined for the cultivation 150710 in the range of 0–125 mg L−1 hLIF.

Scattered light bend occurring after induction with IPTG during a cultivation of E. coli BL21(DE3) producing GFP-GST in TB-medium at 37 °C (ID 140115). The control (ID 140108) was time shifted +0.15 h specifically to enable a direct comparison

Scattered light bend occurring after induction with IPTG during a cultivation of E. coli BL21(DE3) producing hLIF (a) in defined medium ZYP-31 at 37 °C. Control without induction (b). SSC, FSC, OD600 measurement, and IB concentration for the induced cultivations is shown in the plot a. The first three parameters are shown for the control separately (b). The control 150714 was time shifted specifically to enable a direct comparison as follows: ID 150708 + 0.65 h

hLIF (34 kDa) production in E. coli BL21(DE3). An example of a SDS-PAGE gel of the insoluble protein fraction belonging to the cultivation 150709 samples t9-t17 (a), samples t18–t20 (b), and a second loading of selected samples. Soluble protein fraction of selected samples belonging to the cultivations ID 150709-150714 (c, d)

Correlation of Δamp and IB concentration (a), along with SSC and IB concentration (b) during hLIF production in E. coli BL21(DE3) grown in defined medium ZYP-31 at 37 °C
Online 180° scattered light analysis controls
Different controls were performed to examine effects which could have influenced the online scattered light measurement. (1) The same fraction volume of demineralized water (300–800 μL, depending on the present culture volume) was injected instead of IPTG (Fig. 3b). No bend of the signal was observed. (2) OD600 was measured in a 15-min interval before and after induction while no increase in growth rate could be stated after the induction (Fig. 2, Fig. 3a, b). Additionally, it could be noticed that 2 h after induction, OD600 was constant while scattered light was still slightly increasing (Fig. 3a, b). (3) Automatic addition of IPTG (final conc. = 2 mM, via syringe pump and capillary inlet) without automatic sampling was applied to a non-recombinant E. coli BL21 culture. No bend in scattered light signal occurred after IPTG addition (Fig. S14). (4) Automatic addition of IPTG without automatic sampling was applied to the strain E. coli BL21(DE3) producing hLIF. This led to the already described bend as well (Fig. S15).
Analysis of morphological changes during protein overexpression using HRSEM
Populations of induced and non-induced E. coli cells of the cultivation ID 150710 were immobilized onto SiO2 wafers for studying possible alterations in cell morphology. HRSEM images presented in Fig. 6 reveal significant alterations in the morphology of the cells in comparison to the control. The surface of the induced cells appears dented with some swelling at the poles. It should be noted that only a small portion (<3 %, n = 72) of these cells retained their original morphology. In contrast, the non-induced cells maintained their typical rod shape, while few irregular cells were observed among the non-induced cells (∼4 %, n = 123). The measurement of cell dimensions, based on analyzing HRSEM images, shows that the induced cells were significantly wider compared to non-induced cells (Table 2). In contrast, the mean cell length of septated cells did not change significantly. Elongated non-septated cells were found in both control (non-induced) and induced cultures (Fig. S10). Samples taken during cultivation and visualized by light microscopy also confirmed the presence of abnormal elongated cells before induction (Fig. S11).

SEM analysis of induced (3 h after induction) and non-induced E. coli BL21(DE3) cells producing hLIF. Example of 13 induced (a–b) and non-induced (c–d) cells immobilized on the top section of the silicon wafer
RIFTS analysis of porous SiPA optical biosensors
The EOT data is presented starting from the introduction time point of bacterial suspension onto the SiPA. Figure 7a depicts the change of the EOT as function of time in a representative experiment. Figure 7b shows a section of the SiPA with E. coli cells residing inside the pores. Thirty minutes after the IPTG induction point, the rate of EOT change is observed to upswing and deflect from the trend line associated with non-induced control culture. After 3 h cultivation, the extent of difference between ∆EOT of induced and control cultures reached ∼10 nm, representing a major alteration in the optical properties of the bacterial cells (Fig. 7a).

