
Abstract
Using methanol as an alternative non-food feedstock for biotechnological production offers several advantages in line with a methanol-based bioeconomy. The Gram-positive, facultative methylotrophic and thermophilic bacterium Bacillus methanolicus is one of the few described microbial candidates with a potential for the conversion of methanol to value-added products. Its capabilities of producing and secreting the commercially important amino acids l-glutamate and l-lysine to high concentrations at 50 °C have been demonstrated and make B. methanolicus a promising target to develop cell factories for industrial-scale production processes. B. methanolicus uses the ribulose monophosphate cycle for methanol assimilation and represents the first example of plasmid-dependent methylotrophy. Recent genome sequencing of two physiologically different wild-type B. methanolicus strains, MGA3 and PB1, accompanied with transcriptome and proteome analyses has generated fundamental new insight into the metabolism of the species. In addition, multiple key enzymes representing methylotrophic and biosynthetic pathways have been biochemically characterized. All this, together with establishment of improved tools for gene expression, has opened opportunities for systems-level metabolic engineering of B. methanolicus. Here, we summarize the current status of its metabolism and biochemistry, available genetic tools, and its potential use in respect to overproduction of amino acids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is a high societal demand for a sustainable production of special, fine, bulk, and fuel chemicals, including food and health-care compounds. Biotechnological processes will play a prominent role in the coming bioeconomy era by gradually complementing and substituting petrochemical synthesis. In biotechnology, microorganisms are widely used as cell factories and, in particular for high-volume products, raw material costs make up a large part of process costs. In white biotechnology, mainly sugars and molasses are used as carbon sources, and these raw materials are derived from plants, demanding cultivable land which is more and more needed for human nutrition. The possibility to utilize non-food raw materials, such as one-carbon (C1) substrates like methane (CH4) and methanol (CH3OH), as alternative feedstock has therefore gained high scientific interest but is not yet implemented at commercial scale. Major reasons for this are that methanol is still a more expensive substrate than sugar (worldwide methanol prizes at Methanex: https://www.methanex.com/our-business/pricing) and that methanol fermentations can be technically challenging; for example, high O2 requirements and concomitant heat production may cause increased cooling requirements, and careful substrate feeding is needed to avoid toxic formaldehyde accumulation in the cells (see below). However, substantial progress on fermentation technology and construction of production strains should enable commercialization of methanol-based bioprocesses in the near future, as reviewed here and elsewhere (Brautaset et al. 2007; Ochsner et al. 2014b; Schrader et al. 2009).
Methanol is a pure and non-food chemical that is soluble in water, and it is completely utilized during microbial fermentations. Methanol should thus represent an attractive alternative raw material for biotechnological processes from an economic, ecologic, and process point of view. With a worldwide production capacity of more than 53 million tons per year, methanol is one of the most important raw chemicals on earth. The supply of methanol can be based either upon fossil or renewable resources, rendering it a highly flexible and sustainable raw material (Ochsner et al. 2014b). Today, almost all methanol worldwide is produced from syngas, a fuel gas mixture consisting of H2, CO, and CO2, obtained from incomplete combustion of natural gas. In addition, new ways for methanol production directly from natural gas, carbon dioxide, or biogas are being developed, and mega-methanol production facilities (5000 tons per day) are now being constructed in regions rich in natural gas (Brautaset et al. 2007; Schrader et al. 2009).
