
Abstract
L-Ribose isomerase from Cellulomonas parahominis MB426 (CpL-RI) can catalyze the isomerization between L-ribose and L-ribulose, which are non-abundant in nature and called rare sugars. CpL-RI has a broad substrate specificity and can catalyze the isomerization between D-lyxose and D-xylulose, D-talose and D-tagatose, L-allose and L-psicose, L-gulose and L-sorbose, and D-mannose and D-fructose. To elucidate the molecular basis underlying the substrate recognition mechanism of CpL-RI, the crystal structures of CpL-RI alone and in complexes with L-ribose, L-allose, and L-psicose were determined. The structure of CpL-RI was very similar to that of L-ribose isomerase from Acinetobacter sp. strain DL-28, previously determined by us. CpL-RI had a cupin-type β-barrel structure, and the catalytic site was detected between two large β-sheets with a bound metal ion. The bound substrates coordinated to the metal ion, and Glu113 and Glu204 were shown to act as acid/base catalysts in the catalytic reaction via a cis-enediol intermediate. Glu211 and Arg243 were found to be responsible for the recognition of substrates with various configurations at 4- and 5-positions of sugar. CpL-RI formed a homo-tetramer in crystals, and the catalytic site independently consisted of residues within a subunit, suggesting that the catalytic site acted independently. Crystal structure and site-direct mutagenesis analyses showed that the tetramer structure is essential for the enzyme activity and that each subunit of CpL-RI could be structurally stabilized by intermolecular contacts with other subunits. The results of growth complementation assays suggest that CpL-RI is involved in a novel metabolic pathway using L-ribose as a carbon source.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prokaryotes potentially have the ability to use the various sugars as carbon sources in order to enhance their survival. The pathway for the metabolism of sugars such as D-lyxose and L-ribose, which are non-abundant in nature and called rare sugars, to form intermediates of the pentose phosphate pathway has been considered (Izumori et al. 1976). The pentose phosphate pathway, which generates NADPH, is very important for the production of metabolic energy (Gunsalus et al. 1955; Heath et al. 1958). Previous studies reported that a number of microorganisms could survive on medium using D-lyxose as a carbon source (Stevens and Wu 1976; Anderson and Allison 1965; Cho et al. 2007; Kwon et al. 2010; Park et al. 2010; Patel et al. 2011; Choi et al. 2012). However, microorganisms that can grow using L-ribose as a carbon source have not yet been identified. We were the first to report the cloning, amino acid sequence, and overexpression in Escherichia coli (E. coli) of the novel enzyme, L-ribose isomerase from Acinetobacter sp. strain DL-28 (AcL-RI) (Shimonishi and Izumori 1986; Mizanur et al. 2001). This enzyme was expected to be involved in the metabolic pathway for L-ribose. We then determined the crystal structures of AcL-RI (PDB ID: 4Q0P) (Yoshida et al. 2011, 2014). The overall structure of the subunit of AcL-RI showed acupin-type β-barrel with a metal ion coordinated by a Glu and three His residues. Although the subunit structure of AcL-RI was similar to those of the previously reported D-lyxose isomerases from pathogenic E. coli O157:H7 (EcD-LI, PDB ID: 3MPB) (Staalduinen et al. 2010) and Bacillus subtilis strain 168 (BsD-LI, PDB ID: 2Y0O) (Marles-Wright and Lewis 2011), significant differences were observed in their three-dimensional structures and enzymatic properties. The amino acid sequence identities of AcL-RI with EcD-LI and BsD-LI were 18 and 19 %, respectively. AcL-RI forms a tetramer, while EcD-LI and/or BsD-LI form dimers. AcL-RI shows higher activity for D-lyxose (47 % that of L-ribose) (Yoshida et al. 2014), while EcD-LI exhibits a lower activity for L-ribose (1.6 % that of D-lyxose) (Staalduinen et al. 2010). D-Lyxose isomerases from Aerobacter aerogenes (Anderson and Allison 1965), Cohnella laevoribosii (Cho et al. 2007), Providencia stuartii (Kwon et al. 2010; Park et al. 2010), Bacillus licheniformis (Patel et al. 2011), Dictyoglomus turgidum (Choi et al. 2012), and Bacillus subtilis (Marles-Wright and Lewis 2011) have already been characterized but showed low activity for L-ribose.
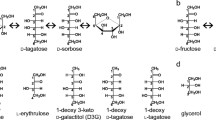
We recently reported the cloning and characterization of another L-ribose isomerase from Cellulomonas parahominis (CpL-RI, 249 amino acids, MW = 27,435, UniProt accession number L0N3Y0) that had a broad substrate specificity (Morimoto et al. 2013). The amino acid sequence identity of CpL-RI with AcL-RI was 75 %. CpL-RI was shown to catalyze reversible isomerization not only between L-ribose and L-ribulose but also between D-lyxose and D-xylulose, D-talose and D-tagatose, L-allose and L-psicose (L-allulose), L-gulose and L-sorbose, and D-mannose and D-fructose (Fig. 1). Due to this enzymatic property, CpL-RI may potentially be used in the production of various rare sugars. CpL-RI also exhibited its maximum enzymatic activity at a higher temperature (40 °C) than that of AcL-RI (30 °C). Therefore, CpL-RI could be a good candidate as the production enzyme of rare sugars, which are relatively scarce or non-abundant in nature and expensive due to inconvenient production methods. Interest in rare sugars has increased due to their physiological functions, particularly their ability to promote human health (Levin 2002; Hayashi et al. 2010). Recently, a rare sugar syrup containing D-psicose has been commercially available as a functional food, Rare Sugar Sweet® (Matsutani Chemical Industry Co,. Ltd., Hyogo, Japan) (Hayashi et al. 2014). L-Ribose has attracted attention as a precursor for the synthesis of L-nucleoside analogs, which have been used as antiviral drugs in the treatment of several viral diseases, such HIV and hepatitis (Gumina et al. 2001; Cheng 2001).