∆EOT plotted as function of time, for control culture and for IPTG induced culture (a). Representative SEM image of SiPA after 3 h experiment (b). White arrows indicate E. coli cells trapped inside the porous array
Discussion
Online 180° scattered light analysis, flow cytometry analysis for inclusion body detection
One key goal of this investigation was to find a simple optical method to monitor the formation of IB online for shake flask cultivations. During the performed experiments, a significant scattered light signal shift (Δamp) was observed in consequence of induction under conditions in which IB are preferentially produced. The temporal match in the course of Δamp and SCC indicates that Δamp is very likely to be correlated to a change in cell optical properties. More considerable technical factors which could have led to a signal shift were excluded by the controls. The increase of IB concentration based on densitometric determination is also supporting the results of online scattered light and SSC. The possibility of the formation of cell clusters which could have caused the increase in Δamp is not consistent with the observed constant FSC since it is known to increase in the case of cell cluster formation (Neumeyer et al. 2013). Flow cytometric analyses to detect IB in vivo were applied by different authors in combination with unspecific DNA and protein staining (Lewis et al. 2004; Medwid et al. 2007). The use of unspecific DNA and/or protein staining in order to discriminate cells from background and cell debris was also required in this study using flow cytometers of the same manufacturer. While Wittrup et al. (1988) also reported the IB associated increase of right angle light scattering (RALS) without using fluorescent dyes, other authors negated this possibility (Medwid et al. 2007; Wållberg et al. 2005).
The aforementioned decrease of Δamp 8.8 h after inoculation is very likely due to a slowdown in growth proved by OD600 measurement (“metabolic burden”) (Hoffmann and Rinas 2004). The growth saturation was also monitored indirectly by online DO measurement which showed an increase in the moment of signal saturation. This slowdown is relevant for data interpretation because the sensor does not discriminate between increasing scattered light caused by proliferation and changes in the optical properties of cells. Therefore, an appropriate reference for Δamp determination would not be a linear function but a “saturating” function in this case. So far, it was not possible to predict the growth behavior online with only one scattered light channel during IB formation. Nevertheless, the point of DO increase could be used as hint whether the estimation of IB concentration is still valid or not. In the context of online monitoring, it would be possible to automatize the described process presented in the section “Online scattered light shift analysis.” In this, a predefined function type (e.g., linear, polynomial) would be adapted continuously to the growth curve using an online algorithm (Nadungodage et al. 2011). The obligatory optimization algorithm to minimize least squares could be a simplex- or PSO (particle swarm optimization)-algorithm (Gálvez and Iglesias 2011). The type of function should be selected according to the growth curve determined in a previous control cultivation.
Online 180° scattered light analysis controls
(1) The addition of demineralized water instead of IPTG disproved the assumption that, e.g., the intake of additional oxygen (by the liquid) during an oxygen-limited phase of the cultivation may have caused an increase in growth rate. Furthermore, it was shown that neither a short stop of the shaker (approx. 2 min) nor the sampling process was causing a signal bend. (2) At-line measurement of OD600 proved that no increase of the growth rate took place after the induction. (3) 2 mM IPTG did not directly influence the growth behavior of the strain E. coli BL21. The non-toxic properties of IPTG at low concentrations were also reported by Lewis et al. (2004). (4) The bend of scattered light shortly after induction which was measured in the absence of sampling showed that this effect was not caused by the sampling process itself.