Methylotrophy, the ability of certain specialized microorganisms to utilize reduced C1 compounds as their sole carbon source for growth, bears the potential to build value from methanol through production of key chemicals. In nature, methylotrophic bacteria can synthesize all their cell constituents from C1 compounds and use them as both carbon and energy sources. This is in contrast to most autotrophs that perform total biosynthesis from carbon dioxide but require a separate energy source. The early interest in biotechnology to use methylotrophs is exemplified best by the large-scale biotechnological processes employing methanol-converting microorganisms as a source of single cell protein (Tannenbaum and Wang 1975). The same factors that made methanol attractive as a substrate for this process make it even more attractive as a source of various products nowadays (Brautaset et al. 2007; Schrader et al. 2009). Nature evolved different solutions to harness methanol for the purpose of energy generation and biomass formation, and phylogenetically, methylotrophs belong to several genera within Alpha- (e.g., Methylobacterium), Beta- (e.g., Methylobacillus), and Gammaproteobacteria (e.g., Methylococcus), as well as within the Firmicutes (e.g., Bacillus), and Verrucomicrobia (e.g., Methylacidiphilum) (Chistoserdova and Lidstrom 2013). Many of these are facultative methylotrophs that are able to grow not only on C1 compounds but also on a generally limited number of multi-carbon compounds. While the metabolism and potential of Methylobacterium extorquens as another potential platform organism will be presented in a separated review (Ochsner et al. 2014b), here, we will focus on methylotrophy and application potential of Bacillus methanolicus.
B. methanolicus is a Gram-positive facultative methylotrophic bacterium which possesses methanol dehydrogenase and the ribulose monophosphate (RuMP) cycle as key enzyme and pathway, respectively, enabling methanol growth (Arfman et al. 1992a, b, 1991, 1989; Dijkhuizen et al. 1988; Schendel et al. 1990), and this organism represents the first example of plasmid-dependent methylotrophy (Brautaset et al. 2004). Methanol is more reduced than sugars, and thus, growth and production with methanol are characterized by a high oxygen demand (Arfman et al. 1992a, b; 1991, 1989; Dijkhuizen et al. 1988; Schendel et al. 1990), and as heat production increases with oxygen consumption, cooling requirements are high compared to those of fermentation of sugar. Importantly in this regard, B. methanolicus has a growth optimum around 50–55 °C which offers reduced cooling requirement compared to utilization of mesophilic methylotrophic bacteria. The isolation and microbiological characterization of thermotolerant Bacilli, selection of classical B. methanolicus mutants overproducing l-lysine, and fed-batch methanol fermentations demonstrating l-glutamate production at 50 °C have been reviewed previously (Brautaset et al. 2007). In the recent years, significant progress has been made on this organism, including development of improved genetic tools; biosynthetic engineering of the aspartate pathway for l-lysine overproduction (Jakobsen et al. 2009; Nærdal et al. 2011); manipulations of the anaplerotic enzyme pyruvate carboxylase, important for replenishing oxaloacetate to the tricarboxylic acid (TCA) cycle (Brautaset et al. 2010); and most importantly genome sequencing of two B. methanolicus model strains MGA3 and PB1 (Heggeset et al. 2012; Irla et al. 2014). Besides gaining insight into the repertoire of genes, pathways, and metabolism, genome-wide transcriptomic and proteomic approaches became possible (Heggeset et al. 2012; Müller et al. 2014). In addition, biochemical analysis furthered our understanding of methylotrophic pathways (Krog et al. 2013b; Markert et al. 2014; Ochsner et al. 2014a; Stolzenberger et al. 2013a, b) and l-glutamate synthesis and degradation (Krog et al. 2013a). Thus, B. methanolicus is on the one hand a promising biocatalyst for conversion of methanol into value-added products at elevated temperature and on the other hand a highly interesting model strain for basic studies aiming at understanding bacterial methylotrophy.