Chemical reactions catalyzed by CpL-RI. Relative activity for each substrate to L-ribose is given in parentheses
To elucidate the molecular basis underlying the unique enzymatic properties of CpL-RI, the determination of its three-dimensional structure is very important. Although we previously determined the crystal structures of AcL-RI in complexes with L-ribose and an inhibitor, ribitol (Yoshida et al. 2014), the enzyme structures free from a substrate and in complexes with other substrate than L-ribose are not available. We here report the crystal structures of CpL-RI alone and in complexes with L-ribose, L-allose, and L-psicose.
Materials and methods
Protein preparation
The CpL-RI gene (Genbank accession number AB729109.2) was cloned into a pQE30 expression vector containing an N-terminal hexa-histidine tag (Qiagen, Venlo, The Netherlands), previously (Morimoto et al. 2013). Using the constructed plasmid (pCpL-RI), CpL-RI was expressed in E. coli JM109 with Super broth medium containing 1.0 mM MnCl2, and purified by the affinity column, HisTrapTM HP (5 ml) (GE Healthcare Bio-Sciences Corp., Piscataway, NJ), and anion-exchange column, RESOURCE Q (6 ml) (GE Healthcare Bio-Sciences Corp) (Morimoto et al. 2013). Purified CpL-RI was dialyzed against a buffer solution (5.0 mM Tris-HCl, pH 8.0) overnight and was concentrated to 30 mg/ml using an Amicon Ultra-4 (30 kDa) (Millipore, Billerica, MA, USA).
Crystallization and X-ray diffraction data collection
Initial crystal screening was performed by the sitting drop vapor diffusion method using the mosquito system (TTP LabTech, Hertfordshire, UK) with Crystal Screen kits 1&2, SaltRX Screen, Index Screen (Hampton Research Corp., CA, USA), and Emerald BioSystems Wizard I - IV (Emerald BioSystems, Inc., WA, USA) by the sitting drop method using 96-well plates (VIOLAMO) (AS ONE Corporation, Osaka, Japan) at 20 °C. To obtain crystals that diffracted well, the crystallization condition was optimized as follows; a protein concentration of 30 mg/ml in 5.0 mM Tris-HCl (pH 8.0), reservoir solution of 0.1 M sodium acetate trihydrate (pH 4.6), and 3.9 M ammonium acetate. Crystals were grown by the hanging-drop vapor diffusion method in a drop mixing 2.0 μl each of the protein solution and reservoir solution against 450 μl of the reservoir solution at 20 °C. A single crystal was mounted by a cryo-loop and flash-cooled in stream of nitrogen gas at −173 °C. X-ray diffraction data were collected using the ADSC Quantum 315r detector system on the BL5A in the Photon Factory (Tsukuba, Japan), and Rigaku R-AXIS VII imaging system on a Rigaku RA-Micro7HF rotating anode (CuKα) with ValiMax optics (40 kV, 30 mA). To obtain complex structures with substrates, 50 % (w/v) sugar solutions (L-ribose, L-allose, and L-psicose) were used as cryoprotectants. Diffraction data were processed using the program HKL2000 (Otwinowski and Minor 1997), CrystalClear system (Rigaku Corp. Tokyo, Japan), and CCP4 program suite (Winn et al. 2011). The collected data and scaling results are listed in Table 1.
The initial phases of CpL-RI were determined by molecular replacement (MOLREP) (Vagin and Teplyakov 1997) using the structure of AcL-RI (PDB ID: 4Q0P) as a probe model. Two molecules (Mol-A and Mol-B) were found in an asymmetric unit. Further model building was performed with the programs Coot (Emsley and Cowtan 2004) and X-fit (McRee 1999). Water molecules were gradually introduced if the peaks above 3.5 σ in the (Fo-Fc) electron density map were in the range of a hydrogen bond. The structure was refined using the programs Refmac5 (Murshudov et al. 1997) and CNS (Brunger 1993) with Engh and Huber stereochemical parameters at a resolution of 1.90 Å. The bound metal ion was refined as Mn2+ because CpL-RI was overexpressed in the medium containing 1.0 mM of MnCl2. The B-factors of Mn2+ were 20.5 (Mol-A) and 21.7 Å2 (Mol-B), which were similar to those of the metal-coordinated amino acid residues. Using the structure of CpL-RI, the complex structures with L-ribose, L-allose, and L-psicose were determined by the isomorphous replacement method and refined at resolutions of 1.95, 1.95, and 2.00 Å, respectively. In a Ramachandran plot (Ramachandran and Sasisekharan 1968), the number of residues in the most favored regions was determined by the program PROCHECK (Laskowski et al. 1992). Refinement statistics are listed in Table 1. Figures 2, 3, and 4 were illustrated by the program PyMol (DeLano 2002).

