
Abstract
Vinyl chloride (VC), a known human carcinogen, is a common and persistent groundwater pollutant at many chlorinated solvent contaminated sites. The remediation of such sites is challenging because of the lack of knowledge on the microorganisms responsible for in situ VC degradation. To address this, the microorganisms involved in carbon assimilation from VC were investigated in a culture enriched from contaminated site groundwater using stable isotope probing (SIP) and high-throughput sequencing. The mixed culture was added to aerobic media, and these were amended with labeled (13C-VC) or unlabeled VC (12C-VC). The cultures were sacrificed on days 15, 32, and 45 for DNA extraction. DNA extracts and SIP ultracentrifugation fractions were subject to sequencing as well as quantitative PCR (qPCR) for a functional gene linked to VC-assimilation (etnE). The gene etnE encodes for epoxyalkane coenzyme M transferase, a critical enzyme in the pathway for VC degradation. The relative abundance of phylotypes was compared across ultracentrifugation fractions obtained from the 13C-VC- and 12C-VC-amended cultures. Four phylotypes were more abundant in the heavy fractions (those of greater buoyant density) from the 13C-VC-amended cultures compared to those from the 12C-VC-amended cultures, including Nocardioides, Brevundimonas, Tissierella, and Rhodoferax. Therefore, both a previously identified VC-assimilating genus (Nocardioides) and novel microorganisms were responsible for carbon uptake. Enrichment of etnE with time was observed in the heavy fractions, and etnE sequences illustrated that VC-assimilators harbor similar Nocardioides-like etnE. This research provides novel data on the microorganisms able to assimilate carbon from VC.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Groundwater contamination by vinyl chloride (VC) remains an important environmental problem and human health concern worldwide. VC is a known human carcinogen (Bucher et al. 2005) and has been found at many US Environmental Protection Agency National Priority List sites in soils, surface water, and groundwater. VC pollution in groundwater originates primarily from the higher-chlorinated ethenes including tetrachloroethene (PCE) and trichloroethene (TCE) (Bradley 2003). Leakage of these chemicals to aquifers, followed by their reduction by certain anaerobic bacteria (e.g., Dehalococcoides spp.), yields the lesser chlorinated ethenes, cis-1,2-dichloroethene and VC. These secondary pollutants are often more problematic than the parent compounds because they tend to accumulate.
Biological degradation is becoming an increasingly common remediation method for groundwater contaminants, either through natural attenuation or enhanced bioremediation. Microbial VC degradation can occur under both anaerobic and aerobic conditions and by co-metabolism or direct VC-assimilation (Mattes et al. 2010). However, linking specific microorganisms and enzymes to in situ activities remains a major challenge. Several bacteria belonging to the phyla Actinobacteria and Proteobacteria are capable of direct aerobic VC consumption and have been isolated from various environments or have been obtained in the laboratory (Coleman et al. 2002b; Danko et al. 2004; Elango et al. 2006; Fathepure et al. 2005; Hartmans and Debont 1992; Hartmans et al. 1985; Jin et al. 2010; Jin and Mattes 2008; Taylor et al. 2007; Verce et al. 2000). While these organisms have greatly contributed to our understanding of VC metabolism, the limitations associated with culture-based methods has likely resulted in an incomplete understanding of VC-assimilating microorganisms at field sites.
To overcome the limitations associated with culture-based methods, molecular testing during groundwater monitoring has become popular. For example, a quantitative PCR (qPCR) assay has been developed (Jin and Mattes 2010, 2011) and applied (Atashgahi et al. 2013; Jin et al. 2010; Patterson et al. 2013) to detect VC-oxidizing bacteria at contaminated sites. This qPCR method targets the functional genes etnC and etnE, which encode the alkene monooxygenase (AkMO) alpha subunit and the epoxyalkane coenzyme M transferase (EaCoMT), respectively. In VC-assimilators, AkMO is responsible for the initial attack on VC to convert it to VC epoxide. The epoxide is then conjugated to CoM by EaCoMT, which is a critical step to the central metabolic pathway. Therefore, presence of etnE gene indicates the potential for VC-assimilation.
To understand the diversity of mixed cultures and associated functional genes, molecular methods can often more accurately target the key active microorganisms. The stable isotope probing (SIP) method is valuable because it can identify the active microorganisms responsible for carbon or nitrogen uptake from the amended substrates (Radajewski et al. 2000). The method involves sample exposure to a stable-isotope-labeled compound and DNA extraction over time. The DNA is then subject to ultracentrifugation, fractionation (to separate label incorporated DNA from the unlabeled DNA), and community analysis (Dumont and Murrell 2005; Lueders et al. 2004; Luo et al. 2009; Madsen 2006; Singleton et al. 2005; Sun et al. 2010; Sun and Cupples 2012).