Growth characteristics
Wild-type strains of B. methanolicus are able to grow at temperatures between 35 and 60 °C with a growth optimum around 50–55 °C (Arfman et al. 1992b; Schendel et al. 1990). Strains can be isolated from soil samples, wastewater treatment systems, and hot springs. The cells are rod shaped and may form oval endospores. While endospores are rarely generated at 50 °C, they readily form in response to a temperature drop from 50 to 37 °C during exponential growth (Schendel et al. 1990). Most strains of B. methanolicus can grow in 20 g/L but not in 50 g/L NaCl (Arfman et al. 1992b), and it has been shown that strains may be adapted to grow in low-cost seawater-based media (Komives et al. 2005). B. methanolicus strains are restricted methylotrophs that are able to utilize a limited number of alternative carbon sources including mannitol and glucose in addition to methanol (Arfman et al. 1992a, b; Schendel et al. 1990). B. methanolicus MGA3 exhibits similar specific growth rates on mannitol and methanol at 50 °C of about 0.30/h in shake flask cultures and 0.40/h in 0.6 L bioreactors; however, the specific growth rate is considerably lower at 37 °C (0.14/h on methanol in 0.6 L bioreactors) (Jakobsen et al. 2006; Müller et al. 2014). Methanol supports growth of B. methanolicus up to concentrations of about 1 M, but tolerance to methanol is inducible and requires prior exposure to low methanol concentrations (Jakobsen et al. 2006).
The ability of B. methanolicus to grow on methanol has been experimentally demonstrated to be dependent on the plasmid pBM19 (Brautaset et al. 2004). Interestingly, the specific growth rate of the pBM19-cured strain MGA3C-A6 on mannitol was found to be significantly higher (0.37/h, in shake flask cultures) than that of its parental wild-type strain MGA3 (0.30/h), implying that the pBM19 plasmid represents a metabolic burden when its genes are not required for growth (Jakobsen et al. 2006). Indeed, when MGA3 cells were grown for 140 generations in minimal medium using mannitol instead of methanol as sole carbon source, 80 % of the cells had lost plasmid pBM19 and were no longer capable of growing on methanol (Jakobsen et al. 2006). Although genetically very similar, B. methanolicus strains MGA3 and PB1 have been shown to display considerable differences with respect to growth, amino acid production, and respiration profiles in fed-batch methanol cultivations (Heggeset et al. 2012). Whereas MGA3 has been shown to secrete 0.4 g/L l-lysine and 60 g/L l-glutamate in optimized fed-batch cultivations (Brautaset et al. 2010), PB1 produced very little l-glutamate (1.6 g/L) and showed a considerably lower maximal biomass concentration, but much higher respiration (CO2 evolution rate) (Heggeset et al. 2012). The underlying cause for these differences is still unknown.
Genome sequences
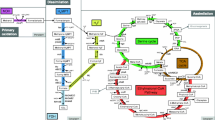
Genome draft assemblies of 3.4 Mbp with low GC contents of 38.5 and 39.0 %, respectively, were obtained for the B. methanolicus strains MGA3 and PB1 (Heggeset et al. 2012). As mentioned above, B. methanolicus MGA3 harbors plasmid pBM19 important for methylotrophy. Also, strain PB1 harbors a plasmid of similar size (20 kbp), denoted pBM20. Similar to pBM19 of strain MGA3, the plasmid was previously shown to encode mdh and five RuMP cycle genes (Brautaset et al. 2004; Jakobsen et al. 2006). Although likely, it is currently not known whether plasmid pBM20 is required for methylotrophic growth of strain PB1. Strain PB1 lacks the second plasmid of 69 kbp, pBM69, with genes of unknown functions present in strain MGA3 (Heggeset et al. 2012), and also, the predicted genes identified on pBM69 are not present on the PB1 chromosome. Surprisingly, chromosomal homologues of all the RuMP cycle genes of pBM19 and pBM20 have been found in both strains with the exception of rpe, encoding ribulose phosphate 3-epimerase, which was not found on the PB1 chromosome. In addition, since the phosphofructokinase gene of pBM20 is