
Abstract
A biogas production plant operating with main and secondary digesters (MD, SD) was analysed for the diversity of bacteria from Clostridium cluster I and its pathogenic members. The plant was run in two parallel lines, both receiving silages, and one, in addition, cattle manure (CM). Quantitative PCR of 16S rRNA genes from directly extracted DNA indicated that cluster I represented 0.2 to 5.6 % of the total bacterial communities. Its prevalence was particularly low in CM and also in SD compared to MD, indicating its decline during fermentation. In contrast, another highly abundant clostridial group, i.e. the “faecal” cluster XIVa, remained quantitatively unaffected during fermentation. A total of 85.1 % of 581,934 rRNA gene sequences gathered by group-specific PCR from the silages, CM and digesters could be assigned to cluster I. All remaining sequences fell into other clostridial groups. The three most dominant operational taxonomic units (OTUs) introduced with CM were from cluster I, and they declined during fermentation. Fermentation with CM significantly increased OTUs of clostridia outside of cluster I but not within. The only OTUs related to pathogens were detected for Clostridium botulinum with 0.18 % of all cluster I sequences in maize silage and less than 0.01 % in the other substrates and digester materials. These OTUs could be assigned to all four established C. botulinum groups, thus, potentially covering all seven neurotoxins. Mouse lethality tests of samples with suspected presence of C. botulinum, however, indicated no toxigenic potential and, thus, no risk associated with the rare occurrence of these OTUs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Anaerobic digestion of organic material leads to the formation of methane-rich biogas. In recent years, the interest in the technical production of biogas in agriculture has increased since the formation of methane from manure or plant material poses an important renewable energy source (Weiland 2010). While the methane molecule itself is exclusively formed by archaea, the preceding activities, including hydrolysis of organic polymeric substances, acidogenesis, acetogenesis and hydrogen production are mainly provided by bacterial consortia, which typically include clostridia among their quantitatively dominant members (Kröber et al. 2009; Li et al. 2013; Sundberg et al. 2013; Ziganshin et al. 2013).
Clostridia represent a highly diverse group of gram-positive, obligatory anaerobic spore-forming bacteria within the phylum Firmicutes. The genus Clostridium itself is a phylogenetically highly heterogeneous group which encompasses bacteria from other spore-forming and non-spore-forming genera and families (Yutin and Galperin 2013). Therefore, Clostridium has been divided into several clusters, including Clostridium cluster I, which represents the genus Clostridium sensu stricto (Collins et al. 1994). Within this cluster I, there are some medically relevant species like Clostridium perfringens, the causative agent of gas gangrene, Clostridium tetani causing the tetanus disease, Clostridium chauvoei which causes blackleg, a highly mortal disease of ruminants, and Clostridium botulinum, the producer of the strong botulinum neurotoxin (BoNT) leading to botulism in humans and animals (Lindstrom et al. 2010; Peck et al. 2011). However, cluster I also contains many nonpathogenic members which can contribute with their metabolic diversity to the degradation of polymeric carbohydrates (Schellenberger et al. 2010), production of solvents (Keis et al. 1995) and organic acids (Schwarz et al. 2012) or of hydrogen (Bowman et al. 2010; Chang et al. 2008).
Bacteria from Clostridium cluster I have been detected in anaerobic sediments (Uz et al. 2007) and bioreactors-producing hydrogen (Park et al. 2014) or methane (Dohrmann et al. 2011) or in the gastrointestinal tract. In the latter, however, Clostridia from cluster XIVa often appear to be more prevalent (Becker et al. 2014; Rinttila et al. 2004; Ritchie et al. 2008; Wise and Siragusa 2007). In fact, clostridia from other clusters, e.g. cluster III, cluster IV or cluster XIVa, have similar metabolic potentials and they have been detected in similar environments like those from cluster I (Nakamura et al. 2009; Shrestha et al. 2011).
Currently, the contribution of specific Clostridium clusters to anaerobic metabolic transformations in the environment and bioreactors is still in an exploratory state and can at present not well be predicted. Considering the potential prevalence of Clostridium cluster I in methane-producing consortia, it is of utmost importance to understand whether pathogenic members of this cluster may also find suitable conditions for growth and, thus, cause unintended risks for humans and animals, especially in farm environments. Indeed, C. chauvoei was detected in biogas plants from farms with proven cases of blackleg (Bagge et al. 2009). For C. botulinum, e.g., pathogen multiplication in silage and the gastrointestinal tract of cattle with botulism has been reported, and potential risks for the contamination of dairy products have been discussed (Lindstrom et al. 2010). Different C. botulinum BoNT types were detected on farms, where bovine and human faeces as well as animal feed and domestic dusts were analysed (Krüger et al. 2012).
For methanogenic bioreactors, cultivation independent analyses of 16S rRNA genes by PCR amplification and DNA sequencing from directly extracted DNA revealed in previous studies the presence of 20 different operational taxonomic units (OTUs) from Clostridium cluster I, but phylogenetic analyses demonstrated that none of them was a close relative of a clostridial pathogen (Dohrmann et al. 2011). The phylogenetic analyses of several metagenomic libraries generated by next-generation high-throughput sequencing from directly extracted DNA, which comprised more than two million gene tags from various biogas reactors, indicated a high abundance of clostridia, but also, a very low presence of genes originating from C. botulinum or its close relatives (Eikmeyer et al. 2013).