The overall objective was to determine the dominant microorganisms involved in carbon uptake from VC from a mixed culture derived from contaminated site groundwater. For this, SIP fractions were subjected to high-throughput sequencing. The relative abundance of phylotypes in fractions obtained from 13C-VC- and 12C-VC-amended mixed cultures were compared. The phylotypes enriched in the fractions from the 13C-VC-amended cultures compared to the fractions from the 12C-VC-amended cultures are considered responsible for incorporating 13C from VC (or VC degradation products). This is the first study to combine SIP and high-throughput sequencing to examine carbon uptake from VC. Along with the information provided by qPCR and clone library analysis of etnE, this study contributes to our understanding of VC degradation in mixed communities and at contaminated sites.
Materials and methods
Site information and groundwater collection
Groundwater was collected in accordance with USEPA/540/S-95/504 on June 9, 2009 from several monitoring wells (RB46D, RB73, RB52I, RB60, RB64I, RB63I, and RB58I) at a site in Carver, MA. The site was contaminated by disposal of material containing PCE at a landfill in 1986, which ultimately resulted in a large, dilute VC plume. Remediation efforts for the plume have involved oxygen and ethene injections and have been described previously (Begley et al. 2009, 2012; Chuang et al. 2010; Jin and Mattes 2010). The groundwater from these wells was composited; 1 L was shipped to the University of Iowa, where it was stored at 4 °C in the dark.
Development of VC-degrading enrichment cultures
Triplicate VC enrichment cultures were constructed in August 2009 by mixing composite groundwater with sterile minimal salts medium (MSM; prepared as described previously (Coleman et al. 2002b)) (1:1), placing 72 mL of the mixture into a 160-mL serum bottle (Wheaton, Millville, NJ), sealing with a butyl rubber stopper and aluminum crimp cap. Approximately 40 μmol VC (99.5 % from Fluka) was added and the bottles were monitored by gas chromatography with flame ionization detection (GC-FID) as described previously (Mattes et al. 2005). VC oxidation commenced after a 63-day lag period. Live culture samples were sent to MSU in January 2010 for the SIP experiment described below.
Analytical methods
Headspace samples of VC (100 μL) were analyzed via gas chromatography (Perkin Elmer) with flame ionization detection and a capillary column (J&W Scientific, DB-624, diameter 0.53 mm). The peak areas were compared to an external standard for VC quantification. Aqueous phase VC concentrations were calculated using a previously reported Henry’s law constant (1.069) (Gossett 1987).
SIP experimental design, DNA extraction, ultracentrifugation, and fractionation
The SIP experimental setup consisted of sterile serum bottles (160 mL), mixed culture (5 mL), minimal salts medium (67 mL MSM) (Hartmans et al. 1985), and VC (∼47 mg L−1 or 120 μmol) (the initial liquid and headspace volumes were 72 and 88 mL). These microcosms were prepared as previously described (Coleman et al. 2002a). Based on the calculated amount of oxygen required for VC oxidation, and on previous VC studies (Coleman et al. 2002b), oxygen limitation was not expected under these conditions. Three abiotic microcosms (controls, obtained via autoclaving) and nine live microcosms were amended with unlabeled VC (hereafter referred to as 12C-VC) (99 %, Specialty Gases of America) (to control for heavy GC microorganisms). An additional nine live microcosms were amended with labeled VC (hereafter referred to as 13C-VC) (13C2 VC, 99 %, Cambridge Isotope Laboratories). The microcosms were sealed, protected from light, and incubated at room temperature (21–23 °C), with shaking (200–300 rpm). VC concentrations were monitored over 45 days and DNA was extracted (from 13C-VC-amended and 12C-VC-amended microcosms) at three time points during the experimental period (days 15, 32, and 45).