non-functional due to a frameshift mutation, PB1 likely recruits Pfk activity needed for the RuMP cycle from the chromosomal gene (Heggeset et al. 2012). Notably, the chromosomes of both strains encode two homologues of methanol dehydrogenase. In strain MGA3, the chromosomally encoded proteins were named Mdh2 and Mdh3 and are 96 % identical to each other, while they share 61 and 62 % overall sequence identity to the pBM19 encoded protein Mdh. The Mdh encoded on the natural plasmid pBM20 of strain PB1 is 93 % identical to Mdh of MGA3. The chromosomally encoded Mdhs, denoted Mdh1 and Mdh2, are 92 and 59 % identical to the Mdh encoded on pBM20. All Mdh enzymes have been biochemically characterized (see below). Genes representing both a cyclic dissimilatory RuMP cycle and a linear tetrahydrofolate pathway for formaldehyde dissimilation were identified in both strains (see Fig. 2; formaldehyde oxidation I (FOI) and formaldehyde oxidation V (FOV), respectively). The cyclic dissimilatory RuMP pathway has an important role in regenerating reducing power [NAD(P)H], and it has been argued that it represents a substitute for the lack of a complete TCA cycle for energetic purposes in many methylotrophic bacteria (Chistoserdova et al. 2009). Both dissimilatory pathways yield CO2 and do not contribute to net biomass formation. Besides their role in generation of reducing power, they are known to regulate toxic formaldehyde levels in the cells during methylotrophic growth (Anthony 1982; Vorholt 2002). The genomes of B. methanolicus MGA3 and PB1 do, however, encode genes representing the complete TCA cycle and the glyoxylate shunt. In addition, genes encoding uptake systems for mannitol and glutamate were found in both strains (Heggeset et al. 2012). B. methanolicus strains are typically auxotrophic for biotin (Komives et al. 2005; Schendel et al. 1990) due to an incomplete biotin biosynthesis operon, where only an assumed non-functional remnant of bioI, encoding the cytochrome P450 hydroxylase generating pimeloyl-ACP, is found on the pBM69 plasmid (Irla et al. 2014).
Genetic tools
The development of genetic tools for B. methanolicus has been a struggle, and still efficient genetic tools represent a major bottleneck for strain engineering of this organism. Although gene delivery by protoplast transformation has been demonstrated (Cue et al. 1997), an electroporation protocol developed by Jakobsen et al. (2006) is the method currently used for transformation of B. methanolicus MGA3 with a limited set of plasmids (Table 1). Transformation frequencies are low, and up to 103–104 cfu are typically obtained per microgram DNA (Brautaset, unpublished). B. methanolicus encodes a restriction endonuclease, BmeTI, an isoschizomer of BclI, that recognizes the DNA sequence 5′-TGATCA 3′, and BmeTI sites are modified to TGm6ATCA by a specific methylase (Cue et al. 1996). It was therefore believed that methylated DNA is more easily established in B. methanolicus MGA3 than unmethylated DNA (Cue et al. 1997); however, experiments have indicated that this is not a bottleneck for transformation efficiency (Brautaset, unpublished). More recently, the Escherichia coli-Geobacillus stearothermophilus-based shuttle vector pNW33N was shown to replicate in B. methanolicus MGA3 (Nilasari et al. 2012) as well as the theta-replicating E. coli-Bacillus subtilis plasmid pHCMC04 (Nguyen et al. 2005) with the xylose-inducible promoter included in the latter vector being functional in this organism (Heggeset, unpublished). All published studies have used the strong and inducible methanol dehydrogenase promoter from plasmid pBM19 for recombinant expression, and typically, a two- to fivefold induction by methanol was observed using vector pTH1mp for homologous gene overexpression (Markert et al. 2014; Stolzenberger et al. 2013a, b). The AT-rich nature of the B. methanolicus DNA makes promoter prediction challenging, and the exact location of the mdh-promoter is not known (Brautaset et al. 2004; Nilasari et al. 2012).