Overall structure of CpL-RI. a Structure of subunit of CpL-RI is illustrated with the bound metal (dark blue sphere) and the metal-coordinated amino acid residues. Selected amino acid residues involved in dimer-dimer interaction are indicated by orange sticks, and the deleted region in mutant form is indicated by a dotted circle. High mobility region between B10 and H6 is indicated by yellow. b The dimer structure of CpL-RI is illustrated viewing from the dimer-dimer interface. c The structure of the tetramer of CpL-RI is illustrated. d The tetramer structure of CpL-RI is illustrated in a surface representation. The amino acid residues involved in the dimer-dimer interactions are shown in red (Color figure online)

Structure of the catalytic site of CpL-RI. a The catalytic site with bound waters (red sphere) is illustrated. Selected interactions among amino acid residues, substrates, water molecules, and a metal ion are indicated by dotted lines. b The catalytic site with a bound L-ribopyranose (green stick) is illustrated. c The catalytic site with a bound L-ribose in a linear form (green stick) is illustrated. d The side view of c is illustrated with the non-polar amino acid residues in the catalytic site. e The catalytic site with a bound L-allose in a linear form (yellow stick) in Mol-A is illustrated. f The catalytic site with a bound L-psicofuranose (blue stick) is illustrated (Color figure online)

Interactions between dimes. Interface among Mol-A (cyan), Mol-C (yellow), and Mol-D (pink) is illustrated with the amino acid residues involving in the dimer-dimer interactions. Intermolecular hydrogen bonds are indicated by dotted lines. The deletion region in the mutant form (V41-G-S-V62) is shown by gray (Color figure online)
Preparation of CpL-RI mutant forms
The Quick change II site-directed mutagenesis kit (Agilent Technology, CA, USA) and plasmid pCpLRI encoding the CpL-RI gene were used to construct the mutant forms E17A, S20W, A24W, and R26A. The primers used for constructions are listed in Supplemental Table S1. To construct a deletion mutant form (V41-G-S-V62), in which 20 amino acid residues of Phe42-Glu61 were replaced by Gly-Ser, PCR reactions were separately carried out for regions upstream and downstream of the deletion site. Each region was amplified using a set of primers (sense2 and anitisense1 for the upstream region, and sense1 and anitisense2 for the downstream region) (Supplemental Table S1). Two amplified PCR products were used as templates for the next PCR step. The final amplified PCR product was digested with BamHI and HindIII and inserted into pQE30, and this constructed plasmid was introduced into E. coli JM109.
Enzyme assay and gel filtration
Enzymatic activity was measured as the accumulation of ketose by the cysteine-carbazole method. The reaction mixture containing 50 mM glycine-NaOH buffer (pH 9.0), 1.0 mM MnCl2, 20 mM L-ribose, and CpL-RI enzyme (1 mg/ml) was incubated at 40 °C for 10 min. One unit of enzyme activity was defined as the amount of enzyme (mg) that produced 1.0 μmol of product per min under the assay conditions.
Gel filtration was carried out using a column of Superdex 200 10/300 GL(GE Healthcare Bio-Sciences Corp) with a solution of 20 mM Tris-HCl buffer and 150 mM NaCl, pH 8.0, in a flow rate of 0.5 ml/min at 4 °C. The standard proteins used for gel filtration were ovalbumin (MW, 43,000; retention time, 15.5 min), conalbumin (MW, 75,000; retention time, 14.0 min), and aldolase (MW, 158,000; retention time, 12.6 min).
Growth complementation experiments
E. coli JM109 cells transformed with pQE30 or pQE30 harboring the CpL-RI gene were grown at 30 °C on M9 minimal medium with 0.4 % L-arabinose or L-ribose, and 1 mM IPTG. The Cellulomonus parahominis MB426 strain was grown at 37 °C on M9 minimal medium with 0.4 % L-ribose.
Results
Crystal structure determination
Four crystal structures were successfully determined: CpL-RI without a substrate, in which water molecules were in the catalytic site (CpL-RI/water), and CpL-RI in complexes with L-ribose (CpL-RI/L-ribose), L-allose (CpL-RI/L-allose), and L-psicose (CpL-RI/L-psicose). Structures were refined to R = 0.189 (R free = 0.219) for CpL-RI/water, R = 0.202 (0.242) for CpL-RI/L-ribose, R = 0.217 (0.255) for CpL-RI/L-allose, and R = 0.221 (0.261) for CpL-RI/L-psicose, with good chemical geometry, as listed in Table 1. Two protein molecules (Mol-A and Mol-B) were detected in an asymmetric unit, and they had almost equivalent structures with root-mean-square deviation (RMSD) of main chain atoms being 0.70 Å (CpL-RI/water), 0.68 Å (CpL-RI/L-ribose), 0.95 Å (CpL-RI/L-allose), and 0.68 Å (CpL-RI/L-psicose). The final (2F o-F c) electron density maps showed that most atoms of protein molecules, metal ions, and solvent molecules fit well (Supplementary Fig. S1). Poor and invisible electron densities were detected in several substrates, His-tag residues, and the loop region (213-219) (Supplementary Fig. S2).