For DNA extraction, at each time point, the entire volume from each mixed culture bottle was centrifuged. Following this, the pellet was washed with MSM and the sample was centrifuged again. The pellet was then resuspended in Tris-EDTA (TE) buffer and DNA was extracted using the Ultra Clean Microbial DNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s procedure. Quantified DNA extracts (∼10 μg) were loaded into Quick-Seal polyallomer tubes (13 by 51 mm, 5.1 mL; Beckman Coulter (Brea, CA) along with a Tris-EDTA (10 mM Tris, 1 mM EDTA, pH 8)-CsCl solution for ultracentrifugation. Prior to sealing (cordless Quick-Seal tube topper; Beckman), the density was determined with a model AR200 digital refractometer (Leica Microsystems Inc., Buffalo Grove, IL) and adjusted by adding small volumes of CsCl solution or TE buffer with a final value of 1.730 g mL−1. The tubes were ultracentrifuged at 178,000×g (20 °C) for 48 h in a StepSaver 70 V6 vertical titanium rotor (8 by 5.1 mL capacity) within a Sorvall WX 80 Ultra Series centrifuge (Thermo Scientific, Waltham, MA). Following ultracentrifugation, the tubes were placed onto a fraction recovery system (Beckman Coulter), and fractions (∼20, 150 μL) were collected. The buoyant density of each fraction was measured, and CsCl was removed by glycogen-assisted ethanol precipitation. The DNA concentration in each fraction was quantified using the Qubit assay (Quant-iT™ dsDNA High-Sensitivity Assay Kit using the Qubit® 2.0 Fluorometer). The abundance of etnE was determined in ultracentrifugation fractions using qPCR. For this, for each time point (days 15, 32, and 45), fractions from one 12C-VC-amended culture and one 13C-VC-amended culture were investigated. In addition, for days 32 and 45, fractions from one 13C-VC-amended culture and fractions from one 12C-VC-amended culture were subjected to Illumina sequencing.
MiSEQ Illumina sequencing and SIP fraction analysis
In all, 94 samples were subjected to high-throughput sequencing (MiSEQ Illumina Sequencing at Michigan State University’s Research Technology and Support Facility, RTSF). This included four DNA extracts (before ultracentrifugation) from the 12C-VC- and 13C-VC-amended cultures at day 32 and at day 45. Ninety additional samples were submitted for Illumina sequencing, and these included thirty individual SIP fractions (following ultracentrifugation) sequenced in triplicate. This involved eight fractions from each of the 13C-VC- and 12C-VC-amended cultures at day 32 (8 fractions × 2 treatments × triplicates = 48). Seven fractions from each of the 13C-VC- and 12C-VC-amended bottles from day 45 were also sequenced (7 fractions × 2 treatments × triplicates = 42).
The fractions were sequenced to determine which organisms were enriched in the heavy fractions and were therefore responsible for VC-assimilation. The fractions were selected based on their buoyant density in comparison to previous SIP studies which illustrated label uptake in fractions with buoyant density values ranging from 1.74 to 1.77 g mL−1 (Sun et al. 2012; Sun and Cupples 2012). In the current study, the fractions selected for sequencing ranged from 1.74 to 1.785 (day 32) and 1.744 to 1.797 g mL−1 (day 45).
PCR and Illumina sequencing were performed at RTSF using a previously reported protocol (Caporaso et al. 2011). Briefly, this involved the amplification of the V4 region of the 16S ribosomal RNA (16S rRNA) gene using a set of multiplex indexed primers. Following amplification, individual reactions were quantified (Picogreen assay), a pool of equimolar amounts of each was made, and these were purified using Ampure XP beads. A final gel purification step was included to ensure that non-specific products were eliminated. The combined library was loaded onto the Illumina MiSEQ Platform using a standard MiSEQ paired end (2 × 250 bp) flow cell and reagent cartridge.
Sequencing data obtained from the MiSEQ platform Laboratory Information Management System were analyzed using Mothur v.1.33.2 (Schloss 2009) using the MiSEQ standard operating procedure (Kozich et al. 2013). The sequence data in the fastq format were processed to remove the barcodes, and these were then aligned to form contiguous sequences. The data were checked for sequencing errors (removing ambiguous bases) and read length (275 bp). The sequences were then aligned to the SILVA database (SILVA version 119) (Pruesse et al. 2007). Additional steps included setting the maximum homopolymer length to 8, checking for chimeras using UCHIME (Edgar et al. 2011), classifying with the Bayesian classifier, and removing unwanted lineages. The sequences were clustered into OTU using a 0.03 cutoff level. Mothur was also used to generate information on phylotypes.
To determine which phylotypes were responsible for label uptake, the most abundant phylotypes in the heavy fractions from the 13C-VC-amended samples from day 32 were determined using the sort function in Excel. These values were then compared to relative abundance of these phylotypes in the fractions from the 12C-VC-amended samples from day 32. Additionally, the relative abundance of these phylotypes was determined in the 13C-VC and 12C-VC fractions from day 45. The relative abundance values were normalized to the mass of DNA in each fraction (Quant-iT™ dsDNA High-Sensitivity Assay Kit using the Qubit® 2.0 Fluorometer) by multiplying the relative abundance by the total mass of DNA in each fraction.