The objective of this study was to apply the new potentials of high-throughput amplicon DNA sequencing of 16S rRNA genes from directly extracted DNA to characterize the diversity of bacteria from Clostridium cluster I at an unprecedented level of sensitivity and search for potential pathogens in a full-scale operating biogas plant. Bacterial community analyses were conducted on three different substrates, i.e. maize silage (MS), whole plant silage (WPS) and cattle manure (CM), entering the plant and on fermenting material from main (MD) and secondary digesters (SD). CM was considered as a potential source of C. botulinum and C. chauvoei because of recent reports on incidences of bovine botulism and blackleg in European farm environments (Bagge et al. 2009; Lindstrom et al. 2010). The architecture of the chosen biogas plant, which consisted of two parallel lines with main and secondary digesters allowed the comparison of bacterial communities in the presence and in the absence of CM. Samples indicating the presence of potential clostridial pathogens by phylogenetic analyses of 16S rRNA genes were further analysed by a reference laboratory for pathogen detection.
Materials and methods
Biogas plant and sampling
The biogas plant, built in 2008, is located in Germany and provides an amount equal to 2.4 MWe to the natural gas grid. The plant consisted of two separate digestion lines each with one main digester (MD) followed by one secondary digester (SD). Each digester had a volume of 4250 m3 and was operated at 40 °C. Each day, both lines received 39 t maize silage (MS) and 15 t of whole plant silage (WPS) from rye. Line I received in addition 43 m3 cattle manure (CM). All substrates (MS, WPS, CM) and slurry samples from main digesters (MD I and MD II) and secondary digesters (SD I and SD II) were collected in June 2012. The material from each sample was split for analyses of the physicochemical process parameters and for molecular analyses of the microbial community composition. Furthermore, additional material of each sample was stored at −20 °C to be available for targeted testing of C. botulinum neurotoxin (BoNT) and other potential pathogens.
Physicochemical analyses
Slurry material from the biogas plant was analysed in accordance to the respective DIN EN protocols for its dry matter (DIN EN 12880 2001) and organic dry matter content (DIN EN 12879 2001), pH (DIN EN 15933 2012) and NH4 +-N content (DIN EN 38406–5 1983). The ratio of volatile organic acids (VOA) and the total inorganic carbon (TIC) VOA/TIC was determined by titration with 0.5 M H2SO4.
Nucleic acid extraction
Sampled material was stored over night at 4 °C, and DNA was extracted on the following day. Each sample was split into two aliquots and both of them were separately analysed. Large particles present in the substrates and slurries from the digesters were separated during the first washing step with sterile phosphate buffered saline (PBS 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4; pH 7.4) by centrifugation at 500×g for 10 min. The supernatants were then centrifuged for 30 min at 11,800×g, and the pellets were washed once with PBS and, after another round of centrifugation, with sterile aqueous 0.85 % (w/v) KCl solution. Samples from the silages were pretreated to extract the microbial cell fraction as follows: 100-g portions were suspended in 700 ml PBS for 40 min at 4 °C in an orbital shaker (GFL, Burgwedel, Germany) at 20 rev min−1. Large plant material was separated by a 2-mm sieve, and the filtrate entered the aforementioned protocol at 500×g centrifugation for 10 min. The nucleic acids were extracted using a phenol-chloroform extraction method described by Lueders et al. (Lueders et al. 2004), modified previously for the analyses of clostridial communities including spore lysis from biogas reactor materials (Method 5b) (Dohrmann et al. 2011). Genomic DNA was then quantified with Quant-iT PicoGreen dsDNA reagent (Life Technologies GmbH, Darmstadt, Germany) following the protocol of the manufacturer.
Quantification of microbial community (qPCR of 16S rRNA genes)
Bacterial and archaeal 16S rRNA gene copy numbers were quantified by separate real-time PCR, conducted in a StepOnePlus Real-Time PCR system (Life Technologies GmbH) using primer and probes described by Yu et.al. (Yu et al. 2005). PCRs were conducted in a volume of 20 μl, containing 2 μl of template DNA, 500 nM of each primer (custom synthesized by Eurofins Genomics, Ebersberg, Germany) and 200 nM of the respective probe (Eurofins) in the Maxima Probe qPCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA) including ROX. The probe to detect Bacteria was labelled with 5′-FAM and 3′-BHQ1 and that for Archaea with 5′-TAMRA and 3′-BHQ2. PCR started with 10 min at 95 °C followed by 45 cycles at 95 °C for 15 s and 60 °C for 60 s. Signals were detected at 60 °C. Standard curves were obtained from 10-fold dilutions of the pGEM-T vector (Promega, Mannheim, Germany) containing the 16S rRNA gene of Bacillus subtilis BD 466 (Escherichia coli positions 8–1513) or the 16S rRNA gene of Methanobacterium oryzae DSM 11106 (E. coli positions 109–1401), respectively. The PCR efficiency Eff was 95 % (R 2 = 0.999) of the standard curves for Bacteria and Eff = 101 % (R 2 = 0.999) for Archaea. Ribosomal 16S rRNA genes of Clostridium cluster I (C. perfringens group) and of cluster XIVa (Clostridium coccoides - Eubacterium rectale group) were quantified by real-time PCR using primers described elsewhere (Rinttila et al. 2004). Each 25 μl PCR reaction contained 2 μl of template DNA, 500 nM of each primer (biomers.net GmbH, Ulm, Germany) in the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific) including ROX. To quantify Clostridium cluster I, MgCl2 was added to the reaction for a final concentration of 4 mM MgCl2. PCR started with 10 min at 95 °C followed by 40 cycles at 95 °C for 15 s, 55 °C for 20 s, 72 °C for 30 s and 30 s at 77 °C for signal detection of Clostridium cluster I or at 80 °C for cluster XIVa, respectively. Melting curves were recorded for the range from 70 to 95 °C. Standard curves were obtained from 10-fold dilutions of the pGEM-T vector (Promega) containing the 16S rRNA gene of C. coccoides DSM 935 (E. coli positions 8–1513) or a synthetic DNA (biomers.net GmbH, Ulm, Germany) corresponding to the 16S rRNA gene rrsA of C. perfringens ATCC 13124 (CP000246) (E. coli positions 55–181), respectively. The PCR efficiency Eff was 91 % (R 2 = 0.999) of the standard curves for Clostridium cluster I and 86 % (R 2 = 0.999) for cluster XIVa.