Overall structure
The structure of the subunit of CpL-RI was very similar to that of AcL-RI with an RMSD of 1.13 Å (Cα atoms). CpL-RI adopted a cupin-type β-barrel structure, having four α-helices (H1, H3, H4, and H7), three 310-helices (H2, H5, and H6), and two large β-sheets (β-sheet 1 and β-sheet 2) formed by 11 β-strands (B1–B11), which were numbered and labeled as represented in Fig. 2a. Two molecules in the asymmetric unit (Mol-A and Mol-B) formed a homo-dimer (Fig. 2b) with 2-fold symmetry, and this dimer formed a homo-tetramer with the symmetry-operated dimer (Mol-C and Mol-D) (Fig. 2c, d). The β-sheet 1 formed by six β-strands (B1, B2, B3, B10, B5, and B8) was located at the interface between Mol-A and Mol-B, contributing to the intermolecular interactions of the dimer. The β-sheet 2 formed by five β-strands (B11, B4, B9, B6, and B7) was located on the molecular surface. Two β-sheets faced each other at the center of the subunit, and the catalytic site was between them with a bound metal ion (Mn2+). Two β-sheets were surrounded by helices and loop regions. Two α-helices, H1 and H3, were located at the interface between dimers (Mol-A/Mol-B and Mol-C/Mol-D) (Fig. 2b, c) and were involved in dimer-dimer interactions. The kinked α-helix, H4, was located at the interface between Mol-A and Mol-B, contributing to the formation of a stable dimer. Other short helices, H2, H5, H6, and H7, were exposed on the molecular surface. A slight difference in structure between CpL-RI and AcL-RI was found at the loop region between B10 and H6 (Fig. 2a, yellow), which was located at the gate for the substrate entrance. This region of CpL-RI, T213E214H215A216H217D218K219, had no electron density, suggesting that they were highly disordered, while that of AcL-RI, T213E214A215A216A217G218N219, adopted a short 310 helix with significant electron densities (Supplemental Fig. S2).
Catalytic site structure
The structure of the catalytic site of CpL-RI/water is shown in Fig. 3a. A bound metal ion in the catalytic site of CpL-RI was coordinated by His106, His108, Glu113, His188, and two waters (W1 and W2), giving an octahedral form of metal coordination. A large space for a substrate was identified between β-sheet 1 and β-sheet 2. Glu113 formed hydrogen bonds with W1 and W2, and Lys111 formed a hydrogen bond with W1. Glu204 directed its side chain to the catalytic center by a salt bridge with Lys93. These charged residues, Glu113, Lys111, Glu204, and Lys93, were sequentially aligned on β-sheet 1 to recognize the hydroxyl groups of the substrate.
In CpL-RI/L-ribose, the bound substrates adopted a pyranose ring form in Mol-A (Fig. 3b) and linear form in Mol-B (Fig. 3c). L-Ribose in Mol-A appeared to be modeled in a 1C4 pyranose ring (L-ribopyranose) with O1 and O5 coordinating to a metal ion, and the O2, O3, and O4 of L-ribopyranose formed hydrogen bonds with Lys93, Lys111, Glu204, Glu211, and Arg243. However, the electron density for L-ribopyranose remained ambiguous, suggesting that the bound substrate partially adopted a linear form (Collins and Ferrier 1995) (Supplemental Fig. S1a). Therefore, it was difficult to describe substrate-enzyme interactions in detail. In Mol-B, O1 and O2 of linear L-ribose coordinated to the metal ion (Fig. 3c). Depending on the structures of the bound L-ribose (pyranose form or linear form), Lys93, Glu113, Glu211, and Arg243 changed their side chain conformations to form favorable interactions with the substrate. Glu113 formed hydrogen bonds with O1 and O2, and Glu204 directed its carboxyl side chain to a hydrogen atom attached to C2. This result suggested that Glu113 and Glu204 might act as acid/base catalysts in the catalytic isomerization reaction via a cis-enediol intermediate. The O2, O3, and O5 of L-ribose were recognized by hydrogen bonds with Lys111, Lys93, and Arg243, respectively. There was no amino acid residue to recognize the O4 of L-ribose, and the O4 in a different configuration (right-handed configuration) was spatially permitted without a steric hindrance. Hydrogen bond interactions between L-ribose and the enzyme were mainly found at the side of the metal ion. Non-polar amino acid residues (Ile40, Phe42, Met63, Ile65, Met95, Cys103, and Phe190) on the opposite side formed hydrophobic interactions with the bound L-ribose, which fixed a proper conformation of linear L-ribose for the catalytic reaction (Fig. 3d).
In CpL-RI/L-allose, the bound L-allose molecules adopted a linear form in both Mol-A and Mol-B (Fig. 3e, Supplemental Fig. S1c, d). The conformation of the bound L-allose and enzyme-ligand interactions were very similar to those found in CpL-RI/L-ribose (Fig. 3c, e) because L-allose had the same configurations of C2, C3, and C4 as L-ribose (Fig. 1). Glu211 and Arg243 formed hydrogen bonds with the O5 and O6 of L-allose, respectively.
In CpL-RI/L-psicose, the bound L-psicose molecules adopted a furanose ring form in Mol-A (Fig. 3f) and linear form in Mol-B (Supplemental Fig. S1f). In Mol-A, the O1 and O2 of L-psicofuranose coordinated to the metal ion, and seven hydrogen bonds were detected between L-psicofuranose and the enzyme: O1–Glu113, O2–Glu113, O2–Lys111, O3–Glu204, O3–Lys93, O4–Glu211, and O6–Arg243. The conformation of the bound L-psicose in Mol-B was almost equivalent to those found in CpL-RI/L-allose, as well as the substrate-enzyme interactions (Supplemental Fig. S1f).
Site-directed mutagenesis analyses for the amino acid residues involved in the tetramer formation
The gel filtration experiment revealed that CpL-RI formed a homo-tetramer in a solution (Morimoto et al. 2013) (Supplemental Fig. S4). The PISA server (Krissinel and Henrick 2007) also showed the formation of a homo-tetramer of CpL-RI in a crystal with 222 symmetry (ΔGint = −91.6 kcal/mol and ΔGdiss = 67.4 kcal/mol), as reported previously for AcL-RI (Yoshida et al. 2014).