Functional gene (etnE) qPCR and clone libraries
Fractions from all three time points (15, 32, and 45 days) from both the 13C-VC- and 12C-VC-amended cultures were used in the etnE qPCR analysis. This resulted in 78 DNA fractions (buoyant density range = 0.981–1.799 g mL−1) being analyzed by qPCR for etnE. An ABI 7000 Sequence Detection System (Applied Biosystems) with a 96-well plate was used for qPCR, as described previously (Jin et al. 2010). Reaction mixtures (25 μL) contained 12.5 μL of Power SYBR Green PCR Master Mix (Applied Biosystems), 750 nM etnE qPCR primers RTE_F (5′-CAGAAYGGCTGYGACATYATCCA-3′) and RTE_R (5′-CSGGYGTRCCCGAGTAGTTWCC-3′) (Jin and Mattes 2010), and 2 μL of DNA extract. Each fraction was analyzed in duplicate.
Standard curves were developed in triplicate using etnE from Nocardioides sp. strain JS614 (Jin et al. 2010) amplified using the CoMF1L (5′-AACTACCCSAAYCCSCGCTGGTACGAC-3′) and CoMR2E (5′-GTCGGCAGTTTCGGTGATCGTGCTCTTGAC-3′) (Coleman and Spain 2003). Reactions (25 μL) contained 12.5 μL Qiagen PCR Master Mix, 0.2 μM of each primer, and 2 ng of total DNA. Genes per microliter of PCR product were estimated using a previously reported equation (Jin and Mattes 2010). ABI 7000 System SDS software (Applied Biosystems) was used to analyze real-time PCR fluorescence data using the auto baseline function. The following information is provided in accordance with MIQE guidelines (Bustin et al. 2009): The fluorescence threshold was set manually (at 0.05505) to optimize qPCR efficiency (102.5 %) and obtain a linear fit of the standard curve (>0.9976). The Y-intercept of the standard curve was 31.96.
A light fraction (denoted as L9, buoyant density = 1.734 g mL−1) and a heavy fraction (denoted as L4, buoyant density = 1.773 g mL−1) from day 45 were selected as representative fractions for the etnE clone libraries. The L4 and L9 fractions were purified as stated above, and etnE was amplified using a touch-down PCR protocol. Reaction mixtures (25 μL) contained Qiagen Taq Core Kit 10X buffer (12.5 μL), 25 mM Mg2+ solution (0.5 μL), 10 mM dNTPs (0.5 μL), Taq polymerase (0.2 μL), the CoM-F1L/CoM-R2E primer set (0.2 μM)(Coleman and Spain 2003), and 1 μL DNA. The thermocycling protocol consisted of an initial denaturation step (94 °C, 5 min), followed by a touch-down phase (20 cycles of 94 °C for 30 s, 65 °C for 45 s (0.5 °C decrease of each cycle), and 72 °C for 2 min), a secondary amplification (10 cycles of 94 °C for 30 s, 55 °C for 45 s, 72 °C for 2 min), and a final extension (72 °C for 15 min). PCR products (891 bp) from L4 and L9 were purified with QIAquick PCR Purification Kit (Qiagen) and cloned with the Invitrogen TA Cloning Kit with an overnight ligation at 4 °C into the pCR®2.1 vector. A 1:1 molar insert to vector ratio was used. Ligations were transformed into One Shot® TOP10 Chemically Competent E. coli. Transformants were analyzed according to the cloning kit instructions. Plasmids were extracted using QIAprep Spin Miniprep Kit and PCR-screened with M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′) primers. Those with the appropriately sized inserts were Sanger-sequenced at the University of Iowa Institute of Human Genetics Genomics Division with the M13F primer. Sequences with good quality (9 sequences from L4 and 10 sequences from L9) were used for alignment via Clustal W (Thompson et al. 2002) and further phylogenetic analysis with MEGA 5 (Tamura et al. 2011).
Genbank accession numbers
The Nocardioides sp. partial 16S rRNA gene was deposited in the NCBI Genbank Database (Accession Number: KJ509930.1). The etnE sequences were also placed in this database (Accession Numbers: KJ509928-KJ509936, KM245084, KM245085). Illumina sequencing data was deposited in the NCBI Sequence Read Archive under BioProject Number SAMN03202071.
Results
VC degradation and total microbial community characterization
VC degradation occurred in both of the 13C-VC-amended and 12C-VC-amended mixed cultures but not in the abiotic controls, confirming biological removal (Supplementary Fig. S1). The DNA extracted during this period (days 15, 32, and 45) was subjected to ultracentrifugation, and the heavy fractions (from day 32 and 45) were submitted for Illumina sequencing. Following Mothur analysis, 9,029,943 sequences were obtained. On average, each sample generated 99,230 ± 36,815 sequences (total sequences, not unique sequences).
