
Abstract
Snake venom serine proteases (SVSPs) act primarily on plasma proteins related to blood clotting and are considered promising for the treatment of several hemostatic disorders. We report the heterologous expression of a serine protease from Crotalus durissus collilineatus, named collinein-1, in Pichia pastoris, as well as the enzymatic comparative characterization of the toxin in native and recombinant forms. The complementary DNA (cDNA) encoding collinein-1 was amplified from cDNA library of C. d. collilineatus venom gland and cloned into the pPICZαA vector. The recombinant plasmid was used to transform cells of KM71H P. pastoris. Heterologous expression was induced by methanol and yielded 56 mg of recombinant collinein-1 (rCollinein-1) per liter of culture. The native collinein-1 was purified from C. d. collilineatus venom, and its identity was confirmed by amino acid sequencing. The native and recombinant enzymes showed similar effects upon bovine fibrinogen by releasing preferentially fibrinopeptide A. Although both enzymes have induced plasma coagulation, native Colinein-1 has shown higher coagulant activity. The serine proteases were able to hydrolyze the chromogenic substrates S-2222, S-2238, and S2302. Both enzymes showed high stability on different pH and temperature, and their esterase activities were inhibited in the presence of Zn2+ and Cu2+. The serine proteases showed similar k cat/K m values in enzyme kinetics assays, suggesting no significant differences in efficiency of these proteins to hydrolyze the substrate. These results demonstrated that rCollinein-1 was expressed with functional integrity on the evaluated parameters. The success in producing a functionally active recombinant SVSP may generate perspectives to their future therapeutic applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Snake venom serine proteases (SVSPs) are considered promising for the treatment of certain hemostatic disorders because of their ability to mimic natural regulatory components, selectively activating or inactivating some factors involved in platelet aggregation, coagulation, and fibrinolysis (Braud et al. 2000).
Snake venom thrombin-like enzymes (SVTLEs) are the prevalent class of serine proteases from Viperidae venoms. SVTLEs have similar activity to that of human thrombin, promoting in vitro blood coagulation. However, thrombin-like enzymes turn the blood incoagulable in vivo by cleaving fibrinogen, without activating factor XIII, that is responsible for the formation of cross-links between fibrin monomers. Thus, only a loose clot is formed, which can be easily degraded by the fibrinolytic system, leading to depletion of plasma fibrinogen (Hutton and Warrel 1993; Serrano and Maroum 2005; Kini 2006).
Thrombin-like enzymes cleave Aα and/or Bβ chain of fibrinogen, releasing fibrinopeptides A (FPA) and/or B (FPB), respectively. Less common than the enzymes that cleave preferably one of fibrinogen chains, some SVTLEs cleave both chains, releasing FPA and FPB in similar proportions (Pirkle 1998). Hence, SVTLEs can be classified as (i) FP-A that preferentially cleaves fibrinogen Aα chain, (ii) FP-B that preferentially cleaves fibrinogen Bβ chain, and (iii) FP-AB, which, as well as thrombin, lead to blood clotting by cleaving both fibrinogen chains (Castro et al. 2004).
Thrombin-like enzymes have been extensively studied, both in basic research and in clinical studies, because of their potential use in the treatment of myocardial infarction, ischemic stroke, among other vascular disorders (Escoubas et al. 2008). Ancrod and batroxobin are the most known examples of SVTLEs that have been used for therapeutic purposes. Batroxobin (Defibrase®, Pentapharm-CH) is a thrombin-like enzyme isolated from Bothrops atrox venom that has been used in patients with deep vein thrombosis, myocardial infarction, angina, ischemia, and among others. Ancrod (Viprinex®, Knoll, Ludwigshafen, Germany), isolated from Calloselasma rhodostoma, was initially indicated for the treatment of ischemic stroke, and its efficiency and safety have been evaluated in recent years (Mackessy 2010). However, ancrod was disapproved in stage III of clinical trials for the treatment of ischemia and is now no longer produced for use in humans with this kind of coagulopathy (Levy et al. 2009).
Despite the great therapeutic potential of SVTLEs, there is still a discrepancy between the vast number of proteins that has been isolated and characterized and the total of proteins that have become effectively a drug. The major difficulty in applied research is the limitations in obtaining toxins to a detailed structural and functional characterization (Vetter et al. 2011). Thus, heterologous protein expression represents an alternative to produce these molecules in both laboratory and industrial scales, what may generate perspectives for their therapeutic application.
SVSPs have been expressed in various systems, such as bacteria (Maeda et al. 1991; Fan et al. 1999; Dekhil et al. 2003, Yang et al. 2003; Zha et al. 2003; Lin et al. 2009; Jiang et al. 2010), yeasts (You et al. 2004; Muanpasitporn and Rojnuckari 2007; Yang et al. 2009), and mammalian cells (Yonamine et al. 2009). However, expression of recombinant SVSPs with functional integrity without refolding processes is only achieved in eukaryotic systems since they are glycoproteins rich in disulfide bonds (Vetter et al. 2011). In this context, we report the production of a recombinant serine protease in Pichia pastoris system, as well as the comparative enzymatic characterization of the native and recombinant proteins in order to validate the heterologous expression process.
Materials and methods
Venom and materials
Crotalus durissus collilineatus venom was extracted from specimens collected in the state of Goiás, Brazil. After extraction, the venom was dried under vacuum at room temperature and stored at −20 °C. The snakes were kept in captivity at the Serpentarium Bioagents of Batatais—SP, Brazil. P. pastoris KM71H strain, pPICZαA expression plasmid, and Zeocin™ were purchased from Invitrogen™ (Carlsbad, USA). Restriction enzymes used in gene cloning and plasmid linearization were purchased from New England Biolabs (Ipswich, USA). Chromogenic substrates were purchased from Chromogenix (Instrumentation Laboratory, Bedford, USA). Bovine fibrinogen (>90 % of clottable protein) and N-p-toluenesulfonyl arginine methyl ester substrate (TAME) were from Sigma-Aldrich (St. Louis, USA). Other reagents used in this study but not specified were all of analytical grade. Other materials and equipment used were described throughout the methodology.
Construction of recombinant expression vector
The clone corresponding to the serine protease was previously obtained from the complementary DNA (cDNA) library of C. d. collilineatus venom gland (Boldrini-França et al. 2009), and its cDNA sequence is available on European Nucleotide Archive database (http://www.ebi.ac.uk/ena/data/view/LN651356). The full-length cDNA encoding mature collinein-1 was amplified by polymerase chain reaction (PCR) with specific forward (5′-CCGCTCGAGAAAAGAGTCATTGGAGGAGATGAATG-3′) and reverse (5′-AAATATGCGGCCGCCGAGGGGCAATTCACAGTTTC-3′) primers. The PCR product was cloned into XhoI and NotI restriction sites of the pPICZαA expression plasmid. The correct in-frame connection of the insert was confirmed by sequencing using ET Dye Terminator kit in the equipment MegaBACE 1000—automated DNA sequencer (GE Healthcare, Chalfont St. Giles, UK).
Transformation of Pichia pastoris
The resulting recombinant plasmid, named pPICZαA-collinein1, was linearized with SacI, and transformed into P. pastoris KM71H cells by electroporation in the equipment GenePulser II (Bio-Rad Laboratories, Hercules, USA) at 1500 V, 25 mF, and 200 Ω. The same procedure was performed with the plasmid without insert, for use as a negative control of expression. After transformation, cells were plated onto YPDS agar medium (1 % yeast extract, 2 % peptone, 2 % dextrose, 1 M sorbitol, 2 % agar) containing 100 mg/mL of Zeocin and incubated at 30 °C for 3–5 days for selection of transformants. For the selection of recombinant colonies containing multiple copies of the gene, cells (200 μL) were plated on YPDS medium containing 500 g/mL Zeocin and incubated under the conditions described above. The recombinant colonies were subjected to colony PCR, and the product was visualized on a 1 % agarose gel to confirm that the insert was incorporated in the yeast genomic DNA.