Mol-A and Mol-B made contact with each other through β-sheet 1 and H4, to form a stable dimer with an interface area of 2304 Å2 (19 % of the monomer surface). Many hydrophobic interactions by Leu69, Pro72, Val74, Leu77, Pro78, Ala79, Ala82, Tyr87, Leu90, Leu139, Phe141, Val181, Val205, Ala221, Pro222, and Leu223 were found at the interface. Although Asp75, Ser88, Asp92, Glu116, Lys179, Arg184, Lys185, and Arg203 were involved in intermolecular hydrogen bonds and/or salt bridge interactions, the dimer was predominantly formed by hydrophobic interactions. EcD-LI and BsD-LI, which have marginally homologous amino acid sequences, but similar three-dimensional structures, were also previously shown to form a stable dimer of Mol-A and Mol-B with hydrophobic interactions (Staalduinen et al. 2010; Marles-Wright and Lewis 2011). Therefore, the dimerization of Mol-A and Mol-B was suggested to provide solubility and structural stability to the enzyme, which was indispensable for enzyme activity.
In the crystals of CpL-RI, two dimers associated with each other to form a tetramer. At the interface, the H1 of Mol-A complementarily bound to the shallow groove formed by the H1, H3, and loops between the β-sheets of Mol-D (Fig. 4). Ser20 and Ala24 in the H1 of Mol-A made contact with the hydrophobic cluster by the Leu21, Leu25, Val118, Leu119, and Val198 of Mol-D, while the Glu17 of Mol-A formed a hydrogen bond with the His98 of Mol-D. Between Mol-A and Mol-C, there was an intermolecular hydrogen bond by Ala24 (Mol-A) and Arg26 (Mol-C), and vice versa. The H3 of Mol-D was located at the edge of the tetramer and contributed to interactions with the H1 of Mol-A (Fig. 4).
To clarify whether the formation of a tetramer was indispensable for the enzymatic activity of CpL-RI, mutant forms were prepared, in which the amino acid residues involved in the dimer-dimer interaction were replaced (Figs. 2d and 4). In these mutant forms, Glu17 was replaced by Ala (E17A), Arg26 by Ala (R26A), Ser20 by Trp (S20W), and Ala24 by Trp (A24W). A mutant form with the deletion of H3 (V41-G-S-V62), in which 20 amino acid residues of Phe42-Glu61 were replaced by Gly-Ser, was also prepared. The replaced Ala residue in E17A and R26A could not form intermolecular hydrogen bonds with His98 (Mol-D), or Ala24 (Mol-C), respectively, leading to fewer intermolecular interactions between dimers. As listed in Table 2, the enzymatic activities of these mutant forms decreased to 15.8 % (E17A) and 21.2 % (R26A), and gel filtration experiments showed that the tetramer is partially dissociated to dimers (Supplemental Fig. S4). The replaced Trp residue in S20W and A24W should have caused steric hindrance at the interface, disturbing the formation of a tetramer. As expected, the gel filtration experiment revealed that tetramers were largely dissociated (Supplemental Fig. S4). It showed that 48.9 % of S20W and 30.3 % of A24W still formed a tetramer at 4 °C in gel filtration; however, in enzyme activity measurement at 40 °C, they are expected to be almost dissociated to dimer or monomer form, being inactive (Table 2).
Growth complementation assays
To investigate whether microorganisms could grow on L-ribose as a carbon source, we attempted to grow E. coli JM109 transformed with the CpL-RI gene and Cellulomonas parahominis MB426 in minimal medium. E. coli generally possesses the metabolic pathway using L-arabinose as a carbon source, in which L-arabinose isomerase catalyzes the isomerization of L-arabinose to L-ribulose, followed by the phosphorylation of L-ribulose to L-ribulose 5-phosphate by L-ribulokinase, and L-ribulose-5-phosphate can be converted to D-xylulose-5-phosphate, an intermediate in the pentose phosphate pathway, by L-ribulose-5-phosphate 4-epimerase (Lee et al. 1986; Schleif 2000) (Fig. 5a). As expected, E. coli JM109 could grow on L-arabinose (Fig. 5b), but E. coli JM109 could not grow on L-ribose, suggesting that it could not use L-ribose as a carbon source (Fig. 5c). It should be noted that E. coli JM109 expressing CpL-RI could grow on L-ribose, as well as L-arabinose (Fig. 5b, c).

Proposed metabolic pathway using L-ribose as a carbon source. a Scheme of the metabolic pathway is illustrated. L-ribose can be isomerized to L-ribulose, and then L-ribulose converted to L-ribulose-5-phosphate, and to D-xylulose-5-phosphate by the enzymes of L-arabinose metabolic pathway. b Both E. coli JM 109 and E. coli JM 109 harboring the gene of CpL-RI could grow on M9 minimal medium with L-arabinose. c E. coli JM 109 harboring the gene of CpL-RI could grow on M9 minimal medium with L-ribose. d Cellulomonas parahominis MB426 could grow on M9 minimal medium with L-ribose
Discussion
Two reaction mechanisms of aldose-ketose isomerization for free monosaccharides have been proposed, acis-enediol intermediate mechanism (Davenport et al. 1991) and hydride-shift mechanism (Whitlow et al. 1991). Two amino acid residues act as acid/base catalysts in a cis-enediol intermediate mechanism, while in a hydride-shift mechanism, a hydrogen ion shifts between C1 and C2 in the presence of an acid/base catalyst. The crystal structures of CpL-RI with bound substrates showed that Glu113 and Glu204 could act as acid/base catalysts in the catalytic isomerization reaction via a cis-enediol intermediate, in which Glu204 transferred a proton between C1 and C2, assisted by Glu113 protonating/deprotonating of O2 (Supplemental Fig. S3). The O1, O2, and O3 of the bound L-psicofuranose could be superimposed onto those of the bound L-allose (Fig. 3e) and L-psicose in Mol-B (Supplemental Fig. S1f). This result indicated that after isomerization from L-allose to L-psicose, the conformational change in the linear L-psicose occurred with rotation around the C3–C4 bond, followed by the nucleophilic attack of O5 to C2 (carbonyl carbon) to form L-psicofuranose (Supplemental Fig. S3).