Group-specific rRNA gene amplicon pyrosequencing of members of Clostridium cluster I
To amplify E. coli positions 691 to 1359 of the 16S rRNA genes of Clostridium cluster I, the forward primer P930 (GTG AAA TGC GTA GAG ATT AGG AA) and the reverse primer P932 (GAT YYG CGA TTA CTA GYA ACT) (Le Bourhis et al. 2005) were selected and modified for 454 pyrosequencing applying the Lib-L emulsion PCR method on the GS FLX Titanium system (Roche, Mannheim, Germany). Forward primers were composed of the Lib-L primer A sequence, the key sequence, a sample specific multiplex identifier to trace the sequences back to the respective sample and P930. Fourteen different forward primers were used in combination with a single reverse primer consisting of Lib-L primer B sequence, the key sequence and P932. For primer sequences, see Supplemental material (Table S1). PCR conditions were 15 min at 95 °C, 30 cycles of 94 °C for 20 s, 58 °C for 30 s, 72 °C for 30 s and 5 min at 72 °C, respectively. The PCR master mix and product purification followed a previously described protocol (Dohrmann et al. 2012). Products from a minimum of three independent replicate amplifications (technical replicates) were pooled for pyrosequencing. Pyrosequencing was conducted by GATC (GATC Biotech AG, Konstanz, Germany). Two samples, namely single replicates of MS and WPS, respectively, failed in the pyrosequencing process.
Amplicon sequence quality evaluation and phylogenetic analyses
Sequences were processed with mothur v.1.32 and v.1.33 (Schloss et al. 2009) following the mothur 454 SOP version February 2014 including qfile, qwindowaverage = 35, qwindowsize = 50, reference = silva.bacteria.fasta, minlength = 150 and average neighbour algorithm for clustering. Chimera sequences (6.6 %) were identified with chimera.uchime (http://drive5.com/uchime) and removed from the dataset. Sequences were grouped in OTUs at similarities of at least 97 % and classified in accordance to the RDP taxonomy with trainset9_032012. Summary.single was applied to calculate library coverage C. To get representative sequences for each OTU, get.oturep was applied with options column, list, name, label = 0.03, weighted = true and method = distance. For assignment of the representative sequences to the respective OTUs, the get.otulist command was used to create a list with all sequences in each OTU. For a detailed taxonomic analysis of the most abundant 20 OTUs, all sequences grouped in it were identified, picked from the primary fasta file and a consensus sequence was calculated for each OTU applying mothur commands align.seqs, filter.seqs and consensus.seqs with cutoff = 50. Consensus sequences were thought to represent the OTU better than a single sequence. Additionally, the length of ca. 620 nt (without primer sequences), in contrast to the 148 nt of the representative sequences generated during the sequence processing, allowed for a more precise evaluation by NCBI megablast (www.ncbi.nlm.nih.gov/blast) and the RDP classifier (Wang et al. 2007).
To compare differences in abundance the number of sequences was rarefied to 34,027 sequences. A t test (Microsoft Excel, Microsoft Corporation, Redmond, WA) was applied to test for significant differences in abundance due to the different digester lines in MDs or SDs, respectively. Differences between MD and SD of each digester line were also evaluated. For all significantly affected OTUs, response ratios were calculated (Hedges et al. 1999). For each OTU identified as sensitive, a consensus sequence was calculated as mentioned before.
For comparisons of the phylogenetic diversity an approximately maximum-likelihood phylogenetic tree was calculated with FastTree Version 2.1.3 SSE3 (Price et al. 2010) and served for the calculation of a weighted UniFrac distance matrix (Lozupone and Knight 2005) with mothur. This matrix was used to calculate nonmetric multidimensional scaling (NMDS) with mothur and for analysis of similarities (ANOSIM) in R using the vegan package (R Core Team 2010).
Search for pathogen-linked 16S rRNA gene sequences within Clostridium cluster I
To identify rRNA gene sequence of C. botulinum, a set of reference sequences was generated from ARB-SILVA SSU 115 (Quast et al. 2013) with the requirements: name = C. botulinum, sequence length >1000, sequence quality >90 and alignment quality >90. These requirements were met by 268 sequences. Reference sequences and OTU representative sequences were aligned and clustered with the respective tools of the RDP pyrosequencing pipeline (Cole et al. 2009). Clusters which contained reference and OTU representative sequences were further analysed for their phylogenetic relation to C. botulinum. For each identified OTU, a consensus sequence was calculated as mentioned above and then analysed by ARB (Ludwig et al. 2004) including the SINA alignment (Pruesse et al. 2007). A maximum-likelihood tree was constructed based on 90 Clostridium cluster I sequences of at least 1300 nucleotides length applying the corresponding 50 % conservation filter and the AxML algorithm. Sequences retrieved in this study from the different substrates and digesters were added to the tree applying the same filter. Similarities between sequences were calculated with the respective tools implemented in ARB. In the same way as described above for C. botulinum, the amplicon dataset was also analysed for the presence of C. tetani, C. chauvoei and C. perfringens, respectively.
Bioassay of C. botulinum neurotoxins
Selected samples were analysed by the mouse lethality test for the presence of active BoNT. This bioassay was performed by an authorized company (Ripac-Labor GmBH, Potsdam, Germany) following the recommendations of the Friedrich-Loeffler-Institut (Dlabola et al. 2013). Retained material as well as the crude material, including large particles, remaining from DNA extraction was analysed directly for the presence of BoNT and after enrichment cultivation of C. botulinum. Thus, the results for each analysed sample were based on four independent analyses.