Due to the high growth temperature, many of the traditional reporter genes used for other bacteria are not functional or function poorly in B. methanolicus. Among these is the green fluorescent protein, GFPuv, which has been successfully expressed in B. methanolicus MGA3 (Fig. 1) (Irla, Markert, and Wendisch, unpublished) (Nilasari et al. 2012), however, only when the temperature was lowered from 50 to 40 °C for at least 30 min before measuring fluorescence in order for the protein to fold properly and generate the chromophore (Nilasari et al. 2012). A promising alternative reporter gene is the lacZ from Bacillus coagulans, encoding a thermotolerant β-galactosidase (Kovacs et al. 2010), which was successfully expressed in B. methanolicus MGA3 and could be used for blue-white screening (Heggeset, unpublished).

GFP fluorescence analysis of a mixture of B. methanolicus MGA3 (pTH1mp) and MGA3 (pTH1mp-gfp) cells (Irla, Markert, and Wendisch, unpublished). Prior to analysis by FACS and fluorescence microscopy (inset shows an unlabeled and a labeled cell), cells of both strains growing exponentially at 50 °C in separate methanol minimal medium cultures were harvested, incubated at 37 °C to facilitate GFP chromophore formation, and mixed in a 1:1 ratio
Methylotrophy of B. methanolicus
Since the discovery of plasmid-dependent methylotrophy (Brautaset et al. 2004), considerable genetic, regulatory, and biochemical knowledge has been generated on the methylotrophic properties of B. methanolicus. These studies have unraveled several unique and surprising traits, making it an interesting methylotrophic model strain, and this knowledge is also valuable for engineering this organism for biotechnological purposes.
Oxidation of methanol to formaldehyde by NAD-dependent methanol dehydrogenase (Mdh)
When grown on methanol a number of B. methanolicus isolates were discovered to catalyze methanol oxidation by an NAD-dependent alcohol dehydrogenase more than two decades ago (Dijkhuizen et al. 1988). Upon purification of the responsible enzyme from cell-free extracts and initial characterization, the enzyme was determined to be a family III alcohol dehydrogenase (Dijkhuizen et al. 1988). Analyses of Mdh from B. methanolicus strain C1 revealed a decameric structure consisting of subunits with a molecular weight of about 43 kDa and one zinc and two magnesium ions per subunit (Arfman et al. 1989; Vonck et al. 1991). The magnesium-dependent NAD-binding domain is formed by the GGGSX2DX2K motif which is conserved in all family III alcohol dehydrogenases, and Mdh contains one tightly but non-covalently bound NAD+ molecule which is reduced in the presence of methanol (Arfman et al. 1997). Mdh from strain C1 and also from MGA3 and PB1 are specific with respect to the cofactor NAD+ as no activity with NADP+ could be determined. Their substrate specificity is more relaxed, and primary alcohols plus isopropanol are generally accepted (Arfman et al. 1989; Krog et al. 2013b).