In other aldose-ketose isomerases, two amino acid residues acting as acid/base catalysts face each other across the metal ion (Kim et al. 2006; Takeda et al. 2010). However, Glu113 and Glu204 of CpL-RI located on the same side of the metal ion, and this location of Glu113 and Glu204 is required to accommodate an aldose with a hydroxyl group (O2) having a left-handed configuration in Fischer’s projection (Fig. 1). This novel catalytic site structure of a sugar isomerase was also found in AcL-RI (Yoshida et al. 2014), EcD-LI (Staalduinen et al. 2010), and BsD-LI (Marles-Wright and Lewis 2011). Thus, CpL-RI, AcL-RI, EcD-LI, and BsD-LI could be classified in a new family of aldose-ketoseisomerases.
In crystal structure of CpL-RI/L-ribose, the O2, O3, and O5 of L-ribose were recognized by hydrogen bonds, but the O4 in a different configuration was spatially permitted without a steric hindrance. This may explain why CpL-RI exhibited activity for D-lyxose, a C4 epimer of L-ribose. In structural comparisons between CpL-RI/L-ribose (Fig. 3c) and CpL-RI/L-allose (Fig. 3e), Glu211 and Arg243 flexibly moved, depending on the bound substrate, to form favorable interactions with the substrate. Thus, Glu211 and Arg243 may have been responsible for the recognition of substrates with various configurations of C4 and C5 (D-lyxose, D-talose, L-allose, L-gulose, and D-mannose). The disordered region (213-219) was proximate to Glu211 and Arg243. In the AcL-RI structure with the observed electron density for the region (213-219), the successive three Ala residues (A215A216A217) formed 310 helix, and Thr213 and Glu214 made contacts with Glu211 and Arg243. Due to the high mobility of this region (213-219), CpL-RI more easily changed the orientations and positions of Glu211 and Arg243 than AcL-RI. These may be the reason for CpL-RI having a broad substrate specificity.
The catalytic site of CpL-RI was at the center of the subunit, and independently consisted of residues within a subunit. The catalytic sites of the other aldose-ketose isomerases consist of the amino acid residues from two or three subunits (Allen et al. 1994; Korndörfer et al. 2000; Takeda et al. 2010; Yoshida et al. 2007). In CpL-RI and AcL-RI, the catalytic sites were expected to act independently; hence, the biological implications of the formation of tetramers were unclear.
The results of site-directed mutagenesis analyses for the formation of oligomers suggested that the formation of a tetramer was important for enzymatic activity and that the dissociation of this tetramer led to the destruction of the catalytic site (Table 2). In CpL-RI and AcL-RI, the intermolecular interactions by H1 (Mol-A), the loops between β-sheets (Mol-D), and H3 (Mol-D) markedly contributed to the stable subunit structure, having a catalytic site independently. In other words, each subunit of CpL-RI was structurally stabilized by intermolecular contacts with other subunits. This may be a biological implication of the formation of tetramers in CpL-RI and AcL-RI. The formation of a stable tetramer was expected to be related to the thermostability of the enzyme (Morimoto et al. 2013). However, differences in the thermostabilities of CpL-RI and AcL-RI could not be explained with respect to the stability of the tetramers, because AcL-RI also formed a stable tetramer with (ΔGint = −92.9 kcal/mol and ΔGdiss = 64.2 kcal/mol), similar to CpL-RI.
E. coli generally possesses the metabolic pathway using L-arabinose as a carbon source (Lee et al. 1986; Schleif 2000) (Fig. 5a). E. coli JM109 expressing CpL-RI could grow on L-arabinose or L-ribose, meaning that it acquired the ability to use L-ribose as a carbon source. L-Ribulose produced from L-ribose by CpL-RI was most likely used in the L-arabinose metabolic pathway (Fig. 5a). Cellulomonas parahominis MB426, which produces L-ribose isomerase, could also grow on L-ribose (Fig. 5d). The whole genome sequence of Cellulomonas parahominis MB426 currently remains unknown. However, Cellulomonas sp. KRMCY2 (NCBI BioProject Accession. PRJNA195883) was reported to have L-arabinose isomerase (WP_024286844), L-ribulokinase (WP_024286842), and L-ribulose-5-phosphate 4-epimerase (WP_024286843) genes, indicating that it utilizes the metabolic pathway using L-arabinose. Thus, Cellulomonas parahominis MB426 may also have the L-ribulokinase and L-ribulose-5-phosphate 4-epimerase genes. If this is the case, Cellulomonas parahominis MB426 may utilize the L-ribose metabolic pathway by which CpL-RI produces L-ribulose from L-ribose, followed by the same steps as L-arabinose pathway (Lee et al. 1986; Schleif 2000) (Fig. 5a). It is advantageous for microorganisms to have a metabolic pathway using rare sugars (L-ribose and D-lyxose) for their survival among different species of bacteria. We proposed that CpL-RI is involved in a novel metabolic pathway using L-ribose as a carbon source. However, further studies are needed to clarify this issue.