Screening of secreting clones and optimum expression conditions
The expression of the rCollinein-1 was carried out as described in the Easy Select™ Pichia Expression manual (Invitrogen™, Carlsbad, USA), with some modifications. Positive clones, selected by colony PCR, were individually inoculated in a 24-deep-well plate containing 3 mL of BMGY medium (1 % yeast extract, 2 % peptone, 1.34 % YNB, 4 × 10–5 biotin, 1 % glycerol, 100 mM phosphate potassium) per well. As a negative control, a clone containing the empty vector pPICZαA was inoculated in the same conditions. The screening plate was incubated at 30 °C under constant stirring of 250 rpm. After 48 h, the cultures were centrifuged at 1500×g, the supernatant was discarded, and cells were resuspended in 2 mL of BMMY medium (1 % yeast extract, 2 % peptone, 1.34 % YNB, 4 × 10–5 % biotin, 1 % methanol, 100 mM potassium phosphate) and incubated at 26 °C under constant stirring to induce expression. For the maintenance of induction, methanol was replaced every 24 h at a final concentration of 0.75 %. Aliquots were collected after 96 h of induction, and the culture supernatants were analyzed by SDS-PAGE 13.5 % stained with silver to determine heterologous expression levels. The clone showing the highest expression level of the recombinant protein was submitted to a further screening of optimal pH and media composition for protein expression. For this, the colony was inoculated in the same conditions described above, except that, after 48 h of biomass generation, cells were centrifuged and resuspended in the following conditions: (i) buffered complex media in different pHs (BMMY, pH 4.0, 6.0, and 7.0); (ii) buffered minimal media in different pHs [BMM (1.34 % YNB, 4 × 10–5 biotin, 1 % methanol, 100 mM potassium phosphate), pH 4.0, 6.0, and 7.0]; and (iii) non-buffered minimal medium [MM (1.34 % YNB, 4 × 10–5 biotin, 1 % methanol)]. After 96 h of induction, culture supernatants were analyzed by 13.5 % SDS-PAGE stained with silver. The conditions that resulted in the higher expression level of the recombinant protein were used to scale up protein expression.
Scale up expression of rCollinein-1
The selected clone was pre-inoculated with 10 mL of BMGY medium and incubated at 30 °C under constant stirring of 250 rpm. After 24 h, the culture was inoculated in 500 mL of BMGY medium and incubated at 30 °C under constant stirring of 250 rpm until reaching an optical density of 2 to 6 in 600 nm. After achieving the desired optical density, the culture was centrifuged at 1500×g, the supernatant was discarded, and the cells were resuspended in 100 mL of BMM medium (1.34 % YNB, 4 × 10–5 biotin, 1 % methanol, 100 mM potassium phosphate pH 7.0) and incubated at 26 °C under constant stirring of 250 rpm. Methanol was replaced every 24 h at a final concentration of 0.75 %, for the maintenance of induction. After 96 h of induction, the culture was centrifuged at 10,000×g and the supernatant was separated, filtered and used for purification of recombinant protein.
Purification of recombinant collinein-1
The recombinant collinein-1 was purified by combining ion-exchange and reverse phase chromatography, accomplished on a fast protein liquid chromatography system (FPLC) Äkta Purifier UPC10 (GE Healthcare, Chalfont St. Giles, UK). The medium supernatant was previously incubated with Tris-HCl 0.02 M, pH 8.0, containing 7 M urea in a ratio of 1:5 (v/v) for 90 min at room temperature. Then, the supernatant was applied in a MonoQ ion exchange column 5/50 GL (GE Healthcare, Chalfont St. Giles, UK), previously equilibrated with Tris-HCl 0.02 M, pH 8.0. Elution was performed with a discontinuous gradient to 1 M NaCl at a flow rate of 1 mL/min. The fraction presenting coagulant activity was subjected to reverse phase C4 column (Shodex™, Munich, Germany), pre-equilibrated with solvent A (0.1 % trifluoroacetic acid). The sample components were eluted with a linear gradient to 100 % of solution B (0.1 % trifluoroacetic acid to 80 % acetonitrile) at a flow rate of 0.8 ml/min. The purity of the recombinant protein was verified by the symmetry of chromatographic peak and polyacrylamide gel electrophoresis (SDS-PAGE) stained with silver. The peak corresponding to the purified rCollinein-1 was frozen at −80 °C, lyophilized and stored at −20 °C for subsequent assays.
Purification of native collinein-1
Purification of native collinein-1 from C. d. collilineatus crude venom was performed by combining gel filtration, ion-exchange and reverse phase chromatographic techniques. All chromatographies were carried out on Fast Protein Liquid Chromatography System (FPLC) Äkta Purifier UPC10 (GE Healthcare, Chalfont St. Giles, UK). Crude venom (50 mg) was dispersed in ammonium formate buffer 0.05 M, pH 3.5, followed by addition of 10 μL of formic acid (FA) to achieve a total solubilization of proteins. The sample was centrifuged at 13,000×g for 10 min, and the supernatant was applied on a Sephacryl S100 HR column (GE Healthcare, Chalfont St. Giles, UK), pre-equilibrated with the same buffer. Elution of venom components was monitored by spectrophotometry at 280 nm, in a flow rate of 0.5 mL/min. Fractions presenting clotting activity were pooled and applied to MonoQ ion exchange column 5/50 GL (GE Healthcare, Chalfont St. Giles, UK), previously equilibrated with Tris-HCl 0.02 M, pH 8.0. Elution was performed with a discontinuous gradient to 1 M of NaCl at a flow rate of 1 mL/min. Fractions with coagulant activity, obtained from ion exchange chromatography, were pooled and subjected to reverse phase chromatography in a C4 column (Shodex™, Munich, Germany), pre-equilibrated with solvent A (0.1 % trifluoroacetic acid). Elution was performed by a stepwise gradient to 100 % solution B (0.1 % trifluoroacetic acid to 80 % acetonitrile) with a flow rate of 0.8 mL/min. The purity of the native protein was verified by the symmetry of chromatographic peak and polyacrylamide gel electrophoresis (SDS-PAGE) stained with silver. Peaks corresponding to purified proteins were frozen at −80 °C, lyophilized, and stored at −20 °C for subsequent assays.
Amino-terminal sequencing
The amino-terminal sequence of the first 58 amino acid residues of native collinein-1 was obtained by Edman degradation method of protein sequencing (Edman and Begg 1967). Purified proteins were directly applied in a glass fiber membrane (Wako Pure Chemicals Industries, Osaka, Japan) and subjected to analysis in a PPSQ-33A automatic protein sequencer (Shimadzu, Columbia, USA). PTH-amino acids were chromatographed on a reverse phase column with isocratic gradient. The quantification and identification of amino acid residues were carried out by comparing the retention times of the standard amino acids (25 pmol), chromatographed before sequencing.
Molecular mass determination
Molecular masses of proteins were determined by mass spectrometry in a SYNAPT G2-S instrument (Waters, Milford, USA), equipped with a nano-electrospray ionization source (nano-ESI), and previously calibrated with sodium iodated. Before the experiment, samples were purified using a C18 Zip-Tip microcolumn (Millipore, Billerica, USA) for eliminating most of the salts and other possible interferences. The elution from the column was made using 10 μL of a mixture of acetonitrile/formic acid 0.1 50/50 (v/v). Four microliter of the purified solution was loaded into a borosilicate emitters (Thermo Fisher Scientific, Waltham, USA). Ionization was performed using a voltage of 2 kV applied on the capillary and 20 V on the inlet cone. The estimated flow rate was 200 nL/min. Spectra were processed (background reduction, smoothing, peak picking) using the software Mass Lynx 4.1 (Waters, Milford, USA).