Deposition of DNA sequences
All DNA sequences retrieved from the different substrates and digester slurries were deposited at the European Nucleotide Archive and can be found under the Accession number PRJEB7129. In addition, consensus sequences of selected OTU are accessible under numbers LN558658 to LN558702 and LN613270.
Results
Quantification of microbial groups
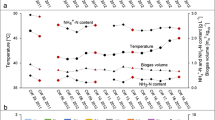
Bacterial 16S rRNA genes detected in total DNA, directly extracted from the different materials, were in a range between 3 × 105 and 8 × 106 copies per ng DNA and generally higher in the materials from digesters than in the substrates entering the biogas plant (Fig. 1a). The low bacterial copy numbers in MS were probably affected by the higher background of total DNA due to the presence of plant residual and fungal DNA (Rossi and Dellaglio 2007). Compared to Bacteria, the copy numbers of Archaea, which included the methanogens, were one to two orders of magnitude lower. The 16S rRNA genes from Clostridium cluster XIVa, i.e. the “faecal clostridia” (see “Materials and methods”; (Song et al. 2004)) represented 1.4 to 10.1 % of the total bacterial 16S rRNA genes in the different materials (Fig. 1b), with highest values for CM. In contrast, the contribution of Clostridium cluster I, potentially including C. botulinum and other clostridial pathogens, was particularly low in CM and, on average, also lower in the two silages. While there was a steep decline of cluster I from the main (MD) to the secondary digesters (SD), the prevalence of cluster XIVa did not significantly change. The contrasting prevalence of both clostridial clusters I and XIVa in the different materials indicated that these groups were not linked to each other.

Copy numbers of qPCR-amplified 16S rRNA genes of Bacteria and Archaea (a) and Clostridia clusters I and XIVa (b), in substrates entering the biogas plant of this study, i.e. maize silage (MS), whole plant silage (WPS) and cattle manure (CM), as well as materials collected from main digesters (MD) and secondary digesters (SD). Digesters indicated with I received MS, WPS and CM, while CM was not included into the substrates fed into digesters indicated with II
Specificity of Clostridium cluster I 16S rRNA gene amplification
A total of 581,934 amplicon sequences, obtained from the substrates fed into the biogas plant and the two stages of the biogas production processes, passed the sequence quality analyses (Table 1). In total, 85.1 % of these sequences fell into Clostridium cluster I, the group targeted with the selected primers. The lowest primer specificities were detected for samples from the two SDs. Overall, a total of 653 different OTUs of Clostridium cluster I were detected. Phylogenetic analyses revealed that all sequences which fell outside of this cluster belonged, at the family rank, to other members of clostridia (Fig. 2).

Taxonomic affiliation of all 16S rRNA gene amplicon sequences detected with the Clostridium-specific PCR. For abbreviations of substrates, see legend of Fig. 1
Diversity and prevalence of 16S rRNA gene amplicons
The 581,934 amplicon sequences could be grouped into 2544 OTUs. For a balanced comparison, the dataset was rarefied to 34,027 sequences for each sample, which resulted in 2541 OTUs remaining. Of these, 318 OTUs contained ten or more sequences, which accounted for 98.5 % of all sequences.
The 20 most abundant OTUs, representing 91.3 % of all sequences, showed at least 97 % DNA sequence identity to previously identified rRNA genes, mainly originating from uncultured bacteria which had been retrieved from various habitats, including the gastrointestinal tract or faeces of humans and animals, composts and biogas plants (Table 2). The most abundant OTU (OTU 1), a member of Clostridium cluster I, represented almost 50 % of all sequences in CM and at least 11 % in the other materials, including those from the digesters. The prevalence of OTU 1 in MD of the line receiving CM declined during anaerobic digestion. In fact, all OTUs with more than 1 % abundance in CM, of which the first three were from cluster I, declined drastically during the digestion process of CM.
Bacteria from OTU 2, closely related to Clostridium saccharoperbutylacetonicum, were highly prevalent in the substrates especially MS, entering the biogas plant, but diminished, as indicated by their lower numbers in both SDs compared to MDs and substrates. In contrast, OTU 5, a Sedimentibacter relative, was more prevalent in the SDs than in the substrates or MDs. Three relatives of Clostridium cluster I, i.e. OTU 3, 6 and 7, which together represented 26.2 % of all sequences, were more abundant in MDs than in SDs or substrates, suggesting an active role in the digestion process. These OTUs were mostly present in WPS, indicating this substrate as a potential source.
Effect of the substrates and the digesters compartment on the clostridial community
Of all 2544 OTUs, only 27 responded significantly to the different conditions in the digesters, i.e. either the presence or absence of CM or the differences prevalent in SD compared to MD. Six OTUs, among them were two from cluster I, responded significantly to both CM and digester compartments (main vs. secondary) (Fig. 3). A comparison between the digesters receiving CM or not, revealed only three responsive OTU in the MDs, but 13 in the SDs. Interestingly, 14 of the 16 responsive genera were more abundant in digesters with CM, suggesting that they might have been involved in the degradation of substrates specific for CM. Only 4 of these 16 responsive OTU originated from Clostridium cluster I, while the other 12 were relatives of Sedimentibacter, Mogibacterium and other non-cultured isolates outside of this cluster. Two of the cluster I sequences declined in their abundance in response to CM. In contrast, all clostridia outside of cluster I increased in presence of CM.

Identification of OTUs which responded significantly different when digesters from line I and line II and/or main and secondary digesters were compared to each other. For each OTU, the most accurate taxonomic affiliation possible, based on RDP analyses, is indicated
NMDS was utilized to visualize the differences in the communities’ phylogenetic structure including all rarefied sequences (n = 34,027 for each sample) (Fig. 4). Each substrate and digester was characterized by distinct clostridial communities, and replicates were, with the exception of SD II, more similar to each other than those of different origin. The analyses indicated that distinct clostridial communities were present in MD and SD and those differences also existed between the digesters receiving CM or not.