References
Allen KN, Lavie A, Glasfeld A, Tanada TN, Gerrity DP, Carlson SC, Farber GK, Petsko GA, Ringe D (1994) Role of the divalent metal ion in sugar binding, ring opening, and isomerization by D-xylose isomerase: replacement of a catalytic metal by an amino acid. Biochemistry 33:1488–1494. doi:10.1021/bi00172a027
Anderson RL, Allison DP (1965) Purification and characterization of D-lyxose isomerase. J Biol Chem 240:2367–2372
Brunger AT (1993) X-PLOR 3.1: A System for X-Ray Crystallography and NMR. Yale University Press, New Haven
Cheng YC (2001) Potential use of antiviral L-nucleoside analogues for the prevention or treatment of viral associated cancers. Cancer Lett 162:S33–S37. doi:10.1016/S0304-3835(00)00650-9
Cho EA, Lee DW, Cha YH, Lee SJ, Jung HC, Pan JG, Pyun YR (2007) Characterization of a novel D-lyxose isomerase from Cohnella laevoribosii RI-39 sp. J Bacteriol 189:1655–1663. doi:10.1128/JB.01568-06
Choi JG, Hong SH, Kim YS, Kim KR, Oh DK (2012) Characterization of a recombinant thermostable D-lyxose isomerase from Dictyoglomusturgidum that produces D-lyxose from D-xylulose. Biotechnol Lett 34:1079–1085. doi:10.1007/s10529-012-0874-y
Collins P, Ferrier R (1995) Monosaccharides: Their Chemistry and Their Roles in Natural Products. Wiley, West Sussex
Davenport RC, Bash PA, Seaton BA, Karplus M, Petsko GA, Ringe D (1991) Structure of the triosephosphate isomerase-phosphoglycolohydroxamate complex: an analogue of the intermediate on the reaction pathway. Biochemistry 30:5821–5826. doi:10.1021/bi00238a002
DeLano WL (2002) ThePyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA, http://www.pymol.org
Emsley P, Cowtan K (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi:10.1107/S0907444904019158
Gumina G, Song GY, Chu CK (2001) L-Nucleosides as chemotherapeutic agents. FEMS Microbiol Lett 202:9–15. doi:10.1111/j.1574-6968.2001.tb10773.x
Gunsalus IC, Horecker BL, Wood WA (1955) Pathways of carbohydrate metabolism in microorganisms. Bacteriol Rev 19:79–128
Hayashi N, Iida T, Yamada T, Okuma K, Takehara I, Yamamoto T, Yamada K, Tokuda M (2010) Study on the postprandial blood glucose suppression effect of D-psicose in borderline diabetes and the safety of long-term ingestion by normal human subjects. Biosci Biotechnol Biochem 74:510–519. doi:10.1271/bbb.90707
Hayashi N, Yamada T, Takamine S, Iida T, Okuma K, Tokuda M (2014) Weight reducing effect and safety evaluation of rare sugar syrup by a randomized double-blind, parallel-group study in human. J Funct Foods 11:152–159. doi:10.1016/j.jff.2014.09.020
Heath EC, Hurwitz J, Horecker BL, Ginsburg A (1958) Pentose fermentation by Lactobacillus plantarum. J Biol Chem 231:1009–1029
Izumori K, Yamanaka K, Elbein AD (1976) Pentose metabolism in Mycobacterium smegmatis: specificity of induction of pentose isomerases. J Bacteriol 128:587–591
Kim K, Kim HJ, Oh DK, Cha SS, Rhee S (2006) Crystal structure of D-psicose 3-epimerase from Agrobacterium tumefaciens and its complex with true substrate D-fructose: a pivotal role of metal in catalysis, an active site for the non-phosphorylated substrate, and its conformational changes. J Mol Biol 361:920–931. doi:10.1016/j.jmb.2006.06.069
Korndörfer IP, Fessner WD, Matthews BW (2000) The structure of rhamnose isomerase from Escherichia coli and its relation with xylose isomerase illustrates a change between inter and intra-subunit complementation during evolution. J Mol Biol 300:917–933. doi:10.1006/jmbi.2000.3896
Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi:10.1016/j.jmb.2007.05.022
Kwon HJ, Yeom SJ, Park CS, Oh DK (2010) Substrate specificity of a recombinant D-lyxose isomerase from Providenciastuartii for monosaccharides. J Biosci Bioeng 110:26–31. doi:10.1016/j.jbiosc.2009.12.011
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1992) PROCHECK v.2: Programs to Check the Stereochemical Quality of Protein Structures. Oxford Molecular Ltd, Oxford
Lee N, Gielow W, Martin R, Hamilton E, Fowler A (1986) The organization of the araBAD operon of Escherichia coli. Gene 47:231–244. doi:10.1016/0378-1119(86)90067-3
Levin GV (2002) Tagatose, the new GRAS sweetener and health product. J Med Food 5:23–36. doi:10.1089/109662002753723197
Marles-Wright J, Lewis RJ (2011) The structure of a D-lyxoseisomerase from the σBregulon of Bacillus subtilis. Proteins 79:2015–2019. doi:10.1002/prot.23028
McRee DE (1999) XtalView/Xfit: a versatile program for manipulating atomic coordinate and electron density. J Struct Biol 125:156–165. doi:10.1006/jsbi.1999.4094