Peptide mass fingerprinting and de novo sequencing
About 3 μg of protein was reduced during 40 min with 10 mM of dithiothreitol (DTT) at 56 °C and alkylated during 30 min with 20 mM of iodoacetamide at room temperature, in the dark. After alkylation, iodoacetamide was inactivated by incubating the sample for 10 min with DTT at a final concentration of 21 mM. Proteins were digested with trypsin at a final enzyme/substrate concentration of 1:50 (w/w) for 16 h at 37 °C. A second digestion was performed by adding trypsin to a final enzyme/substrate concentration of 1:100 (w/w) and acetonitrile (ACN) to a final concentration of 80 % (v/v). The samples were then incubated at 37 °C for 3 h, and the reaction was stopped by adding formic acid (FA) to a final concentration of 0.2 % (v/v). Solutions containing tryptic peptides (1 μL) were spotted with 1 μL of α-cyano-4-hydroxycinnamic acid (CHCA) (10 mg/mL in 0.2 % FA and 50 % ACN). Samples were analyzed by MALDI-TOF/TOF in an Ultraflex II (Bruker Daltonics, Billerica, USA) mass spectrometer, equipped with a SmartBeam laser. The laser power was adjusted to 35 % with an incidence of 500–1000 shots, in order to generate a satisfactory signal/noise ratio (S/N >3). For ion detection, the analyzer was previously calibrated with Peptide Standard II from Bruker Daltonics (Billerica, USA) (m/z 700–3200) and operated in positive reflected mode. Peptides were also fragmented and analyzed in an amaZon Ion Trap mass spectrometer (Bruker Daltonics, Billerica, USA). The enzymes were subjected to reduction, alkylation, and digestion, as described above, and the resulting peptides (2 μL) were diluted in 9 μL of a solution containing 0.1 % FA and 50 % ACN (v/v). The solution containing peptides was ionized by electrospray, and triply charged peptides were subjected to fragmentation by electron transfer dissociation (ETD). MS spectra were analyzed by FlexAnalysis 3.0 and BioTools 3.1 software (Bruker Daltonics, Billerica, USA) for the deduction of the amino acid sequence.
Accession numbers of nucleotide and amino acid sequence data
The nucleotide sequence data reported in this paper will be available from the European Nucleotide Archives (ENA) browser at http://www.ebi.ac.uk/ena/data/view/LN651356 (accession number LN651356) and the protein sequence data will appear in the UniProt Knowledgebase under the accession numbers C0HJR5 (collinein-1) and C0HJR6 (collinein-2).
Multiple sequence alignments
Multiple alignment of the partial amino acid sequences of collinein-1 and 2 was performed by MultiAlin algorithm (Corpet 1988). The translated amino acid sequence of collinein-1 (based on its cDNA encoding sequence) was used to predict the probable positions of tryptic peptides. This sequence was also aligned with nine homologous SVSPs from GenBank and UniProtKB/Swiss-Prot using ClustalW2 program (Larkin et al. 2007).
Fibrinogenolytic activity
The proteolytic activity upon fibrinogen was evaluated as described by Rodrigues et al. (2000). Bovine fibrinogen solution (50 μL, 1.5 mg/mL) was incubated with 10 μg of protein at 37 °C for different time intervals (5, 15, 30, 60, 120, and 240 min). Fibrinogen solution without enzymes was incubated under the same conditions and used as a negative control. Reactions were stopped with 25 μL of stop solution (Tris-HCl 0.06 M, pH 6.8, 0.001 % bromophenol blue, 10 % glycerol, and 10 % β-mercaptoethanol). Samples were heated at 100 °C for 10 min and analyzed by electrophoresis in polyacrylamide gel with SDS 13.5 % (SDS-PAGE) stained with Coomassie Blue R-350.
Identification of fibrinopeptides
Fibrinopeptides originated by proteolytic activity of the serine proteases were identified as described by Magalhães el al. (2003), with some modifications. Fibrinopeptides were generated by incubating the enzymes (10 μg) with 1 mL of bovine fibrinogen solution (3 mg/ml in Tris-HCl 0.05 M, pH 7.5, containing 0.05 M CaCl2), at 37 °C for different time intervals (5, 30, 60, and 120 min). Thrombin was incubated with fibrinogen for 120 min as a positive control, and fibrinogen solution without enzyme was used as negative control. Insoluble proteins were precipitated with 2 % trichloroacetic acid (TCA), and the supernatant was subjected to reverse phase chromatography on a Grace™ Vydac™ C18 column (0.46 × 25 cm) (Thermo Fisher Scientific, Waltham, USA), pre-equilibrated with solvent A (0.1 % trifluoroacetic acid). Fibrinopeptides were eluted by a segmented gradient of acetonitrile (0–15 % in 5 column volumes and 15–30 % in 15 column volumes).
Coagulant activity
The assay was performed as described by Assakura et al. (1992), with some modifications. To determine the minimum coagulant dose (MCD), different amounts of enzyme (10, 15, 20, 30, and 40 μg) were applied in 150 μL of citrated bovine plasma, and the time of fibrin clot formation was determined by the photometric coagulometer Quick Timer II (Drake, São José do Rio Preto, BR). The MCD was defined as the amount of enzyme required to coagulate the bovine-citrated plasma in 60 s at 37 °C and was estimated by logarithmic regression of the dose-response curve.
Proteolytic activity upon chromogenic substrates
The proteolytic activity of serine proteases was evaluated with the following chromogenic substrates: S-2251 for plasmin (H-D-Val-Leu-Lys-pNA•2HC), S-2222 for factor Xa [Bz-IIe-Glu(γ-OR)-Gly-Arg-pNA•HCl], S2238 for thrombin (H-D-Phe-Pip-Arg-pNA•2HCl), and S-2302 for plasma kallikrein (H-D-Pro-Phe-Arg-pNA•2HCl). Substrate hydrolysis was determined by incubating the enzymes (4 μg) with 0.4 mM of substrates in Tris-HCl 0.05 M, pH 7.5, containing 0.05 M CaCl2 for 40 min at 37 °C. Substrate hydrolysis was spectrophotometrically monitored at 405 nm using Versa Max Microplate reader (Molecular Devices, Sunnyvale, USA). Assays were performed in series of three replicates, and the data were fitted with standard errors using GraphPad Prism software, version 5.0 (GraphPad Software, San Diego, USA).
Enzyme kinetics
The kinetic parameters were determined by monitoring the esterase activity of the enzymes upon N-p-toluenesulfonyl arginine methyl ester (TAME) substrate, according to Hummel (1959). Enzymes (5 μg) were incubated with different concentrations of TAME (0.125 to 2 mM) for 30 min at 37 °C, and the activity was monitored at 247 nm. The enzymatic activity (V 0) was expressed in millimole of product formed per minute, considering the molar extinction coefficient of 540 M−1 cm−1 for the product. The K m and V max values were determined from the hyperbolic kinetic curve of Michaelis-Menten. The turnover (k cat) and catalytic efficiency (k cat/K m) values were calculated from the K m and V max. The experiments were performed in a series of three repetitions each, and the data were fitted with the standard errors using the software GraphPad Prism, version 5.0 (GraphPad Software, San Diego, USA).
Enzyme stability assay
The stability of the serine proteases was evaluated by pre-incubating the enzymes at different temperatures (25, 37, 45, 60, 80 and 100 °C) or pH buffers (3.5, 5.5, 7.5, 9.5 and 10,5) for 30 min, followed by the evaluation of enzymatic activity upon TAME, as described in “Enzyme kinetics” section. One unit of esterase activity (U) was defined as the increase of 0.01 units of absorbance at 247 nm, and the results were expressed as units per milligram of protein.
Influence of divalent ions on esterase activity
The effect of divalent cations on the esterase activity of the serine proteases was evaluated by pre-incubating the enzymes with 10 mM of different ions (Ca2+, Cu2+, Ba2+, Zn2+, and Mg2+) for 30 min at 37 °C. The esterase activity was evaluated as described in “Enzyme kinetics” section. The esterase activity of the enzymes without divalent cations was used as a control. One unit of esterase activity (U) was defined as the increase of 0.01 units of absorbance at 247 nm, and the results were expressed as units per milligram of protein.
Statistical analysis
The statistical analysis of variance (ANOVA) and Tukey test were performed using the software GraphPad Prism, version 5.0 (GraphPad Software, San Diego, USA), with significance level of 5 % (p < 0.05).
Results
Heterologous expression and purification of rCollinein-1
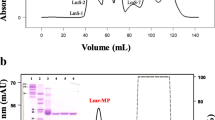
The cDNA encoding the serine protease, named collinein-1, was deposited on European Nucleotide Archive (access number LN651356). The ORF encodes a protein of 262 amino acid residues, including a signal peptide (18 amino acid residues), a pro-peptide (6 amino acid residues), and a serine protease domain with 238 amino acid residues. All P. pastoris colonies transformed with the cDNA encoding mature collinein-1 showed efficiency in expressing and secreting the soluble recombinant protein after 96 h of methanol induction. The clone that secreted the highest amount of rCollinein-1 was selected for screening of optimal expression conditions. The highest level of recombinant protein expression was achieved at pH 7.0, whereas at pH 4.0, it was not possible to observe the presence of the recombinant protein in culture supernatants. On the other hand, addition of yeast extract and peptone to the culture medium did not affect the expression of the recombinant collinein-1, since this protein is present at similar levels in cells grown both in minimal and complex media (data not shown). The optimal expression conditions were utilized to scale up rCollinein-1 expression. The recombinant protein was purified from the culture supernatant by a combination of ion exchange and reverse-phase chromatography (Fig. 1), resulting in a yield of 56 mg of rCollinein-1 per liter of culture, with a recovery rate of 15.38 % of esterase activity (Table 1). When analyzed by mass spectrometry, the recombinant protein presented a molecular mass of 28,868 Da.