Based on the purification factors, it was estimated that the enzyme from strain C1 may constitute up to about 20 % of the total soluble protein of this microorganism upon growth on methanol (Arfman et al. 1989). A possible reason for such high expression levels might be the relatively low catalytic efficiency of the enzyme (Krog et al. 2013b; Ochsner et al. 2014a) (Table 2). The activity of the enzyme can be stimulated in vitro by a factor of 3 to 8 in the presence of a soluble dimeric activator protein called Act (molecular weight of about 25 kDa) (Arfman et al. 1991; Krog et al. 2013b; Ochsner et al. 2014a) (Table 2); however, the activity of the Act-stimulated enzyme is still only about 0.2–0.4 U/mg purified enzyme. Act belongs to the class of NUDIX hydrolases and cleaves NAD+ and ADP-ribose (ADPR) efficiently (Hektor et al. 2002). The investigation of the influence of Act on different Mdh mutants led to the proposal of an Mdh reaction mechanism and a potential mode of action of the activator protein. It was proposed that in the absence of Act, the reaction follows a ping-pong-type reaction mechanism whereby the electrons of the substrate are transferred to the bound NAD+ molecule and subsequently used to reduce a free coenzyme NAD+ to NADH (Arfman et al. 1997). However, when Act is present, the latter may cleave the bound cofactor NAD+ molecule, resulting in hydrolytic removal of the nicotinamide mononucleotide (NMN) moiety (Hektor et al. 2002). It has been proposed that the electrons are then directly transferred to the free coenzyme NAD+ during the reaction and the mechanism switches to a ternary complex mechanism. Notably, a mutation of the serine in the GGGSX2DX2K motif was shown to mimic the effect of Act by destroying the NAD+-binding capacity of Mdh (Hektor et al. 2002). Recently, a study focusing on the activation of Mdh and Mdh-like alcohol dehydrogenases from different microorganisms showed that the in vitro activation of several alcohol dehydrogenases by different NUDIX hydrolases is possible (Ochsner et al. 2014a). In addition, the serine mutation in the NAD+-binding motif also led to an activated state in two additional Mdh-like alcohol dehydrogenases from organisms other than B. methanolicus. Thus, it seems that the ability of alcohol dehydrogenases to be activated is not unique to the Mdh/Act couple of B. methanolicus. However, since gene deletion studies are currently not possible in B. methanolicus, the biological relevance of activation of Mdh by NUDIX hydrolase Act remains to be investigated.
Transcriptome and proteome analysis showed that expression of mdh (which is encoded on the pBM19 plasmid; see above) is high during growth on methanol and also on the alternative carbon substrate mannitol (Heggeset et al. 2012; Müller et al. 2014) (Table 3). The two Mdh-like enzymes in B. methanolicus strains MGA3 and PB1 introduced above, which are encoded on the chromosome (Heggeset et al. 2012), are also activated by Act in vitro, similar to the plasmid-encoded ones (Krog et al. 2013b) (Table 3). However, while the abovementioned serine mutation leads to an increased activity in Mdh-type enzymes (Hektor et al. 2002; Ochsner et al. 2014a), the same mutation almost abolishes the methanol-oxidizing activity in Mdh2-type enzymes (Ochsner et al. 2014a). Similar to Mdh, also the Mdh2-type enzymes exclusively use NAD+ as co-substrate. Notably, it has been shown recently that a single amino acid exchange in Mdh and Mdh2 of MGA3 resulted in enzyme derivatives which also use NADP+ as a cofactor for oxidation of methanol to formaldehyde in addition to NAD+ (Ochsner et al. 2014a). The biological function of the chromosomally encoded Mdhs is still unclear. No upregulation of these enzymes on transcriptome or proteome level was observed during growth on methanol (Heggeset et al. 2012; Müller et al. 2014) (Table 3), which makes it questionable if these enzymes are important for methylotrophic growth of B. methanolicus.
Dissimilation of formaldehyde to CO2
In the first studies on the metabolism of B. methanolicus strain C1, the failure of methanol-grown cells to oxidize formate and the missing activities of formaldehyde and formate dehydrogenases led to the assumption that B. methanolicus does not possess a linear oxidation pathway for formaldehyde to CO2 (Arfman et al. 1989). It was rather assumed that oxidation of formaldehyde takes place via the dissimilatory RuMP cycle (Chistoserdova 2011) (Fig. 2). This notion was supported by the detection of high activities of glucose-6-phosphate isomerase, glucose-6-phosphate dehydrogenase, and 6-phosphogluconate dehydrogenase (Arfman et al. 1989). However, a later study provided direct experimental evidence that a linear oxidation of formaldehyde to CO2 must exist in B. methanolicus MGA3, as production of 13C-labeled formate from 13C-labeled methanol and 13C-labeled CO2 from 13C-labeled formate was observed (Pluschkell and Flickinger 2002). These findings were also consistent with the identification of genes encoding a potential, linear tetrahydrofolate (THF)-based oxidation pathway in the genomes of B. methanolicus MGA3 and PB1 (Heggeset et al. 2012). The genes include folD (encoding methylenetetrahydrofolate dehydrogenase/cyclohydrolase), fhs (encoding formate-tetrahydrofolate ligase), fdhA (encoding formate dehydrogenase α-chain), and fdhD (encoding formate dehydrogenase family accessory protein). While transcriptome and proteome analyses of methanol-grown cells showed no upregulation of the proteins potentially involved in this pathway (Heggeset et al. 2012; Müller et al. 2014) (Table 3), these omics approaches, however, revealed upregulation of glucose-6-phosphate dehydrogenase 2 (Zwf2), an enzyme of the dissimilatory RuMP pathway, in cells grown on methanol (Heggeset et al. 2012; Müller et al. 2014) (Fig. 2, Table 3), strengthening a role of the latter for formaldehyde oxidation. Taken together, B. methanolicus possesses at least two pathways for formaldehyde dissimilation. Additional experimental data including metabolome data will be required to further substantiate pathway operation and to fully understand the distinct biological functions of alternative pathways during methylotrophic growth.