NMDS ordination based on weighted UniFrac distances derived from FastTree phylogeny (stress = 0.11; R 2 = 0.95). For abbreviations, see legend of Fig. 1
In an additional step, all samples were characterized for their physicochemical properties (Supplemental material; Table S2), and ANOSIM revealed that significant effects (p < 0.05) on the clostridial diversity were linked to the amount of organic dry matter (oDM)(R = 0.96) and the concentrations of VOA (R = 0.92) and NH+ 4 (R = 0.92) (Supplemental material, Table S3).
Presence of C. botulinum and other potentially pathogenic members of cluster I
Phylogenetic analyses of the 581,934 amplicon DNA sequences retrieved in this study did not indicate the presence of OTUs related to C. perfringens, C. tetani or C. chauvoei, respectively. In contrast, 4 OTUs were identified with sequence identity of above 97 % to C. botulinum (Table 3). The highest prevalence of sequences, i.e. 150, was found with MS, and phylogenetic analyses assigned this OTU 0066 to C. botulinum group III (BoNT types C or D) (Fig. 5). Sequences of OTU 0541 assigned to group II (BoNT types B, E or F) were detected in CM, and sequences of OTU 0208 assigned to group I (BoNT types A, B or F) were found in all digesters. The two sequences of OTU 1209, one detected in MS and the other in the SD of line II (no CM), belonged to group IV (Clostridium argentinense, BoNT type G). While the overall contribution of C. botulinum related 16S rRNA gene sequences in MS represented 0.18 % of Clostridium cluster I, the occurrence in other materials was below 0.01 %.

Maximum likelihood tree of Clostridium cluster I showing the affiliation of 16S rRNA genes obtained from OTU consensus sequences. Groups I to IV indicate the four distinct groups of C. botulinum strains. Reference sequences for identification of C. botulinum are indicated in bold; OTUs of this study identified as close C. botulinum relatives in red
Three samples with different C. botulinum groups from different substrates, i.e. MS, CM and MD I, were selected for mouse lethality tests. None of these tests indicated the presence of BoNT-producing bacteria, either by direct analyses or after incubation for enrichment of C. botulinum in appropriate growth media (Table 3). Thus, the indication for the presence of toxigenic C. botulinum by 16S rRNA gene-based analyses was not confirmed.
Discussion
The biogas plant selected for this study was in full operation during the time the samples were collected. The population sizes of Bacteria and Archaea, as indicated by domain specific qPCR of their 16S rRNA genes, were similar to those reported for other commercial biogas plants run with similar substrates, including manure and silages (Nettmann et al. 2008) (Sundberg et al. 2013). The analyses of the three substrates entering the biogas plant of this study revealed in CM lower overall abundance for Clostridium cluster I compared to both silages, while, as expected, the contribution of “faecal” cluster XIVa sequences was higher in the manure. The relatively high incidence of both clostridial clusters in silages, in the range of 1 to 6 % of all bacterial genes, was in fact surprising, considering that in contrast to manure (Lu et al. 2014), clostridia generally are not expected to be among the dominant bacterial community members in such materials (Duniere et al. 2013; McGarvey et al. 2013). Clostridia in silages may be introduced with soil (Duniere et al. 2013) or manure used crop fertilization.
Bacterial diversity in this study was analysed by sequencing 16S rRNA gene fragments, but in contrast to other studies in which universal bacterial primers were applied (Bengelsdorf et al. 2013; Kröber et al. 2009; Sundberg et al. 2013), this study applied, for higher sensitivity, group specific primers targeting Clostridium cluster I. The selected primers, however, also amplified rRNA genes from some phylogenetic neighbours of cluster I, originating from some incerta sedis families of the Clostridiales. This primer inaccuracy had already been observed in a preceding study based on the sequencing of 66 amplicons (Dohrmann et al. 2011); while in this study, almost 9000 times more sequences were analysed. This inaccuracy was, in fact, quite useful in this study when the response of single OTUs to the conditions in the digesters receiving CM and those without were compared to each other since they apparently revealed different patterns of responses between Clostridium cluster I and other clostridial groups.
Due to the architecture of the biogas plants selected for this study, with two parallel lines each consisting of one main and one secondary digester and both receiving the same basic substrates, it was possible to investigate the effect of CM addition on the clostridial diversity in a comparative approach. The CM impact on the community structure in fact became evident in NMDS analyses where the communities from digesters receiving CM were clearly distinct from those without CM. However, at the OTU level, the clostridia could rarely be assigned to a specific substrate. This was mainly linked to the fact that, generally, the OTUs from CM also occurred in one or both of the silages. Therefore, it was not surprising that the three dominant OTUs from cluster I of CM also occurred in digesters which did not receive CM (see Table 2). Only three OTUs, of which two, however, were the second and third most dominant OTU from cluster I, responded significantly to the presence of CM in the main digesters. These two exhibited opposing responses and their responses could be linked to their abundance in CM: OTU 4 represented 20 % of the clostridial population in CM and was more abundant in MD I, while OTU 3 was nearly absent in CM (0.004 %), but well represented in WPS (14.6 %); it was more abundant in MD II. The distinction of clostridial clusters as well as OTU in this study was based on phylogenetic analyses; but for clostridia, these do not correlate with differences in carbon source utilization (Gupta and Gao 2009; Rainey and Stackebrandt 1993), which can be considered as a main factor structuring the communities in the digesters.