Purification of rCollinein-1. a Ion exchange chromatography of culture medium in a MonoQ 5/50 GL Column (GE Healthcare, UK), previously equilibrated with Tris-HCl 0.02 M, NaCl 1 M, pH 8.0. Elution was performed in a discontinuous gradient of NaCl 1 M. b Reverse phase of fraction 6 in a C4 column (Shodex, Japan). Elution was performed by a stepwise gradient of solution B (0.1 % TFA, 80 % ACN). Inserted panel: SDS-PAGE (13.5 %) stained with silver of culture supernatant (1), fraction 6 from ion exchange chromatography, (2) and purified rCollinein-1 (3). All chromatographic steps were accomplished on a fast protein liquid chromatography system (FPLC) Äkta Purifier UPC10 (GE Healthcare, UK). When analyzed by mass spectrometry, rCollinein-1 presented a molecular mass of 28,868 Da
Purification of native collinein-1
The first chromatographic step on a size exclusion column resulted in six eluted fractions, named S1 to S6 (Fig. 2a). Peak S3 that showed coagulant activity resulted in seven fractions (Q1—Q7) when chromatographed in an ion exchange column (Fig. 2b). Fraction Q4 presented coagulant activity and, when submitted to a reverse phase chromatography, resulted in two sharp and symmetrical chromatographic peaks (Fig. 2c), both having the ability to coagulate plasma. When analyzed by polyacrylamide gel electrophoresis (SDS-PAGE), both peaks revealed a single band (Fig. 2c), indicating that the protocol outlined here resulted in the purification of two serine protease isoforms, which were named collinein-1 (peak 1) and collinein-2 (peak 2). When analyzed by mass spectrometry, collinein-1 and collinein-2 presented molecular masses of 29,474 and 28,388 Da, respectively.

Purification of two serine protease isoforms from C. d. collilineatus venom. a Molecular exclusion of C. d. collilineatus crude venom on a Sephacryl S100 HR column (GE Healthcare, UK). Fractions were eluted with ammonium formate buffer 0.05 M, pH 3.5. b Ions exchange of fraction S3 on a MonoQ 5/50 GL Column (GE Healthcare, UK), previously equilibrated in Tris-HCl 0.02 M, NaCl 1 M, pH 8.0. Elution was performed in a discontinuous gradient of NaCl 1 M. c Reverse Phase of Q4 fraction in a C4 column (Shodex, Japan). Proteins were eluted by a stepwise gradient of solution B (0.1 % TFA, 80 % ACN). Inserted panel: SDS-PAGE (13.5 %) stained with silver of purified collinein-1 (1) and collinein-2 (2). When analyzed by mass spectrometry, collinein-1 and collinein-2 presented molecular masses of 29,275 and 28,388 Da, respectively
Protein sequencing and multiple sequence alignment
The combination of de novo and N-terminal sequencing and mass fingerprinting allowed the identification of 140 residues of rCollinein-1, 164 residues of native collinein-1, and 89 residues of collinein-2. Multiple sequence alignment showed 100 % of amino acid identity between native and recombinant collinein-1 (Fig. 3). On the other hand, the partial sequence of collinein-2 presented a conservative substitution at position 64 and 4 non-conservative substitutions at positions 115, 156, 159, and 181 when compared to collinein-1.

Multiple alignment of the partial amino acid sequences of the native and recombinant serine proteases. Sequences were obtained by a combination of de novo and N-terminal sequencing and peptide mass fingerprinting. Partial amino acid sequences of collinein-1 and collinein-2 were submitted to UniProt Knowledgebase under the accession numbers C0HJR5 and C0HJR6, respectively. The deduced amino acid sequence of collinein-1 (based on the nucleotide sequence deposited on European Nucleotide Archives under accession number LN651356) was used to predict the probable positions of tryptic peptides. Black boxes represent amino acids conserved in all sequences. Amino acids in black are conserved in at least two sequences. Amino acids in gray correspond to residues not conserved. Multiple sequence alignment was performed by MultiAlin algorithm. Although the sequences are not complete, it is possible to assume that the recombinant protein corresponds to the native collinein-1
The deduced amino acid sequence of collinein-1 showed sequence identity ranging from 77 to 99 % to other known snake venom serine proteases from GenBank and UniProtKB/Swiss-Prot databases (Fig. 4). The multiple sequence alignment demonstrated the conservation of the cysteines involved in the six intra-molecular disulfide bonds that are characteristic of SVSPs. Furthermore, the residues His43, Asp88, and Ser184 that are responsible for the catalytic activity, as well as the amino acid residues located near the active site, are highly conserved in all analyzes sequences.

Multiple sequence alignment of collinein-1 with other SVSPs. The deduced sequence of collinein-1 was aligned with nine SVSPs present in GenBank and UniProtKB/Swiss-Prot databases by ClustalW2. Residues marked in black correspond to the catalytic triad His42, Asp87, and Ser184. Residues marked in gray correspond to substrates recognition sites. Residues in box correspond to the theoretical N-glycosylation site. Symbols indicate the conserved Cys residues. Cys residues indicated by the same symbol represent the formation of intramolecular disulfide bonds. Sequences are indicated by the names of the enzymes and the species to which they belong: Collinein-1_Cdc, collinein-1 from Crotalus durissus collilineatus (UniProtKB/Swiss-Prot: C0HJR5); B2.1_Cdt, B1.3_Cdt, and B1.4_Cdt, gyroxin-like isoforms B2.1 (GenBank: AY954040.1), B1.3 (GenBank: EU360951.1), and B1.4 (GenBank: EU360952.1) from Crotalus durissus terrificus, respectively; Crotalase_Ca, SP5_Ca and SP9_Ca, crotalase (UniProtKB/Swiss-Prot: F8S114.1), and serine proteinases 5 (GenBank: AEJ31999.1) and 9 (UniProtKB/Swiss-Prot: J3RYA3.1) from Crotalus adamanteus, respectively; SP6_Sce, serine proteinase 6 from Sistrurus catenatus edwardsi (GenBank: ABG26972.1); bilineobin_Ab, bilineobin from Agkistrodon bilineatus (UniProtKB/Swiss-Prot: Q9PSN3.1); Stejnobin_Ts, Trimeresurus stejnegeri. Ididentity in relation to collinein-1
Comparative enzymatic and functional characterization of collinein-1 in native and recombinant forms
When evaluated for proteolytic action upon fibrinogen chains, both native and recombinant collinein-1 completely cleaved the Aα chain with 5 min of reaction (Fig. 5). The proteins also showed activity upon Bβ chain; however, this chain is only completely cleaved after 4 h of incubation with the enzymes. Both serine proteases were able to virtually release all fibrinopeptide A from fibrinogen with only 5 min of reaction while fibrinopeptide B was only completely released after 30 min of reaction. After 120 min of reaction, both enzymes, as well as thrombin, released a third peptide, named peptide C (Fig. 6). The estimated values of MCD were 0.225 mg/μL to collinein-1 and 0.08 mg/μL to rCollinein-1. Native and recombinant collinein-1 showed a similar catalytic activity upon synthetic substrates for plasmin (S-2251), factor Xa (S-2222), thrombin (S-2238), and plasma kallikrein (S-2302), although the activity of rCollinein-1 was slightly higher for substrates S-2238 and S-2302. Both serine proteases hydrolyzed substrates that have arginine at P1 position (S-2222, S-2238, and S-2302), while substrate S-2251 that has a lysine at this position did not undergo significant hydrolysis by any of the enzymes (Fig. 7).