Analysis of morphological changes during protein overexpression using HRSEM
The observed morphological changes can be considered to relate with the increase of Δamp and SSC alongside with the increase of intracellular IB concentration. Alterations of cell morphology during overexpression of proteins were little investigated in the literature regarding E. coli. Nevertheless, different authors reported changes during the production of specific proteins. These changes comprise a slight increase in cell size (Castellanos-Mendoza et al. 2014), a larger fraction of non-dividing cells (Kyle et al. 2012), spherical cell formation (Fioroni et al. 2014), and elongated non-septated cells (Gubellini et al. 2011; Kyle et al. 2012). Apart from SDS-PAGE gels, the existence of intracellular IB could be supported by solid chunks arising from cells in which the turgor was completely collapsed, primarily found at the cell poles (Fig. S12). The same effect was already illustrated in HRSEM pictures of E. coli housing IB of HCcp3 which forms aggregates with the genomic DNA (Sun et al. 2013). The observed rubber boat-like cell shapes also correspond to the localization of IB found in literature, like the Aβ40 peptide determined via thioflavin-S staining (Espargaró et al. 2012). The deformation of the cells following their induction is suspected to be associated with physiological stress caused by the IB overexpression (Hoffmann and Rinas 2004; Gubellini et al. 2011). This stress may compromise the integrity of the cell wall, as several cells exhibit cytoplasm leakage while some completely collapsed (Fig. S12). Similar visual consequences were reported after intentional damage of E. coli’s cell wall using oil terpenes (Zengin and Baysal 2014). Regarding the interpretation of the determined cell dimensions, these collapsed cells could simulate the increase in cell width because of the flattening of the cell body on the surface of the SiPA biosensor. Some evidence for a constant cell size is given by a steady FSC signal (Hewitt and Nebe-Von-Caron 2004) and a constant FSC coefficient of variation (CV) during the complete cultivation (data not shown). This statement is based on the effect of the FSC CV to decrease when cells become more spheroidal (e.g., due to protein accumulation). The observed cell elongation is typically caused by mutants lacking in certain penicillin-binding proteins (PBP) in the presence of β-lactam antibiotics (Laubacher et al. 2013; Nelson and Young 2000; Rolinson 1980). Due to the existence of elongated non-septated cells before induction, there is no evidence to associate cell elongation with the formation of IB after induction. In summary, we suggest that IB concentration and cell shape are potential parameters responsible for causing the increase in Δamp and SSC. The increase of SSC or rather right angle light scatter (RALS) in consequence of an increasing intracellular concentration of IB was observed before by Wittrup et al. (1988). In this study, RALS was also given as explanation using the proteins VGH1 and VGH2 in opposition to a cell size increase. A more profound analysis of the cell size or bio-volume course during process could be realized by impedance measurement in addition to scattered light measurement (Kubitschek and Friske 1986; Wu et al. 2012).
RIFTS analysis of porous SiPA optical biosensors
Data in Fig. 7a suggest that bacteria induced for producing IB have a higher cellular RI compared to non-induced cells (control). Furthermore, if we consider that the proliferation rate of IB-producing cells is somewhat compromised relative to the control cells then the actual contribution of the IB to the cells’ RI may be higher. The increase in bacterium cellular RI is attributed to the formation of IB, as the latter consist of tightly packed protein molecules forming a dense matter. The RI of condensed proteins was reported to increase by the ratio dn = 0.19 dC where n is the RI and C is protein concentration (mg mL−1) (Margreiter et al. 2008; Crisman and Randolph 2009). Hence, we suggest that in control cultures, the gradual rise in EOT is exclusively caused by bacteria proliferation, while for bacteria producing IB, the more profound EOT increase is ascribed to the formation of highly dense protein aggregates within the cells.
In this work, we demonstrate that online measurement of 180° scattered light in shake flasks allows monitoring of changes in the optical properties of E. coli cells, associated with the formation of inclusion bodies (IB) of GFP-GST and hLIF. A reproducible linear correlation between the IB concentration and a shift (Δamp) in scattered light signal intensity was observed during protein production. These findings were supported by a significant increase in SSC starting shortly after induction using flow cytometry. Changes in cell shape going along with physiological stress caused by protein overexpression were also observed in this context. This parameter could be jointly responsible for the increasing shift in scattered light. Thus, further investigations should focus on the ability of the sensor to discriminate between interior structures and cell shape. A further advance in the development of this specific multisensor platform for shake flask cultivations is present in the integration of additional optical channels like near infrared (NIR) and UV-A. As NIR reflectance measurement is less sensitive to absorption of light by medium compounds and cytoplasm, changes in cell wall topology or intracellular complexity could be identified more effectively comparing the signal of different channels. We also present an additional novel approach for rapid screening and characterizing technique of sampled cell population for IB formation. Bacteria cells residing within SiPA are monitored in real time via RIFTS and upon IB induction. These studies reveal that the measured EOT is highly sensitive to the induction of IB; within 30 min post-induction, a significant increase in the EOT is observed in comparison to control cells (non-induced). This behavior is ascribed to the increase of the cellular RI during the formation of the highly packed protein domains within the cells. The advantages of photonic arrays for bacteria cell studies offer access to a rarely used but insightful cellular property, i.e., the RI, thus introducing a fundamental contribution to the study of intracellular bacterial processes. Optical methods for online monitoring of IB production are presented that can speed up process development and parameter optimization for recombinant protein production industry, therefore facilitating many screening processes.