Indications for the presence of C. botulinum, but not of other pathogens from Clostridium cluster I were obtained from all samples analysed in this study. However, its content of the community of Clostridium cluster I was very small, ranging from 0.18 % in MS to 0.003 % in WPS. Considering the contribution of the cluster I sequences to the total bacterial community, as established by qPCR of their 16S rRNA gene, the detection of a single C. botulinum 16S rRNA gene would require the analysis of a background of 270,000 to 370,000 bacterial genes in MDs, and, a background of 2 to 8 million sequences from SDs. It should be noted that the species name “C. botulinum” is defined on the ability of these bacteria to form the botulinum neurotoxin (BoNT) rather than on phylogenetic grounds (Peck et al. 2011). Therefore, C. botulinum groups I to IV are distinguished by distinct phylogenetic lineages (Hutson et al. 1996), and each of these groups is characterized by a potential to produce different BoNT types, e.g. group I produces A, B or F, and group III, C or D ((Collins and East 1998), see also Fig. 5). Interestingly, the suspected C. botulinum OTUs detected in this study encompassed representatives of all four groups, thus indicating a potential that all seven BoNT types could theoretically be produced. In a farm environment, including silages, other studies revealed the presence of C. botulinum BoNT types A, B, C, D and E (Krüger et al. 2012; Lindstrom et al. 2010; Notermans et al. 1981). Even though the rare occurrence of sequences indicating the presence of the four C. botulinum groups and the fact that the two fermentation lines in the biogas plant were not represented by any replicate digesters do not allow any detailed statistical analysis of the environmental conditions which would affect the prevalence of these groups, it appears clear that none of them multiplied on its path through the biogas plants, independent of whether CM was supplied or not.
While the limitation of the 16S rRNA gene-based approach lies in its inability to ultimately confirm the presence of C. botulinum BoNT, the beauty of this approach is that it allows the testing of a large number of environmental samples without sacrificing laboratory animals. Due to the chosen high-throughput DNA sequencing approach of the 16S rRNA gene amplicons from cluster I, unprecedented detection thresholds of up to one gene copy in a background of two million bacterial genes could be achieved. All three samples, which included together the presence of all four C. botulinum groups, were tested negative in mouse lethality tests. Since the toxicity analyses also included previous enrichment steps, it can be concluded that no vegetative cells or spores of C. botulinum, as defined by their capacity to produce BoNT, were present in the materials analysed. Thus, OTUs assigned to the four C. botulinum groups originated from species in which the BoNT encoding genes must have been absent, dysfunctional or not expressed. The presence of silent BoNT genes in C. botulinum strains is not unusual (Dineen et al. 2003; Hutson et al. 1996). Non-toxigenic C. botulinum derivates could be a result of the instability of BoNT encoding genomic regions, which are typically linked to insertion sequences, transposons or other mobile genetic elements (Brüggemann 2005; Hill et al. 2009).
In summary, this study demonstrates how environmental samples can be analysed independent of cultivation based on high-throughput 16S rRNA gene amplicon sequencing for the diversity of Clostridium cluster I sequences and search with high sensitivity for the presence of suspected bacterial pathogens without sacrificing laboratory animals. The abundance and diversity of 16S rRNA amplicons retrieved in this study suggest that bacteria from Clostridium cluster I, in contrast to other clostridia, played only a minor role in the fermentation of the organic material, including CM, in the selected full-scale operating biogas plant, and no risk was identified for an unintended presence or proliferation of pathogens from Clostridium cluster I.
References
Bagge E, Lewerin SS, Johansson KE (2009) Detection and identification by PCR of Clostridium chauvoei in clinical isolates, bovine faeces and substrates from biogas plant. Acta Vet Scand 51:8, doi:10-1186/1751-0147-51.8
Becker A, Hesta M, Hollants J, Janssens GPJ, Huys G (2014) Phylogenetic analysis of faecal microbiota from captive cheetahs reveals underrepresentation of Bacteroidetes and Bifidobacteriaceae. BMC Microbiol 1443. doi:10.1186/1471-2180-14-43
Bengelsdorf FR, Gerischer U, Langer S, Zak M, Kazda M (2013) Stability of a biogas-producing bacterial, archaeal and fungal community degrading food residues. FEMS Microbiol Ecol 84(1):201–212. doi:10.1111/1574-6941.12055
Bowman KS, Dupre RE, Rainey FA, Moe WM (2010) Clostridium hydrogeniformans sp. nov. and Clostridium cavendishii sp. nov., hydrogen-producing bacteria from chlorinated solvent-contaminated groundwater. Int J Syst Evol Microbiol 60:358–363. doi:10.1099/ijs. 0.013169-0
Brüggemann H (2005) Genomics of clostridial pathogens: implication of extrachromosomal elements in pathogenicity. Curr Opin Microbiol 8(5):601–605. doi:10.1016/j.mib.2005.08.006
Chang JJ, Chou CH, Ho CY, Chen WE, Lay JJ, Huang CC (2008) Syntrophic co-culture of aerobic Bacillus and anaerobic Clostridium for bio-fuels and bio-hydrogen production. Int J Hydrogen Energy 33(19):5137–5146. doi:10.1016/j.ijhydene.2008.05.021
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145. doi:10.1093/nar/gkn879