Fibrinogenolytic activity of native (a) and recombinant (b) collinein-1. The catalytic activity of the serine proteases upon fibrinogen was analyzed by SDS-PAGE 13.5 % stained with Coomassie Blue R-350. MM molecular marker; Cont-, negative control

Reverse phase chromatography of fibrinopeptides released by the catalytic activity of native (a) and recombinant (b) collinein-1 upon fibrinogen. Fibrinopeptides generated by incubation of 10 mg of the enzymes with bovine fibrinogen were subjected to reverse phase chromatography on a Vydac C18 column (0.46 × 25 cm). Fibrinopeptides were eluted by a segmented gradient of acetonitrile (0–15 % in 5-column volumes and 15–30 % in 15-column volumes). Peak A corresponds to fibrinopeptide A and peak B to fibrinopeptide B. Peak C* indicate a peptide corresponding to the 49 N-terminal residues of Bβ chain

Catalytic activity of native and recombinant collinein-1 upon chromogenic substrates The substrate hydrolysis was determined spectrophotometrically at 405 nm after 40 min of reaction at 37 °C. One unit of activity was defined as the increase of absorbance in 0.01 unit. *p < 0.05 compared to the native protein; **p < 0.001 compared to the native protein
The enzymatic activity of both serine proteases upon TAME substrate followed the kinetic model of Michaelis-Menten. Kinetic parameters for the enzymes are shown in Table 2. Native collinein-1 showed a lower K m value comparing to the recombinant protein, demonstrating a higher affinity for the substrate. However, native and recombinant collinein-1 did not significantly differ in efficiency to hydrolyze TAME. The evaluation of esterase activity of native and recombinant collinein-1 in view of variations in temperature and pH demonstrated that these enzymes are highly resistant, since their activity was not affected even after incubation at 100 °C and on extremely acid (pH 3.5) and basic (pH 10.5) buffers (Fig. 8a, b). The assays conducted to assess the influence of divalent ions on the esterase activity of the serine proteases demonstrated a significant decrease in their activity in the presence of Zn2+ and their almost complete inhibition in the presence of Cu2+ (Fig. 8c). The presence of Ba2+ resulted in a decrease in the activity of native collinein-1 while the recombinant enzyme was resistant to the interference of this metal ion. On the other hand, Ca2+ and Mg2+ did not significantly influence the esterase activity of the enzymes.

Stability of native and recombinant collinein-1 at different temperatures (a), pHs (b), and effect of divalent ions on their esterase activity (c). The stability of the enzymes was evaluated by esterase activity upon TAME. The proteins were pre-incubated for 30 min at different temperatures or with divalent ions, and the esterase activity was monitored at 247 nm after 30 min of reaction. One unit of activity was defined as the increase of absorbance in 0.01 unit
Discussion
Several venomous and poisonous animals produce toxins that target the hemostatic system. Snake venoms presents a variety of proteins and peptide, such as phospholipase A2, L-amino acid oxidases, C-type lectins, 5′-nucleotidase, disintegrins, metalloproteases, and serine proteases that act on coagulation resulting in hemostatic disorders that facilitate prey capture. Serine proteases are widely found in venoms of snakes from Viperidae and Crotalidae families and may represent up to 20 % of the total venom toxins (Braud et al. 2000).
The mechanisms by which snake venoms serine proteases affect blood clotting were not yet fully understood. Thus, investigations regarding the structure, function, and mechanisms of action of these enzymes may contribute to the discovery of new targets and drugs for the treatment of physiological dysfunctions related to blood clotting. Furthermore, the development of biotechnological strategies for protein production on laboratory scale is important to enable the deepening of functional studies, since purification of toxins from snake venom is usually a labor-intensive and low-yield process (Vetter et al. 2011).
This paper describes the heterologous expression of a serine protease previously cloned from C. d. collilineatus venom gland (Boldrini-França et al. 2009). Moreover, this study highlights the purification of the native serine protease and reports the comparative enzymatic characterization of this native and recombinant enzyme, validating, therefore, the heterologous expression method.
The heterologous expression described here resulted in a high yield in terms of quantity and functionality of the recombinant protein (Table 1). Protein expression in P. pastoris typically results in recombinant molecules properly processed and folded, since yeasts are eukaryotic organisms that make many of post-translational modifications performed by higher eukaryotes (Cregg et al. 2000; Lin-Cereghino and Cregg 2000). Therefore, studies that compare heterologous proteins expressed in yeasts with those obtained from natural sources have shown little or no difference in their structural and functional properties (Schilling et al. 2002; You et al. 2004; Guiraudie-Capraz et al. 2005; Shukla et al. 2007). Batroxobin from Bothrops atrox moojeni is the only recombinant SVSP produced in P. pastoris that was enzymatically and functionally compared to its native form. Recombinant batroxobin is a potent in vitro and in vivo pro-coagulant agent and might be an effective substitute to the native protein (You et al. 2004). Despite the scarcity of validation data for recombinant SVSPs expressed in yeast, this eukaryote system has been proven effective in heterologous production of this class of enzymes.
In spite of the high efficiency of P. pastoris expression system in producing the heterologous collinein-1, some additional steps were required to improve the expression conditions. P. pastoris survive in a relatively wide pH range (pH 3.0–7.0), which allows a considerable flexibility in adjusting this parameter in the moment of induction. pH may affect the activity of secreted proteases, thereby increasing the stability of the recombinant protein (Macauley-Patrick et al. 2005). The medium pH that resulted in the highest expression of collinein-1 was pH 7.0, whereas no heterologous protein expression was observed at pH 4.0. No optimal pH screening for SVSP expression in P. pastoris was reported so far, but this class of enzymes was already efficiently expressed in yeast at pH 5.0 (You et al. 2004) and at neutral pH (Yu et al. 2007).
The stability of the recombinant protein can also be influenced by amino acid supplements in the culture medium (Macauley-Patrick et al. 2005). In the present study, peptone and yeast extract did not affect the yield of rCollinein-1. Medium supplements provide an ideal chemical environment for P. pastoris growth; however, they may impair the reproducibility of large-scale expression and industrial fermentations (Macauley-Patrick et al. 2005). Thus, the medium of choice for collinein-1 large-scale expression was the minimal medium without supplementation.
Recombinant collinein-1 was tagged with polyhistidine in the C-terminal of the molecule, in order to facilitate its purification by immobilized metal affinity chromatography (IMAC). However, rCollinein-1 did not interact with Ni-agarose column, precluding its purification by this chromatographic method (data not shown). The incapacity of the recombinant protein to interact with the nickel column can be due to (i) internal location of polyhistidine tag in the tridimensional structure of the protein or (ii) cleavage of polyhistidine tag by a proteolytic enzymes from P. pastoris or autoproteolysis. The hypothesis of polyhistidine tag loss by proteolytic activity is supported by the lower molecular mass of rCollinein-1 (28,868 Da), when compared to its native form (29,274 Da). Nevertheless, the tag loss and the possible cleavage site must be further confirmed by structural analysis and complete protein sequencing. Due to the impossibility of purifying rCollinein-1 in a single IMAC step, a new purification protocol was standardized based on ion-exchange and reverse phase chromatographic techniques (Fig. 1), resulting in a high protein yield (56 mg per liter of culture) and activity recovery.
The methodology employed in the purification of native serine protease resulted in the isolation of two isoforms, named collinein-1 and 2, which present molecular masses of 29,474 and 29,388 Da, respectively (Fig. 2). These isoforms differ from each other by five amino acid substitutions in their primary structure. On the other hand, the alignment of the partial sequences of collinein-1 and rCollinein-1 resulted in 100 % of identity (Fig. 3). So, we can assume that the recombinant protein corresponds to the native collinein-1. The calculation of collinein-1 yield for this purification process demonstrated that the serine protease is present in the venom at low concentrations, representing only 0.63 % of the total venom toxin (Table 1). Boldrini-França et al. (2010) determined that serine proteases represent only 0.5 to 1.9 % of the total toxins in the proteome of C. d. collilineatus venom. These data are consistent with the income presented in this work, indicating no significant loss of protein during the isolation process.