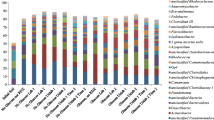
Illumina sequencing of the 16S rRNA gene was also performed on four total DNA extracts (before ultracentrifugation, two from day 32, and two from day 45). The most abundant phylotypes were determined (relative abundance of 1 %) for each culture (Fig. 1). Similar phylotypes were observed at both time points for the 13C-VC- and 12C-VC-amended mixed cultures. In all four cultures, the phylotype Gp4 was present at a high relative abundance (9.9–30.8 %). In addition, the phylotypes Aquabacterium, Sediminibacterium, Nocardioides, and unclassified Comamonadaceae were all more abundant compared to the other phylotypes. The most abundant phylotypes classified within eight phyla (Proteobacteria, Actinobacteria, Acidobacteria, Bacteroidetes, Firmicutes, Flavobacteria, Gemmatimonadetes, and Verrucomicrobia). The most dominant phyla included Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, and Flavobacteria.

Microbial composition at the genus level (unless unclassified) of the four VC enrichment cultures. Only those with a relative abundance of 1 % or more in at least one of the cultures are included. The data were obtained from total DNA extracts (before ultracentrifugation)
Identification of VC-assimilators
To identify the microorganisms responsible for the uptake of 13C from VC (or VC degradation products), DNA extracts from two time points (days 32 and 45) were subject to ultracentrifugation, fractioning, and sequencing. For this, the most abundant phylotypes in the heavy fractions from the 13C-VC-amended samples were determined and compared to the relative abundance of these phylotypes in the fractions from the 12C-VC-amended samples (Fig. 2). In all, four phylotypes showed dominance in the 13C-VC-amended cultures compared to the 12C-VC-amended cultures, including Rhodoferax, Nocardioides, Tissierella, and Brevundimonas. As stated previously, the Illumina relative abundance data were normalized by the amount of DNA in each fraction. Only low levels of enrichment were noted from the phylotypes Tissierella and Brevundimonas at both time points. The other two phylotypes (Nocardioides and Rhodoferax) were enriched at a higher level, with Rhodoferax illustrating the highest level of enrichment. The Nocardioides sequences from these cultures were compared to those in GenBank (Supplementary Fig. S2).

Normalized abundance of dominant phylotypes in ultracentrifugation fractions from the labeled (13C) VC-amended culture compared to their abundance in the unlabeled (12C) VC-amended culture from day 32 (first column) and day 45 (second column). The axis represents the relative abundance of each phylotype (determined by Illumina sequencing) normalized by the amount of DNA in that fraction (determined by Qubit). Error bars represent the standard deviations from triplicate values of Illumina data
The relative abundance of each of these four phylotypes was determined from the total DNA extract sequencing data. These data indicate that only Nocardioides was a dominant community member (4.1–18.7 %). The other three phylotypes illustrated only a low relative abundance in the community (<0.08 %).
Functional gene (etnE) abundance in SIP fractions
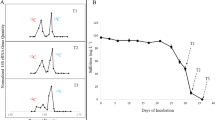
To provide evidence that functional genes associated with VC-assimilation were also enriched in the heavy fractions, we quantified etnE abundance by qPCR (Fig. 3 and Supplementary Table S1). Indeed, as VC degradation proceeded, increased etnE abundance was observed among heavier fractions (buoyant density = 1.720–1.780 g mL−1) from the 13C-VC-fed culture. Meanwhile, the majority of the etnE abundance in the fractions from the unlabeled VC amended cultures occurred within a lower buoyant density range (buoyant density = 1.700–1.740 g mL−1). It is unclear why the etnE gene abundance values were lower at day 45 compared to those at days 15 and 32.

etnE copies after fractionation from 13C-VC-amended (triangles, labeled) and 12C-VC-amended (diamonds, unlabeled) cultures at day 15 (a), day 32 (b), and day 45 (c). The etnE abundance represents the average of duplicate samples and the error bars depict the standard deviation
Phylogenetic analysis of etnE in SIP fractions
Clone libraries were constructed with etnE PCR products from one light fraction (L4) and one heavy fraction (L9) to compare the sequence diversity of 13C-enriched etnE with the etnE in the unlabeled fraction. An etnE phylogenetic tree (Supplementary Fig. S3) revealed that the etnE in these two fractions were similar and that they grouped with the etnE from Nocardioides sp. JS614 (bootstrap value of 99 %).