References
Ami D, Natalello A, Taylor G, Tonon G, Maria Doglia S (2006) Structural analysis of protein inclusion bodies by Fourier transform infrared microspectroscopy. Biochim Biophys Acta BBA—Proteins Proteomics 1764:793–799. doi:10.1016/j.bbapap.2005.12.005
Bleasdale JKA, Nelder JA (1960) Plant population and crop yield. Nature 188:342–342. doi:10.1038/188342a0
Castellanos-Mendoza A, Castro-Acosta RM, Olvera A, Zavala G, Mendoza-Vera M, García-Hernández E, Alagón A, Trujillo-Roldán MA, Valdez-Cruz NA (2014) Influence of pH control in the formation of inclusion bodies during production of recombinant sphingomyelinase-D in Escherichia coli. Microb Cell Factories 13:137. doi:10.1186/s12934-014-0137-9
Crisman RL, Randolph TW (2009) Refolding of proteins from inclusion bodies is favored by a diminished hydrophobic effect at elevated pressures. Biotechnol Bioeng 102:483–492. doi:10.1002/bit.22082
Drezek R, Dunn A, Richards-Kortum R (1999) Light scattering from cells: finite-difference time-domain simulations and goniometric measurements. Appl Opt 38:3651. doi:10.1364/AO.38.003651
Espargaró A, Sabate R, Ventura S (2012) Thioflavin-S staining coupled to flow cytometry. A screening tool to detect in vivo protein aggregation. Mol Biosyst 8:2839. doi:10.1039/c2mb25214g
Fioroni M, Dworeck T, Rodriguez-Ropero F (2014) ß-barrel channel proteins as tools in nanotechnology. Springer Netherlands, Dordrecht
Gálvez A, Iglesias A (2011) Efficient particle swarm optimization approach for data fitting with free knot-splines. Comput Aided Des 43:1683–1692. doi:10.1016/j.cad.2011.07.010
Gubellini F, Verdon G, Karpowich NK, Luff JD, Boel G, Gauthier N, Handelman SK, Ades SE, Hunt JF (2011) Physiological response to membrane protein overexpression in E. coli. Mol Cell Proteomics 10:M111.007930–M111.007930. doi:10.1074/mcp.M111.007930
Hedhammar M, Stenvall M, Lönneborg R, Nord O, Sjölin O, Brismar H, Uhlén M, Ottosson J, Hober S (2005) A novel flow cytometry-based method for analysis of expression levels in Escherichia coli, giving information about precipitated and soluble protein. J Biotechnol 119:133–146. doi:10.1016/j.jbiotec.2005.03.024
Hewitt CJ, Nebe-Von-Caron G (2004) The application of multi-parameter flow cytometry to monitor individual microbial cell physiological state. In: Physiological stress responses in bioprocesses. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp. 197–223
Hoffmann F, Rinas U (2004) Stress induced by recombinant protein production in Escherichia coli. In: Physiological stress responses in bioprocesses. Springer Berlin Heidelberg, Berlin, Heidelberg, 73–92
Ignatova Z, Gierasch LM (2004) Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc Natl Acad Sci 101:523–528. doi:10.1073/pnas.0304533101
Jones JJ, Bridges AM, Fosberry AP, Gardner S, Lowers RR, Newby RR, James PJ, Hall RM, Jenkins O (2004) Potential of real-time measurement of GFP-fusion proteins. J Biotechnol 109:201–211. doi:10.1016/j.jbiotec.2003.10.039
Kensy F, Zang E, Faulhammer C, Tan R-K, Büchs J (2009) Validation of a high-throughput fermentation system based on online monitoring of biomass and fluorescence in continuously shaken microtiter plates. Microb Cell Factories 8:31. doi:10.1186/1475-2859-8-31
Kraft M, Knüpfer U, Wenderoth R, Pietschmann P, Hock B, Horn U (2007) An online monitoring system based on a synthetic sigma32-dependent tandem promoter for visualization of insoluble proteins in the cytoplasm of Escherichia coli. Appl Microbiol Biotechnol 75:397–406. doi:10.1007/s00253-006-0815-6