The deduced amino acid sequence of collinein-1 was aligned with other homologous SVSPs from database to assess the conservative domains (Fig. 4). All aligned sequences present 12 cysteine residues in conserved positions, indicating that the 6 disulfide bond and molecular scaffold typical of SVSPs are preserved. The catalytic triad, composed by His43, Asp88, and Ser184 residues, as well as amino acid residues located close to the active site, is highly conserved in these toxins. The enzymatic activity performed by the catalytic triad is probably related to some pathophysiological properties of SVSPs, such as effects on blood coagulation and immune system (Hedstrom 2002a, b). The replacement of the catalytic residue His57 by an arginine in a serine protease homologue from Trimeresurus jerdonii led to the loss of its proteolytic activity and, therefore, to its effects on blood coagulation (WU 2008). The residue Asp178 is preserved in all aligned serine proteases, as well as in thrombin and kallikrein. This residue interacts with the basic amino acid at the P1 position of substrate, being associated to the primary specificity of trypsin-like enzymes (Stubbs and Bode 1993).
All aligned SVSPs present a putative N-glycosylation site (Asn-X-Thr) in conserved position. SVSPs are usually glycoproteins presenting variable contents of N- and O-linked glycosylations in non-homologous positions, which can shift the molecular mass of these enzymes in up to 40 kDa (Serrano and Maroun 2005). SVSPs are known to present carbohydrate motif composed by fucose, hexose, sialic acid, N-acetylglucosamine (GlcNAc), mannose, and N-acetylneuraminic acid (NauAc) (Pirkle 1998; Koh et al. 2001; Wang et al. 2001; Bortoleto et al. 2002; Oyama and Takahashi 2003; Costa et al. 2009; Lin et al. 2011). On the other hand, N-glycosylations carried out by yeast are generally polysaccharides composed of 100 or more mannose residues. These differences in glycosylation performed by yeasts and higher eukaryotes may potentially interfere in the correct folding, function, and stability of the recombinant molecule.
Native and recombinant collinein-1 showed a proteolytic activity upon fibrinogen similar to that of thrombin, releasing both fibrinopeptides A and B (Fig. 6). The majority of the thrombin-like serine proteases differ from thrombin by preferentially cleaves one of the fibrinogen chains. As like as collinein-1, most thrombin-like enzymes preferentially cleaves the peptide bond Arg15-Gly16 of the Aα chain, being classified as FP-A, such as crotalase from Crotalus adamanteus (Markland et al. 1982), batroxobin from B. atrox (Nishida et al. 1994) and ancrod from C. rhodostoma (Nolan et al. 1976). There is only one report of a serine protease isolated from C. d. collilineatus venom (Oliveira et al. 2009). This enzyme, named Cdc-3, differs from collinein-1 by cleaving preferentially the fibrinogen Bβ chain, being classified as a FP-B.
After 120 min of reaction, it is possible to identify the onset of a third peptide, named peptide C. TLE-B and TLE-P enzymes from Lachesis muta muta (Magalhães et al. 2003) and leucurobina from Bothrops leucurus (Magalhães et al. 2007) also release the peptide C after prolonged incubation time with fibrinogen. Previous mass spectrometry analysis demonstrated that this peptide is a result of the cleavage of the Arg42-Ala43 peptide bond of the Bβ chain of human fibrinogen (Magalhães et al. 2003, 2007). So, peptide C may represent the 49 N-terminal residues of bovine fibrinogen since this cleavage site is conserved at positions Arg49-Ala50 in this molecule.
The mechanism by which thrombin-like enzymes induce blood clotting is closely related to the ability of these enzymes to cleave Aα and Bβ chains of fibrinogen, leading to the formation of insoluble fibrin monomers (Lu et al. 2005; Serrano and Maroun 2005; Kini 2011). Both native and recombinant collinein-1 were able to clot bovine plasma in a dose-dependent manner, but rCollinein-1 exhibited a coagulant activity almost three times greater than the native protein. A possible explanation for this disparity might be the influence of differences in glycosylation promoted by snakes and P. pastoris, since, in some cases, the oligosaccharides may delimit the entrance of the catalytic cleft, thus exerting influence on accessibility to active site (Zhu et al. 2005; Mackessy 2010).
Although the conversion of fibrinogen into a fibrin clot is the most important activity of thrombin-like serine proteases, certain enzymes may also act on other substrates associated to fibrino(geno)lysis, prothrombin, and protein C activation and conversion of kininogen into kinin (Castro et al. 2004; Serrano and Maroun 2005). Regarding the catalytic activity of native and recombinant collinein-1 upon synthetic substrates, both enzymes were able to hydrolyze not only the substrate for thrombin but also those for factor Xa and plasma kallikrein (Fig. 7). Native and recombinant collinein-1 preferably cleaved substrates that have an arginine at P1, while the substrate that presents a lysine in this position (S2251 for plasmin) did not undergo significant proteolysis by these enzymes. Furthermore, native and recombinant collinein-1 presented the higher catalytic activity upon the substrate for plasma kallikrein, indicating that phenylalanine at P2 and proline at P3 play an important role on the hydrolysis of Arg-pNA bond.
Presumably, the most remarkable result of this work is the high stability of native and recombinant collinein-1 at different pH and temperature. These enzymes completely retained their esterase activity upon TAME even after pre-incubation at 100 °C and with extremely acid and alkaline buffers (Fig. 8a, b). Several reports have demonstrated that SVSPs are resistant to heat and/or pH denaturation (Costa et al. 2009; Barros et al. 2011; Menaldo et al. 2012). However, none of these studies has reported the full maintenance of the enzymatic activity after pre-treating the enzymes on extreme pH and temperature conditions. The resistance of SVSPs to variations in temperature and pH is probably due to their three-dimensional structure, which is stabilized by six disulfide bonds, what increases the threshold energy required to generate significant conformational changes in the molecular scaffold. In addition, some SVSPs, such as BJ-48 from Bothrops jararacussu, protease A from Bothrops jararaca, and brevinase from Agkistrodon blomhoffi brevicaudus, completely or partially lost their thermal stability when deglycosylated, indicating that the carbohydrate motif play important role in their resistance to heat denaturation (Silva-Junior et al. 2007; Paes-Leme et al. 2008; Lee et al. 1999).
Divalent ions may also influence the enzymatic activity of SVSPs, probably by promoting small changes in their three-dimensional structure. Esterase activity of both native and recombinant collinein-1 was significantly decreased in the presence of Zn2+ and Cu2+ while Ba2+ only influenced the activity of the native enzyme. On the other hand, the enzymatic activity of both serine proteases was not influenced by Ca2+ and Mg2+ (Fig. 8c). Similar results were reported for gyroxin from Crotalus durissus terrificus, which is partially and completely inhibited by Ba2+ and Cu2+, respectively. Gyroxin has also undergone the influence of Mn2+, which was not tested for collinein-1 (Barros et al. 2011).
Native and recombinant collinein-1 did not significantly differ in efficiency to hydrolyze TAME, as demonstrated by kinetic assays (Table 2). The similarities of the enzymatic properties observed for collinein-1in native and recombinant forms may reflect the conservation of crucial structures for catalytic activity. The low divergence of the enzymatic activities of the native and recombinant collinein-1 reveals the efficiency of the system of choice in producing correctly folded and functionally active snake venom serine protease.
Over the past few years, the number of studies that use P. pastoris system to produce bioactive molecules has substantially increased. This is reflected in the growing number of protein that has been industrially produced in P. pastoris as an alternative to some traditional employed methods, such as Escherichia coli and Saccharomyces cerevisiae. P. pastoris is one of the most productive eukaryotic expression systems currently available and has been used in the production of a diversity of functional proteins in soluble form, which are easily purified from the culture medium (Macauley-Patrick et al. 2005; Damasceno et al. 2012).
The interest in developing biotechnological strategies for heterologous expression of SVSPs is due to the low yield of these toxins when purified from the venom, precluding their detailed functional characterization. Thus, the success in producing an enzymatically active recombinant SVSP is important not only to probe the biological and functional characterization of these enzymes but also to enable their future molecular manipulations and therapeutic applications. The high stability of rCollinein-1, combined with the maintenance of native enzymatic properties, sheds light on perspectives in applying this toxin as possible biotechnological agents or as models for developing new drugs for the treatment of a various hemostatic disorders.