A percent identity matrix (Supplementary Table S2) further showed that etnE was highly conserved among the light (L4) and heavy (L9) fractions, varying from 94.7 to 100 % identical to each other. An exception is clone L4-10, which was 94.7 to 95.2 % identical to the remaining sequences. Sequences from the light fraction (L4) were 97.8 to 100 % identical to each other, while sequences within the heavy fraction were 99.2 to 99.9 % identical to each other.
Discussion
In this study, VC-assimilating microorganisms were investigated in a mixed culture derived from contaminated site groundwater using SIP, high-throughput sequencing, and qPCR. Although others have isolated VC-assimilating microorganisms (Coleman et al. 2002b; Danko et al. 2004; Elango et al. 2006; Fathepure et al. 2005; Hartmans and Debont 1992; Hartmans et al. 1985; Jin et al. 2010; Jin and Mattes 2008; Taylor et al. 2007; Verce et al. 2000), the microorganisms responsible for VC-assimilation within a mixed culture is more challenging to determine. Such research is important because there is likely a greater diversity of VC-assimilators in the environment than is currently represented in pure culture.
Here, four phylotypes were responsible for 13C uptake (Fig. 2). As with many other SIP studies, it is unclear if these phylotypes were involved in label uptake directly from VC or from VC degradation products. VC-assimilators are known to use an alkene monooxygenase to attack VC (Mattes et al. 2010) which forms VC epoxide (chlorooxirane). This intermediate is very unstable in aqueous systems (∼1.6 min half-life). It can spontaneously rearrange into chloroacetaldehyde (Barbin et al. 1975). It can also undergo hydrolysis to glycolaldehyde. It is possible that rearrangement or hydrolysis of VC epoxide occurred in the mixed culture and that label uptake by bacteria that are not true VC-assimilators may have also occurred.
From the Proteobacteria, the phylotypes Brevundimonas (Alphaproteobacteria) and Rhodoferax (Betaproteobacteria) were observed in the heavy fractions from the 13C-VC-amended cultures at both time points. Previously identified VC-assimilators in this phylum include Pseudomonas (Gammaproteobacteria) (Danko et al. 2004; Verce et al. 2000), Ochrobactrum (Alphaproteobacteria) (Danko et al. 2004), and Ralstonia (Betaproteobacteria) (Elango et al. 2006). In the current study, sequences corresponding to the genera Pseudomonas and Ralstonia were present but were not enriched in the heavy fractions.
From the phylum Firmicutes, one phylotype (Tissierella) was observed in the heavy fractions from the 13C-VC-amended cultures at both time points. This phylum (Firmicutes) has not yet been associated with VC-assimilation but has been previously reported in sites contaminated with chlorinated solvents (Miller et al. 2007).
Similar to previous research (Coleman et al. 2002b), the current study indicated that the phylotype Nocardioides (Actinobacteria) was responsible for carbon assimilation from VC. This phylotype was observed in the heavy fractions from the 13C-VC-amended cultures at both day 32 and day 45. Additionally, Nocardioides was a significant community member in all four cultures (4.1, 6.2, 8.9, and 18.7 %). The other previously reported VC-assimilating phylotype (Mycobacterium) within the Actinobacteria (Coleman et al. 2002b; Fathepure et al. 2005; Hartmans and Debont 1992; Hartmans et al. 1985; Jin et al. 2010; Jin and Mattes 2008; Taylor et al. 2007) was observed in the culture only at low levels (relative abundance 0.001–0.079 %) and was not found in the heavy fractions, indicating that it was not responsible for carbon assimilation from VC. Overall, of the four enriched phylotypes, Nocardioides is the only microorganism previously linked to VC-assimilation.
The shift in etnE abundance toward the heavier fractions during VC degradation indicates the accumulation of 13C in etnE. The strongest shift was at day 32; therefore, this time likely represents the greatest label uptake from VC. The shift is less clear at day 45, possibly indicating label cross feeding. Also, the limited shift at day 15 suggests that label uptake was minor early in the incubation. These observations guided the sequencing efforts to day 32 and day 45 samples. Each of the 19 unique etnE sequences retrieved from both light (L4) and heavy (L9) fractions formed a clade (a grouping) with the etnE from Nocardioides sp. JS614. This pattern contrasts with previous studies where VC- or ethene-degrading isolates were primarily Mycobacterium spp. (Chuang et al. 2010). The data suggests that potentially greater etnE diversity is present within the ethene- and VC-assimilating Nocardioides spp. or that Nocardioides-like etnE sequences are more widely distributed in the environment than Mycobacterium-like etnE. It is interesting to note that although the VC-assimilators identified by SIP are relatively diverse in comparison to known isolates, the etnE genes harbored by these bacteria appear to be relatively conserved.