Collins MD, East AK (1998) Phylogeny and taxonomy of the food-borne pathogen Clostridium botulinum and its neurotoxins. J Appl Microbiol 84(1):5–17
Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandezgarayzabal J, Garcia P, Cai J, Hippe H, Farrow JAE (1994) The phylogeny of the genus Clostridium—proposal of 5 new general and 11 new species combinations. Int J Syst Bacteriol 44(4):812–826
DIN EN 12879 (2001) Characterization of sludges—determination of loss on ignition of dry mass. Beuth Verlag, Berlin
DIN EN 12880 (2001) Characterization of sludges—determination of dry residue and water content. Beuth Verlag, Berlin
DIN EN 15933 (2012) Sludge, treated biowaste and soil—determination of pH. Beuth Verlag, Berlin
DIN EN 38406–5 (1983) German standard methods for the examination of water, waste water and sludge; cations (group E); determination of ammonia-nitrogen (E5). Beuth Verlag, Berlin
Dineen SS, Bradshaw M, Johnson EA (2003) Neurotoxin gene clusters in Clostridium botulinum type A strains: sequence comparison and evolutionary implications. Curr Microbiol 46(5):345–352. doi:10.1007/s00284-002-3851-1
Dlabola J, Aue A, Gessler F, Köhler B, Neubauer H, Repp A, Seyboldt C (2013) Nachweis von Clostridium botulinum Neurotoxin in Rinderkot und Silage. Empfehlungen zur Durchführung des Maus Bioessays. Amtstierärztlicher Dienst und Lebensmittelkontrolle, vol 20. Verlag Alpha Informations-GmbH, Lampertheim, pp 120–126
Dohrmann AB, Baumert S, Klingebiel L, Weiland P, Tebbe CC (2011) Bacterial community structure in experimental methanogenic bioreactors and search for pathogenic clostridia as community members. Appl Microbiol Biotechnol 89(6):1991–2004. doi:10.1007/s00253-010-2955-y
Dohrmann A, Küting M, Jünemann S, Jaenicke S, Schlüter A, Tebbe CC (2012) Importance of rare taxa for bacterial diversity in the rhizosphere of Bt- and conventional maize varieties. ISME J 7(1):37–49. doi:10.1038/ismej.2012.77
Duniere L, Sindou J, Chaucheyras-Durand F, Chevallier I, Thevenot-Sergentet D (2013) Silage processing and strategies to prevent persistence of undesirable microorganisms. Anim Feed Sci Technol 182:1–15. doi:10.1016/j.anifeedsci.2013.04.006
Eikmeyer FG, Rademacher A, Hanreich A, Hennig M, Jaenicke S, Maus I, Wibberg D, Zakrzewski M, Pühler A, Klocke M, Schlüter A (2013) Detailed analysis of metagenome datasets obtained from biogas-producing microbial communities residing in biogas reactors does not indicate the presence of putative pathogenic microorganisms. Biotechnol Biofuels 6:49. doi:10.1186/1754-6834-6-49
Gupta RS, Gao B (2009) Phylogenomic analyses of clostridia and identification of novel protein signatures that are specific to the genus Clostridium sensu stricto (cluster I). Int J Syst Evol Microbiol 59:285–294. doi:10.1099/ijs. 0.001792-0
Hedges LV, Gurevitch J, Curtis PS (1999) The meta-analysis of response ratios in experimental ecology. Ecology 80(4):1150–1156. doi:10.2307/177062
Hill KK, Xie G, Foley BT, Smith TJ, Munk AC, Bruce D, Smith LA, Brettin TS, Detter JC (2009) Recombination and insertion events involving the botulinum neurotoxin complex genes in Clostridium botulinum types A, B, E and F and Clostridium butyricum type E strains. BMC Biol 7:66. doi:10.1186/1741-7007-7-66
Hutson RA, Zhou YT, Collins MD, Johnson EA, Hatheway CL, Sugiyama H (1996) Genetic characterization of Clostridium botulinum type A containing silent type B neurotoxin gene sequences. J Biol Chem 271:10786–10792
Keis S, Bennett CF, Ward VK, Jones DT (1995) Taxonomy and phylogeny of industrial solvent-producing clostridia. Int J Syst Bacteriol 45(4):693–705
Kröber M, Bekel T, Diaz NN, Goesmann A, Jaenicke S, Krause L, Miller D, Runte KJ, Viehover P, Pühler A, Schlüter A (2009) Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J Biotechnol 142(1):38–49. doi:10.1016/j.jbiotec.2009.02.010
Krüger M, Grosse-Herrenthey A, Schrödl W, Gerlach A, Rodloff A (2012) Visceral botulism at dairy farms in Schleswig Holstein, Germany—prevalence of Clostridium botulinum in feces of cows, in animal feeds, in feces of the farmers, and in house dust. Anaerobe 18(2):221–223. doi:10.1016/j.anaerobe.2011.12.013
Le Bourhis AG, Saunier K, Dore J, Carlier JP, Chamba JF, Popoff MR, Tholozan JL (2005) Development and validation of PCR primers to assess the diversity of Clostridium spp. in cheese by temporal temperature gradient gel electrophoresis. Appl Environ Microbiol 71(1):29–38. doi:10.1128/aem. 71.1.29-38.2005
Li A, Chu YN, Wang XM, Ren LF, Yu J, Liu XL, Yan JB, Zhang L, Wu SX, Li SZ (2013) A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol Biofuels 6:3. doi:10.1186/1754-6834-6-3
Lindstrom M, Myllykoski J, Sivela S, Korkeala H (2010) Clostridium botulinum in cattle and dairy products. Crit Rev Food Sci 50(4):281–304. doi:10.1080/10408390802544405
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71(12):8228–8235. doi:10.1128/aem. 71.12.8228-8235.2005
Lu XM, Lu PZ, Zhang H (2014) Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 98(6):2657–2665. doi:10.1007/s00253-013-5211-4
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüßmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32(4):1363–1371. doi:10.1093/nar/gkh293
Lueders T, Manefield M, Friedrich MW (2004) Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ Microbiol 6(1):73–78. doi:10.1046/j.1462-2920.2003.00536.x
McGarvey JA, Franco RB, Palumbo JD, Hnasko R, Stanker L, Mitloehner FM (2013) Bacterial population dynamics during the ensiling of Medicago sativa (alfalfa) and subsequent exposure to air. J Appl Microbiol 114(6):1661–1670. doi:10.1111/jam.12179