References
Assakura MT, Furtado MD, Mandelbaum FR (1992) Biochemical and biological differentiation of the venoms of the lancehead vipers (Bothrops atrox, Bothrops asper, Bothrops marajoensis and Bothrops moojeni). Comp Biochem Physiol 102:727–732. doi:10.1016/0305-0491(92)90071-X
Barros LC, Soares AM, Costa FL, Rodrigues VM, Fuly AL, Giglio JR, Gallacci M, Thomazini-Santos IA, Barraviera SRCS, Barraviera B, Ferreira-Junior RS (2011) Biochemical and biological evaluation of gyroxin isolated from Crotalus durissus terrificus venom. J Venom Anim Toxins 17:23–33. doi:10.1590/S1678-91992011000100004
Boldrini-França J, Rodrigues RS, Fonseca FP, Menaldo DL, Ferreira FB, Henrique-Silva F, Soares AM, Hamaguchi A, Rodrigues VM, Otaviano AR, Homsi-Brandeburgo MI (2009) Crotalus durissus collilineatus venom gland transcriptome: analysis of gene expression profile. Biochimie 91:586–595. doi:10.1016/j.biochi.2009.02.001
Boldrini-Franca J, Correa-Netto C, Silva MMS, Rodrigues RS, De La Torre P, Perez A, Soares AM, Zingali RB, Nogueira RA, Rodrigues VM, Sanz L, Calvete JJ (2010) Snake venomics and antivenomics of Crotalus durissus subspecies from Brazil: assessment of geographic variation and its implication on snakebite management. J Proteome 73:1758–1776. doi:10.1016/j.jprot.2010.06.001
Bortoleto RK, Murakami MT, Watanabe L, Soares AM, Arni RK (2002) Purification, characterization and crystallization of Jararacussin-I, a fibrinogen-clotting enzyme isolated from the venom of Bothrops jararacussu. Toxicon 40:1307–1312. doi:10.1016/S0041-0101(02)00140-X
Braud S, Bon C, Wisner A (2000) Snake venom proteins acting on hemostasis. Biochimie 82:851–859. doi:10.1016/S0300-9084(00)01178-0
Castro HC, Zingali RB, Albuquerque MG, Pujol-Luz M, Rodrigues CR (2004) Snake venom thrombin-like enzymes: from reptilase to now. Cell Mol Life Sci 61:843–856. doi:10.1007/s00018-003-3325-z
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890. doi:10.1093/nar/16.22.10881
Costa FLS, Rodrigues RS, Izidoro LFM, Menaldo DL, Hamaguchi A, Homsi-Brandeburgo MI, Fuly AL, Soares SG, Selistre-De-Araujo HS, Barraviera B, Soares AM, Rodrigues VM (2009) Biochemical and functional properties of a thrombin-like enzyme isolated from Bothrops pauloensis snake venom. Toxicon 54:725–735. doi:10.1016/j.toxicon.2009.05.040
Cregg JM, Lin-Cereghino JL, Shi J, Higgins DR (2000) Recombinant protein expression in Pichia pastoris. Mol Biotechnol 16:23–52. doi:10.1385/MB:16:1:23
Damasceno LM, Huang CJ, Batt CA (2012) Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol 93:31–39. doi:10.1007/s00253-011-3654-z
Dekhil H, Wisner A, Marrakchi N, El Ayeb M, Bon C, Karoui H (2003) Molecular cloning and expression of a functional snake venom serine proteinase, with platelet aggregating activity, from the Cerastes cerastes viper. Biochemistry 42:10609–10618. doi:10.1021/bi034790b
Edman P, Begg G (1967) A protein sequenator. Eur J Biochem 1:80–91. doi:10.1111/j.1432-1033.1967
Escoubas P, Quinton L, Nicholson GM (2008) Venomics: unraveling the complexity of animal venoms with mass spectrometry. J Mass Spectrom 43:279–295. doi:10.1002/jms.1389
Fan CY, Qian YC, Yang SL, Gong Y (1999) Cloning, sequence analysis and expression in E. coli of the cDNA of the thrombin-like enzyme (pallabin) from the venom of Agkistrodon halys pallas. Biochem Mol Biol Int 47:217–225. doi:10.1080/15216549900201223
Guiraudie-Capraz G, Clot-Faybesse O, Pageat P, Malosse C, Cain AH, Ronin C, Nagnan-Le MP (2005) Heterologous expression of piglet odorant-binding protein in Pichia pastoris: a comparative structural and functional characterization with native forms. J Biotechnol 117:11–19. doi:10.1016/j.jbiotec.2005.01.005
Hedstrom L (2002a) An overview of serine proteases. Curr Protoc Protein Sci. doi:10.1002/0471140864.ps2110s26
Hedstrom L (2002b) Serine protease mechanism and specificity. Chem Rev 102:4501–4524. doi:10.1021/cr000033x
Hummel BCW (1959) A modified spectrophotometric determination of chymotrypsin, trypsin, and thrombin. Can J Biochem Cell Biol 37:1393–1399. doi:10.1139/o59-157
Hutton RA, Warrell DA (1993) Action of snake venom components on the haemostatic system. Blood Rev 7:176–189. doi:10.1016/0268-960X(93)90004-N
Jiang X, Xu J, Yang Q (2010) Soluble expression, purification, and characterization of Gloydius shedaoensis venom gloshedobin in Escherichia coli by using fusion partners. Appl Microbiol Biotechnol 85:635–642. doi:10.1007/s00253-009-2141-2
Kini RM (2006) Anticoagulant proteins from snake venoms: structure, function and mechanism. Biochem J 97:377–387. doi:10.1042/BJ20060302
Kini RM (2011) Toxins in thrombosis and haemostasis: potential beyond imagination. J Thromb Haemost 9:195–208. doi:10.1111/j.1538-7836.2011.04279.x
Koh YS, Chung KH, Kim DS (2001) Biochemical characterization of a thrombin-like enzyme and a fibrinolytic serine protease from snake (Agkistrodon saxatilis) venom. Toxicon 39:555–560. doi:10.1016/S0041-0101(00)00169-0
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi:10.1093/bioinformatics/btm404
Lee JW, Seu JH, Rhee IK, Jin I, Kawamura Y, Park W (1999) Purification and characterization of brevinase, a heterogeneous two-chain fibrinolytic enzyme from the venom of Korean snake, Agkistrodon blomhoffii brevicaudus. Biochem Biophys Res Commun 260:665–670. doi:10.1006/bbrc.1999.0977
Levy DE, Del Zoppo GJ, Demaerschalk BM, Demchuk AM, Diener HC, Howard G, Kaste M, Pancioli AM, Ringelstein EB, Spatareanu C, Wasiewski WW (2009) Ancrod in acute ischemic stroke: results of 500 subjects beginning treatment within 6 hours of stroke onset in the ancrod stroke program. Stroke 40:3796–3803. doi:10.1161/STROKEAHA.109.565119
Lin Y, Yu X, He Q, Li H. Li D, Song X, Wang Y, Wen H, Deng H, Deng J (2009) Expression and functional characterization of chitribrisin, a thrombin-like enzyme, in the venom of the Chinese green pit viper (Trimeresurus albolabris). Protein Expr Purif 67.48–52. doi: 10.1016/j.pep.2009.03.005
Lin CW, Chen JM, Wang YM, Wu SW, Tsai IH, Khoo KH (2011) Terminal desialylated multiantennary complex-type N-glycans carried on acutobin define the glycosylation characteristics of the Deinagkistrodon acutus venom. Glycobiology 21:530–542. doi:10.1093/glycob/cwq195
Lin-Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66. doi:10.1111/j.1574-6976.2000.tb00532.x
Lu Q, Clemetson JM, Clemetson KJ (2005) Snake venoms and hemostasis. J Thromb Haemost 3:1791–1799. doi:10.1111/j.1538-7836.2005.01358.x
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270. doi:10.1002/yea.1208
Mackessy S (2010) Thrombin-like enzymes in snake venoms. In: Kini RM, Clemetson KJ, Markland FS, Mclane MA, Morita T (eds) Toxins and hemostasis, 1st edn. Springer, London, pp 518-558
Maeda M, Satoh S, Suzuki S, Niwa M, Itoh N, Yamashina I (1991) Expression of cDNA for batroxobin, a thrombin-like snake venom enzyme. J Biochem (Tokyo) 109:632–637