In summary, the microorganisms responsible for assimilating 13C from VC within a mixed culture derived from contaminated site groundwater were identified. SIP analysis and Illumina sequencing indicated that Nocardioides was a dominant culture phylotype as well as a key 13C assimilator. Therefore, both a previously identified VC-assimilating genus (Nocardioides) a novel microorganisms (Rhodoferax, Tissierella, and Brevundimonas) were responsible for carbon uptake from VC. Interestingly, Rhodoferax was enriched to the highest level and could therefore represent a particularly important novel VC degrader. Despite the diversity of newly discovered VC-assimilators, the functional gene etnE associated with VC-assimilation was relatively conserved in this mixed culture.
References
Atashgahi S, Maphosa F, Dogan E, Smidt H, Springael D, Dejonghe W (2013) Small-scale oxygen distribution determines the vinyl chloride biodegradation pathway in surficial sediments of riverbed hyporheic zones. FEMS Microbiol Ecol 84(1):133–142
Barbin A, Bresil H, Croisy A, Jacquignon P, Malaveille C, Montesano R, Bartsch H (1975) Liver-microsome-mediated formation of alkylating-agents from vinyl bromide and vinyl chloride. Biochem Biophys Res Commun 67(2):596–603
Begley JF, Hansen E, Wells AK, Fogel S, Begley GS (2009) Assessment and monitoring tools for aerobic bioremediation of vinyl chloride in groundwater. Remediat J 20(1):107–117
Begley JF, Czarnecki M, Kemen S, Verardo A, Robb AK, Fogel S, Begley GS (2012) Oxygen and ethene biostimulation for a persistent dilute vinyl chloride plume. Ground Water Monit Remediat 32(1):99–105
Bradley PM (2003) History and ecology of chloroethene biodegradation: A review. Bioremediat J 7(2):81–109
Bucher JR, Cooper G, Haseman JK, Jameson CW, Longnecker M, Kamel F, Maronpot R, Matthews HB, Melnick R, Newbold R (2005) Report on Carcinogens, 11th edn. Public Health Service National Toxicology Program, Research Triangle Park, NC, US Department of Health and Human Services
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522
Chuang AS, Jin YO, Schmidt LS, Li YL, Fogel S, Smoler D, Mattes TE (2010) Proteomic analysis of ethene-enriched groundwater microcosms from a vinyl chloride-contaminated site. Environ Sci Technol 44(5):1594–1601
Coleman NV, Spain JC (2003) Distribution of the coenzyme M pathway of epoxide metabolism among ethene- and vinyl chloride-degrading Mycobacterium strains. Appl Environ Microb 69(10):6041–6046
Coleman NV, Mattes TE, Gossett JM, Spain JC (2002a) Biodegradation of cis-dichloroethene as the sole carbon source by a beta-proteobacterium. Appl Environ Microb 68(6):2726–2730
Coleman NV, Mattes TE, Gossett JM, Spain JC (2002b) Phylogenetic and kinetic diversity of aerobic vinyl chloride-assimilating bacteria from contaminated sites. Appl Environ Microb 68(12):6162–6171
Danko AS, Luo MZ, Bagwell CE, Brigmon RL, Freedman DL (2004) Involvement of linear plasmids in aerobic biodegradation of vinyl chloride. Appl Environ Microb 70(10):6092–6097
Dumont MG, Murrell JC (2005) Stable isotope probing - linking microbial identity to function. Nat Rev Microbiol 3(6):499–504
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200
Elango VK, Liggenstoffer AS, Fathepure BZ (2006) Biodegradation of vinyl chloride and cis-dichloroethene by a Ralstonia sp strain TRW-1. Appl Microbiol Biotechnol 72(6):1270–1275
Fathepure BZ, Elango VK, Singh H, Bruner MA (2005) Bioaugmentation potential of a vinyl chloride-assimilating Mycobacterium sp., isolated from a chloroethene-contaminated aquifer. FEMS Microbiol Lett 248(2):227–234
Gossett JM (1987) Measurement of Henrys Law constants for C1 and C2 chlorinated hydrocarbons. Environ Sci Technol 21(2):202–208
Hartmans S, Debont JAM (1992) Aerobic vinyl chloride metabolism in Mycobacterium aurum L1. Appl Environ Microb 58(4):1220–1226
Hartmans S, Debont JAM, Tramper J, Luyben K (1985) Bacterial degradation of vinyl chloride. Biotechnol Lett 7(6):383–388
Jin YO, Mattes TE (2008) Adaptation of aerobic, ethene-assimilating Mycobacterium strains to vinyl chloride as a growth substrate. Environ Sci Technol 42(13):4784–4789
Jin YO, Mattes TE (2010) A quantitative PCR assay for aerobic, vinyl chloride- and ethene-assimilating microorganisms in groundwater. Environ Sci Technol 44(23):9036–9041