Nakamura N, Gaskins HR, Collier CT, Nava GM, Rai D, Petschow B, Russell WM, Harris C, Mackie RI, Wampler JL, Walker DC (2009) Molecular ecological analysis of fecal bacterial populations from term infants fed formula supplemented with selected blends of prebiotics. Appl Environ Microbiol 75(4):1121–1128. doi:10.1128/aem. 02359-07
Nettmann E, Bergmann I, Mundt K, Linke B, Klocke M (2008) Archaea diversity within a commercial biogas plant utilizing herbal biomass determined by 16S rDNA and mcrA analysis. J Appl Microbiol 105(6):1835–1850. doi:10.1111/j.1365-2672.2008.03949.x
Notermans S, Dufrenne J, Oosterom J (1981) Persistence of Clostridium botulinum type B on a cattle farm after an outbreak of botulism. Appl Environ Microbiol 41(1):179–183
Park JH, Lee SH, Yoon JJ, Kim SH, Park HD (2014) Predominance of cluster I Clostridium in hydrogen fermentation of galactose seeded with various heat-treated anaerobic sludges. Bioresour Technol 157:98–106. doi:10.1016/j.biortech.2014.01.081
Peck MW, Stringer SC, Carter AT (2011) Clostridium botulinum in the post-genomic era. Food Microbiol 28(2):183–191. doi:10.1016/j.fm.2010.03.005
Price MN, Dehal PS, Arkin AP (2010) FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5(3) doi:10.1371/journal.pone.0009490
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35(21):7188–7196. doi:10.1093/nar/gkm864
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D141–D145. doi:10.1093/nar/gks1219
R Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rainey FA, Stackebrandt E (1993) 16S rDNA analysis reveals phylogenetic diversity among the polysaccharolytic clostrida. FEMS Microbiol Lett 113(2):125–128. doi:10.1016/0378-1097(93)90256-2
Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A (2004) Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol 97(6):1166–1177. doi:10.1111/j.1365-2672.2004.02409.x
Ritchie LE, Steiner JM, Suchodolski JS (2008) Assessment of microbial diversity along the feline intestinal tract using 16S rRNA gene analysis. FEMS Microbiol Ecol 66(3):590–598. doi:10.1111/j.1574-6941.2008.00609.x
Rossi F, Dellaglio F (2007) Quality of silages from Italian farms as attested by number and identity of microbial indicators. J Appl Microbiol 103(5):1707–1715. doi:10.1111/j.1365-2672.2007.03416.x
Schellenberger S, Kolb S, Drake HL (2010) Metabolic responses of novel cellulolytic and saccharolytic agricultural soil bacteria to oxygen. Environ Microbiol 12(4):845–861. doi:10.1111/j.1462-2920.2009.02128.x
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541. doi:10.1128/aem. 01541-09
Schwarz KM, Kuit W, Grimmler C, Ehrenreich A, Kengen SWM (2012) A transcriptional study of acidogenic chemostat cells of Clostridium acetobutylicum—cellular behavior in adaptation to n-butanol. J Biotechnol 161(3):366–377. doi:10.1016/j.jbiotec.2012.03.018
Shrestha M, Shrestha PM, Conrad R (2011) Bacterial and archaeal communities involved in the in situ degradation of 13C-labelled straw in the rice rhizosphere. Environ Microbiol Rep 3(5):587–596. doi:10.1111/j.1758-2229.2011.00267.x
Song YL, Liu CX, Finegold SA (2004) Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol 70(11):6459–6465. doi:10.1128/aem. 70.11.6459-6465.2004
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH, Sørensen SJ, Karlsson A (2013) 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol 85(3):612–626. doi:10.1111/1574-6941.12148
Uz I, Chauhan A, Ogram AV (2007) Cellulolytic, fermentative, and methanogenic guilds in benthic periphyton mats from the Florida Everglades. FEMS Microbiol Ecol 61(2):337–347. doi:10.1111/j.1574-6941.2007.00341.x
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267. doi:10.1128/aem. 00062-07
Weiland P (2010) Biogas production: current state and perspectives. Appl Microbiol Biotechnol 85(4):849–860. doi:10.1007/s00253-009-2246-7
Wise MG, Siragusa GR (2007) Quantitative analysis of the intestinal bacterial community in one- to three-week-old commercially reared broiler chickens fed conventional or antibiotic-free vegetable-based diets. J Appl Microbiol 102(4):1138–1149. doi:10.1111/j.1365-2672.2006.03153.x
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89(6):670–679. doi:10.1002/bit.20347
Yutin N, Galperin MY (2013) A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15(10):2631–2641. doi:10.1111/1462-2920.12173
Ziganshin AM, Liebetrau J, Proter J, Kleinsteuber S (2013) Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl Microbiol Biotechnol 97(11):5161–5174. doi:10.1007/s00253-013-4867-0
Acknowledgments
We thank the owner of the biogas plant analysed in this study for providing the sample materials. We thank Karin Trescher (Thünen Institute) for the excellent technical assistance and Kirsten Löwe (HAWK) and Astrid Näther (Thünen Institute) for the discussion. We would also like to acknowledge the support of Martin Bolz from the Thünen IT facility for the maintenance of the Linux-based computer systems. The work was supported financially by the Lower Saxony Ministry of Food, Agriculture, Consumer Protection and Regional Development, Hannover, Germany.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 124 kb)
Rights and permissions
About this article

Cite this article
Dohrmann, A.B., Walz, M., Löwen, A. et al. Clostridium cluster I and their pathogenic members in a full-scale operating biogas plant. Appl Microbiol Biotechnol 99, 3585–3598 (2015). https://doi.org/10.1007/s00253-014-6261-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6261-y