Kubitschek HE, Friske JA (1986) Determination of bacterial cell volume with the Coulter Counter. J Bacteriol 168:1466–1467
Kyle S, James KAR, McPherson MJ (2012) Recombinant production of the therapeutic peptide lunasin. Microb Cell Factories 11:28. doi:10.1186/1475-2859-11-28
Laubacher ME, Melquist AL, Chandramohan L, Young KD (2013) Cell sorting enriches Escherichia coli mutants that rely on peptidoglycan endopeptidases to suppress highly aberrant morphologies. J Bacteriol 195:855–866. doi:10.1128/JB.01450-12
Lewis G, Taylor IW, Nienow AW, Hewitt CJ (2004) The application of multi-parameter flow cytometry to the study of recombinant Escherichia coli batch fermentation processes. J Ind Microbiol Biotechnol 31:311–322. doi:10.1007/s10295-004-0151-8
Li Z, Kessler W, van den Heuvel J, Rinas U (2011) Simple defined autoinduction medium for high-level recombinant protein production using T7-based Escherichia coli expression systems. Appl Microbiol Biotechnol 91:1203–1213. doi:10.1007/s00253-011-3407-z
Margreiter G, Messner P, Caldwell KD, Bayer K (2008) Size characterization of inclusion bodies by sedimentation field-flow fractionation. J Biotechnol 138:67–73. doi:10.1016/j.jbiotec.2008.07.1995
Massad-Ivanir N, Mirsky Y, Nahor A, Edrei E, Bonanno-Young LM, Ben Dov N, Sa’ar A, Segal E (2014) Trap and track: designing self-reporting porous Si photonic crystals for rapid bacteria detection. Analyst 139:3885. doi:10.1039/C4AN00364K
Medwid R, Krebs L, Welch S (2007) Evaluation of Escherichia coli cell disruption and inclusion body release using nucleic acid binding fluorochromes and flow cytometry. Biotechniques 43:777–782. doi:10.2144/000112621
Mirsky Y, Nahor A, Edrei E, Massad-Ivanir N, Bonanno LM, Segal E, Sa’ar A (2013) Optical biosensing of bacteria and cells using porous silicon based, photonic lamellar gratings. Appl Phys Lett 103:033702. doi:10.1063/1.4813740
Nadungodage CH, Xia Y, Li F, Lee JJ, Ge J (2011) StreamFitter: a real time linear regression analysis system for continuous data streams. In: Yu JX, Kim MH, Unland R (eds) Database Systems for advanced applications. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp. 458–461
Nelson DE, Young KD (2000) Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol 182:1714–1721. doi:10.1128/JB.182.6.1714-1721.2000
Nemecek S, Marisch K, Juric R, Bayer K (2008) Design of transcriptional fusions of stress sensitive promoters and GFP to monitor the overburden of Escherichia coli hosts during recombinant protein production. Bioprocess Biosyst Eng 31:47–53. doi:10.1007/s00449-007-0143-y
Neumeyer A, Hübschmann T, Müller S, Frunzke J (2013) Monitoring of population dynamics of Corynebacterium glutamicum by multiparameter flow cytometry: population dynamics of Corynebacterium glutamicum. Microb Biotechnol 6:157–167. doi:10.1111/1751-7915.12018
Patkar A, Vijayasankaran N, Urry DW, Srienc F (2002) Flow cytometry as a useful tool for process development: rapid evaluation of expression systems. J Biotechnol 93:217–229
Rolinson GN (1980) Effect of beta-lactam antibiotics on bacterial cell growth rate. J Gen Microbiol 120:317–323
Schmidt-Hager J, Ude C, Findeis M, John GT, Scheper T, Beutel S (2014) Non-invasive online biomass detector system for cultivation in shake flasks. Eng Life Sci. doi:10.1002/elsc.201400026