Jin YO, Mattes TE (2011) Assessment and modification of degenerate qPCR primers that amplify functional genes from etheneotrophs and vinyl chloride-assimilators. Lett Appl Microbiol 53(5):576–580
Jin YO, Cheung S, Coleman NV, Mattes TE (2010) Association of missense mutations in epoxyalkane Coenzyme M transferase with adaptation of Mycobacterium sp strain JS623 to growth on vinyl chloride. Appl Environ Microb 76(11):3413–3419
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina Sequencing Platform. Appl Environ Microb 79(17):5112–5120
Lueders T, Wagner B, Claus P, Friedrich MW (2004) Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic interactions with fungi and protozoa in oxic rice field soil. Environ Microbiol 6(1):60–72
Luo C, Xie S, Sun W, Li X, Cupples AM (2009) Identification of a novel toluene-degrading bacterium from the candidate phylum TM7, as determined by DNA stable isotope probing. Appl Environ Microb 75(13):4644–4647
Madsen EL (2006) The use of stable isotope probing techniques in bioreactor and field studies on bioremediation. Curr Opin Biotechnol 17(1):92–97
Mattes TE, Coleman NV, Gossett JM, Spain JC (2005) Physiological and molecular genetic analyses of vinyl chloride and ethene biodegradation in Nocardioides sp. strain JS614. Arch Microbiol 183:95–106
Mattes TE, Alexander AK, Coleman NV (2010) Aerobic biodegradation of the chloroethenes: pathways, enzymes, ecology, and evolution. FEMS Microbiol Rev 34(4):445–475
Miller TR, Franklin MP, Halden RU (2007) Bacterial community analysis of shallow groundwater undergoing sequential anaerobic and aerobic chloroethene biotransformation. FEMS Microbiol Ecol 60(2):299–311
Patterson BM, Aravena R, Davis GB, Furness AJ, Bastow TP, Bouchard D (2013) Multiple lines of evidence to demonstrate vinyl chloride aerobic biodegradation in the vadose zone, and factors controlling rates. J Contam Hydrol 153:69–77
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Gloeckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35(21):7188–7196
Radajewski S, Ineson P, Parekh NR, Murrell JC (2000) Stable-isotope probing as a tool in microbial ecology. Nature 403(6770):646–649
Schloss PD (2009) A high-throughput DNA sequence aligner for microbial ecology studies. PLOS 4(12):1–9
Singleton DR, Powell SN, Sangaiah R, Gold A, Ball LM, Aitken MD (2005) Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl Environ Microb 71(3):1202–1209
Sun WM, Cupples AM (2012) Diversity of five anaerobic toluene-degrading microbial communities investigated using stable isotope probing. Appl Environ Microb 78(4):972–980
Sun W, Xie S, Luo C, Cupples AM (2010) Direct link between toluene degradation in contaminated-site microcosms and a Polaromonas strain. Appl Environ Microb 76(3):956–959
Sun W, Sun X, Cupples AM (2012) Anaerobic methyl tert-butyl ether-degrading microorganisms identified in wastewater treatment plant samples by stable isotope probing. Appl Environ Microb 78(8):2973–2980
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Taylor AE, Dolan ME, Bottomley PJ, Semprini L (2007) Utilization of fluoroethene as a surrogate for aerobic vinyl chloride transformation. Environ Sci Technol 41(18):6378–6383
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX Curr Protoc Bioinformatics. John Wiley & Sons, Inc., Hoboken, NJ
Verce MF, Ulrich RL, Freedman DL (2000) Characterization of an isolate that uses vinyl chloride as a growth substrate under aerobic conditions. Appl Environ Microb 66(8):3535–3542
Acknowledgments
We thank James Begley and Bioremediation Consulting, Inc. for coordinating the sampling of Carver, MA groundwater, and Yang Oh Jin for initial development and maintenance of the VC-degrading culture used in this study. This work was funded by a collaborative NSF Grant (number 1233154) awarded to T. E. Mattes and A. M. Cupples.
Conflict of interest
The authors have no conflict of interest with the methods and data described in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 295 kb)
Rights and permissions
About this article

Cite this article
Paes, F., Liu, X., Mattes, T.E. et al. Elucidating carbon uptake from vinyl chloride using stable isotope probing and Illumina sequencing. Appl Microbiol Biotechnol 99, 7735–7743 (2015). https://doi.org/10.1007/s00253-015-6606-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6606-1