Magalhães A, Ferreira RN, Richardson M, Gontijo S, Yarleque A, Magalhães HPB, Bloch C, Sanchez EF (2003) Coagulant thrombin-like enzymes from the venoms of Brazilian and Peruvian bushmaster (Lachesis muta muta) snakes. Comp Biochem Physiol B 136:255–266. doi:10.1016/S1096-4959(03)00202-1
Magalhães A, Magalhães HPB, Richardson M, Gontijo S, Ferreira RN, Almeida AP, Sanchez EF (2007) Purification and properties of a coagulant thrombin-like enzyme from the venom of Bothrops leucurus. Comp Biochem Physiol A 146:565–575. doi:10.1016/j.cbpa.2005.12.033
Markland FS, Kettner C, Schiffman S, Shaw E, Bajwa SS, Reddy KNN, Kirakossian H, Patkos GB, Theodor I, Pirkle H (1982) Kallikrein-like activity of crotalase, a snake venom enzyme that clots fibrinogen. Proc Natl Acad Sci U S A 79:1688–1692. doi:10.1073/pnas.79.6.1688
Menaldo DL, Bernardes CP, Santos-Filho NA, Moura LA, Fuly AL, Arantes EC, Sampaio SV (2012) Biochemical characterization and comparative analysis of two distinct serine proteases from Bothrops pirajai snake venom. Biochimie 94:2545–2558. doi:10.1016/j.biochi.2012.07.007
Muanpasitporn C, Rojnuckarin P (2007) Expression and characterization of a recombinant fibrinogenolytic serine protease from green pit viper (Trimeresurus albolabris) venom. Toxicon 49:1083–1089. doi:10.1016/j.toxicon.2007.01.015
Nishida S, Fujimura Y, Miura S, Ozaki Y, Usami Y, Suzuki M, Titani K, Yoshida E, Sugimoto M, Yoshioka A, Fukui H (1994) Purification and characterization of bothrombin, a fibrinogen-clotting serine protease from the venom of Bothrops jararaca. Biochemistry 33:1843–1849. doi:10.1021/bi00173a030
Nolan C, Hall LS, Barlow GM (1976) Ancrod, the coagulating enzyme from Malayan pit viper (Agkistrodon rhodostoma) venom. Methods Enzymol 45:205–213. doi:10.1016/S0076-6879(76)45020-6
Oliveira DGL, Murakami MT, Cintra ACO, Franco JJ, Sampaio SV, Arni RK (2009) Functional and structural analysis of two fibrinogen-activating enzymes isolated from the venoms of Crotalus durissus terrificus and Crotalus durissus collilineatus. Acta Biochim Biophys Sin 41:21–29. doi:10.1093/abbs/gmn003
Oyama E, Takahashi H (2003) Purification and characterization of a thrombin like enzyme, elegaxobin II, with Lys-bradykinin releasing activity from the venom of Trimeresurus elegans (Sakishima Habu). Toxicon 41:559–568. doi:10.1016/S0041-0101(02)00363-X
Paes-Leme AF, Prezoto BC, Yamashiro ET, Bertholim L, Tashima AK, Klitzke CF, Camargo ACM, Serrano SMT (2008) Bothrops protease A, a unique highly glycosylated serine proteinase, is a potent, specific fibrinogenolytic agent. J Thromb Haemost 6:1363–1372. doi:10.1111/j.1538-7836.2008.02995.x
Pirkle H (1998) Thrombin-like enzymes from snake venoms: an update inventory. Thromb Haemost 79:675–683
Rodrigues VM, Soares AM, Guerra-Sá R, Fontes MRM, Giglio JR (2000) Structural and functional characterization of neuwiedase, a nonhemorrhagic fibrin(ogen)olytic metalloprotease from Bothrops neuwiedi snake venom. Arch Biochem Biophys 381:213–224. doi:10.1006/abbi.2000.1958
Schilling S, Hoffmann T, Rosche F, Manhart S, Wasternack C, Demuth HU (2002) Heterologous expression and characterization of human glutaminyl cyclase: evidence for a disulfide bond with importance for catalytic activity. Biochemistry 41:10849–10857. doi:10.1021/bi0260381
Serrano SMT, Maroun RC (2005) Snake venom serine proteinases: sequence homology vs. substrate specificity, a paradox to be solved. Toxicon 45:1115–1132. doi:10.1016/j.toxicon.2005.02.020
Shukla AK, Haase W, Reinhart C, Michel H (2007) Heterologous expression and comparative characterization of the human neuromedin U subtype II receptor using the methylotrophic yeast Pichia pastoris and mammalian cells. Int J Biochem Cell Biol 39:931–942. doi:10.1016/j.biocel.2007.01.016
Silva-Junior FP, Floriano P, Guedes HLM, Garvey LC, Aguiar AS, Bourguignon SC, Di-Cera E, Giovanni-de-Simone S (2007) BJ-48, a novel thrombin-like enzyme from the Bothrops jararacussu venom with high selectivity for Arg over Lys in P1: role of N-glycosylation in thermostability and active site accessibility. Toxicon 50:18–31. doi:10.1016/j.toxicon.2007.02.018
Stubbs MT, Bode W (1993) A player of many parts: the spotlight falls on thrombin structure. Thromb Res 69:1–58. doi:10.1016/0049-3848(93)90002-6
Vetter I, Davis JL, Rash LD, Anangi R, Mobli M, Alewood PF, Lewis RJ, King GF (2011) Venomics: a new paradigm for natural products-based drug discovery. Amino Acids 40:15–28. doi:10.1007/s00726-010-0516-4
Wang YM, Wang SR, Tsai IH (2001) Serine protease isoforms of Deinagkistrodon acutus venom: cloning, sequencing and phylogenetic analysis. Biochem J 354:161–168. doi:10.1042/0264-6021:3540161
Wu J, Jin Y, Zhong S, Chen R, Zhu S, Wang W, Lu Q, Xiong Y (2008) A unique group of inactive serine protease homologues from snake venom. Toxicon 52:277–284. doi:10.1016/j.toxicon.2008.05.013
Yang Q, Li M, Xu J, Bao Y, Lei X, An L (2003) Expression of gloshedobin, a thrombin-like enzyme from the venom of Gloydius shedaoensis, in Escherichia coli. Biotechnol Lett 25:101–104. doi:10.1023/A:1021950815453
Yang D, Peng M, Yang H, Yang Q, Xu J (2009) Expression, purification and characterization of Gloydius shedaoensis venom gloshedobin as Hsp70 fusion protein in Pichia pastoris. Protein Expr Purif 66:138–142. doi:10.1016/j.pep.2009.03.003
Yonamine CM, Prieto-Da-Silva ARB, Magalhães GS, Rádis-Baptista G, Morganti L, Ambiel FC, Chura-Chambi RM, Yamane T, Camillo MAP (2009) Cloning of serine protease cDNAs from Crotalus durissus terrificus venom gland and expression of a functional Gyroxin homologue in COS-7 cells. Toxicon 54:110–120. doi:10.1016/j.toxicon.2009.03.022
You WK, Choi WS, Koh YS, Shin HC, Jang Y, Chung KH (2004) Functional characterization of recombinant batroxobin, a snake venom thrombin-like enzyme, expressed from Pichia pastoris. FEBS Lett 571:67–73. doi:10.1016/j.febslet.2004.06.060
Yu X, Li Z, Xia X, Fang H, Zhou C, Chen H (2007) Expression and purification of ancrod, an anticoagulant drug, in Pichia pastoris. Protein Expr Purif 55:257–261. doi:10.1016/j.pep.2007.07.002
Zha XD, Ren B, Liu J, Xu KS (2003) cDNA cloning and high-level expression of a thrombin-like enzyme from Agkistrodon acutus venom. Methods Find Exp Clin Pharmacol 25:253–257. doi:10.1358/mf.2003.25.4.769672
Zhu ZL, Liang Z, Zhang TY, Zhu ZQ, Xu WH, Teng MK, Niu LW (2005) Crystal structures and amydolytic activities of two glycosylated snake venom serine proteinases. J Biol Chem 280:10524–10529. doi:10.1074/jbc.M412900200
Acknowledgments
The authors acknowledge the financial support of Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Núcleo de Apoio à Pesquisa em Toxinas Animais da Universidade de São Paulo (NAP-TOXAN-USP).
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Boldrini-França, J., Rodrigues, R.S., Santos-Silva, L.K. et al. Expression of a new serine protease from Crotalus durissus collilineatus venom in Pichia pastoris and functional comparison with the native enzyme. Appl Microbiol Biotechnol 99, 9971–9986 (2015). https://doi.org/10.1007/s00253-015-6836-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6836-2