Sun S, Liu M, Dong F, Fan S, Yao Y (2013) A histone-like protein induces plasmid DNA to form liquid crystals in vitro and gene compaction in vivo. Int J Mol Sci 14:23842–23857. doi:10.3390/ijms141223842
Tomala M, Lavrentieva A, Moretti P, Rinas U, Kasper C, Stahl F, Schambach A, Warlich E, Martin U, Cantz T, Scheper T (2010) Preparation of bioactive soluble human leukemia inhibitory factor from recombinant Escherichia coli using thioredoxin as fusion partner. Protein Expr Purif 73:51–57. doi:10.1016/j.pep.2010.04.002
Ude C, Schmidt-Hager J, Findeis M, John GT, Scheper T, Beutel S (2014) Application of an online-biomass sensor in an optical multisensory platform prototype for growth monitoring of biotechnical relevant microorganism and cell lines in single-use shake flasks. Sensors 14:17390–17405. doi:10.3390/s140917390
Upadhyay P, Patra A, Mukhopadhyay R, Panda A (2001) Real time detection and quantification of inclusion bodies expressed in Escherichia coli by impedance measurements. Biotechnol Lett 23:839–843. doi:10.1023/A:1010550015147
Wållberg F, Sundström H, Ledung E, Hewitt CJ, Enfors S-O (2005) Monitoring and quantification of inclusion body formation in Escherichia coli by multi-parameter flow cytometry. Biotechnol Lett 27:919–926. doi:10.1007/s10529-005-7184-6
Wittrup KD, Mann MB, Fenton DM, Tsai LB, Bailey JE (1988) Single-cell light scatter as a probe of refractile body formation in recombinant Escherichia coli. Nat Biotechnol 6:423–426. doi:10.1038/nbt0488-423
Wu Y, Benson JD, Almasri M (2012) Micromachined Coulter counter for dynamic impedance study of time sensitive cells. Biomed Microdevices 14:739–750. doi:10.1007/s10544-012-9655-6
Yablonovitch E (1987) Inhibited spontaneous emission in solid-state physics and electronics. Phys Rev Lett 58:2059–2062. doi:10.1103/PhysRevLett.58.2059
Zengin H, Baysal A (2014) Antibacterial and antioxidant activity of essential oil terpenes against pathogenic and spoilage-forming bacteria and cell structure-activity relationships evaluated by SEM microscopy. Molecules 19:17773–17798. doi:10.3390/molecules191117773
Acknowledgments
We would like to thank all members of our research groups for providing feedback and suggestions. This work was partially supported by the Russell Berrie Nanotechnology Institute (RBNI) and the Lorry I. Lokey Interdisciplinary Center for Life Sciences and Engineering (LS&E). SiPA samples were prepared at the Micro-Nano Fabrication Unit (MNFU), Technion.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
No research involving human participants and/or animals was carried out.
Funding
This study was supported by the Bundesministerium für Wirtschaft und Technologie via Arbeitsgemeinschaft industrieller Forschungsvereinigungen “Otto von Guericke” e. V. within the Zentrale Innovationsoffensive Mittelstand-initiative. Additional funding for this study was provided by the Deutsche Technion-Gesellschaft.
Consent to submit
The authors declare that they agree each with submission of this manuscript. The authors declare that the manuscript has not been submitted to more than one journal for simultaneous consideration.
Electronic supplementary material
ESM 1
(PDF 29087 kb)
Rights and permissions
About this article

Cite this article
Ude, C., Ben-Dov, N., Jochums, A. et al. Online analysis of protein inclusion bodies produced in E. coli by monitoring alterations in scattered and reflected light. Appl Microbiol Biotechnol 100, 4147–4159 (2016). https://doi.org/10.1007/s00253-016-7403-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7403-1