Mizanur RM, Takata G, Izumori K (2001) Cloning and characterization of a novel gene encoding L-ribose isomerase from Acinetobacter sp. strain DL-28 in Escherichia coli. Biochim Biophys Acta 1521:141–145. doi:10.1016/S0167-4781(01)00290-1
Morimoto K, Terami Y, Maeda Y, Yoshihara A, Takata G, Izumori K (2013) Cloning and characterization of the L-ribose isomerase gene from Cellulomonas parahominis MB426. J Biosci Bioeng 115:377–381. doi:10.1016/j.jbiosc.2012.10.021
Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst D Biol Crystallogr 53:240–255. doi:10.1107/S0907444996012255
Otwinowski Z, Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Method in Enzymology 276: Macromolecular Crystallography part A 307–326. doi:10.1016/s0076-6879(97)76066-x
Park CS, Kwon HJ, Yeom SJ, Oh DK (2010) Mannose production from fructose by free and immobilized D-lyxose isomerases from Providenciastuartii. Biotechnol Lett 32:1305–1309. doi:10.1007/s10529-010-0300-2
Patel DH, Wi SG, Lee SG, Lee DS, Song YH, Bae HJ (2011) Substrate specificity of the Bacillus licheniformis lyxose isomerase YdaE and its application in in vitro catalysis for bioproduction of lyxose and glucose by two-step isomerization. Appl Environ Microbiol 77:3343–3350. doi:10.1128/AEM. 02693-10
Ramachandran GN, Sasisekharan V (1968) Conformation of polypeptides and protein. Advan Protein Chem 23:283–437
Schleif R (2000) Regulation of the L-arabinose operon of Escherichia coli. Trends Genet 16:559–565. doi:10.1016/S0168-9525(00)02153-3
Shimonishi T, Izumori K (1986) A new enzyme, L-ribose isomerase from Acinetobacter sp. J Ferment Bioeng 81:493–497. doi:10.1016/0922-338X(96)81468-1
Staalduinen LM, Park CS, Yeom SJ, Adams-Cioaba MA, Oh DK, Jia Z (2010) Structure-based annotation of a novel sugar isomerase from the pathogenic E. coli O157:H7. J Mol Biol 401:866–881. doi:10.1016/j.jmb.2010.06.063
Stevens FJ, Wu TT (1976) Growth on D-lyxose of a mutant strain of Escherichia coli K12 using a novel isomerase and enzymes related to D-xylose metabolism. J Gen Microbiol 97:257–265. doi:10.1099/00221287-97-2-257
Takeda K, Yoshida H, Izumori K, Kamitori S (2010) X-ray structures of Bacillus pallidusD-arabinose isomerase and its complex with L-fucitol. Biochim Biophys Acta 1804:1359–1368. doi:10.1016/j.bbapap.2010.01.018
Vagin A, Teplyakov A (1997) MOLREP: An automated program for molecular replacement. J Appl Cryst 30:1022–1025. doi:10.1107/S0907444909042589
Whitlow M, Howard AJ, Finzel BC, Poulos TL, Winborne E, Gilliland GL (1991) Ametalmediated hydride shift mechanism for xylose isomerase based onthe 1.6 Å Streptomyces rubiginosusstructures with xylitol and D-xylose. Proteins Struct Funct Genet 9:153–173. doi:10.1002/prot.340090302
Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi:10.1107/S0907444910045749
Yoshida H, Yamada M, Ohyama Y, Takada G, Izumori K, Kamitori S (2007) The structures of L-rhamnose isomerase from Pseudomonas stutzeri in complexes with L-rhamnose and D-allose provide insights into broad substrate-specificity. J Mol Biol 365:1505–1516. doi:10.1016/j.jmb.2006.11.004
Yoshida H, Teraoka M, Yoshihara A, Izumori K, Kamitori S (2011) Overexpression, crystallization and preliminary X-ray diffraction analysis of L-ribose isomerase from Acinetobacter sp. strain DL-28. Acta Crystallogr Sect F Struct Biol Cryst Commun 67:1281–1284. doi:10.1107/S1744309111030351
Yoshida H, Yoshihara A, Teraoka M, Terami Y, Takata G, Izumori K, Kamitori S (2014) X-ray structure of a novel L-ribose isomerase acting on a non-natural sugar L-ribose as its ideal substrate. FEBS J 281:3150–3164. doi:10.1111/febs.12850
Acknowledgments
We thank Dr. A. Itoh and Mr. K. Yube for their technical assistant with DNA sequencing. Y. Terami is supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (JSPS) Research Fellows. This study was supported in part by Grants-in-Aid for Scientific Research (25440028, 23770122, 22780068, 26450097, 22580088) from JSPS, and by the funds from the Kagawa University New Research Areas and Collaborative Research Organization. This research was performed with the approval of the Photon Factory Advisory Committee and National Laboratory for High Energy Physics (2012G550 and 2013G506) Japan.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1714 kb)
Rights and permissions
About this article

Cite this article
Terami, Y., Yoshida, H., Uechi, K. et al. Essentiality of tetramer formation of Cellulomonas parahominis L-ribose isomerase involved in novel L-ribose metabolic pathway. Appl Microbiol Biotechnol 99, 6303–6313 (2015). https://doi.org/10.1007/s00253-015-6417-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6417-4