Schematic representation of the carbon metabolism in B. methanolicus MGA3. Enzymes encoded on the chromosome (C) or on the plasmid pBM19 (P) are indicated. Green arrows indicate significant upregulation of proteins on methanol. Red arrows correspond to enzymes downregulated on methanol and thus upregulated on mannitol. Dashed arrows indicate several reactions. If more than one protein is predicted to catalyze a reaction, the regulated protein is indicated by color coding. Only proteins with a log2 fold change greater than 1 and a P value below 0.05 were considered (s. Table 3). Pathways: MO methanol oxidation; FOV formaldehyde oxidation V; FOI formaldehyde oxidation I; RuMP ribulose monophosphate cycle; EMP glycolysis; TCA tricarboxylic acid cycle; Lys lysine biosynthesis; Glu glutamate biosynthesis. Proteins: Mdh methanol dehydrogenase; FolD bifunctional methylenetetrahydrofolate dehydrogenase (NADP+)/methenyltetrahydrofolate cyclohydrolase; Fhs formate-tetrahydrofolate ligase; Fdh putative formate dehydrogenase; FdhA formate dehydrogenase alpha chain; Hps 3-hexulose-6-phosphate synthase; Phi 6-phospho-3-hexuloisomerase; Pfk phosphofructokinase; Fba fructose-bisphosphate aldolase; GlpX fructose-bisphosphatase/sedoheptulose-bisphosphatase; Tkt transketolase; Rpe ribulose-phosphate 3-epimerase; RpiB ribose 5-phosphate isomerase; Pgi glucose-6-phosphate isomerase; Zwf glucose-6-phosphate dehydrogenase; Pgl 6-phosphogluconolactonase; Gnd 6-phosphogluconate dehydrogenase; TpiA triose-phosphate isomerase; Gap glyceraldehyde-3-phosphate dehydrogenase; Pgk phosphoglycerate kinase; GpmI phosphoglycerate mutase (2,3-disphosphoglycerate independent); Eno phosphopyruvate hydratase; Pyk pyruvate kinase; PdhA pyruvate dehydrogenase (acetyl transferring) (E1), alpha subunit; PdhB pyruvate dehydrogenase (acetyl transferring) (E1), beta subunit; Pyc pyruvate carboxylase; CitY citrate (Si)-synthase; AcnA aconitate hydratase; Icd isocitrate dehydrogenase (NADP+); OdhA 2-oxoglutarate dehydrogenase (succinyl transferring) (E1); OdhB dihydrolipoyllysine-residue succinyltransferase (E2); KorA 2-oxoglutarate synthase, alpha subunit; KorB 2-oxoglutarate synthase, beta subunit; SucC succinyl-CoA ligase (ADP forming), beta subunit; SucD succinyl-CoA ligase (ADP forming), alpha subunit; SdhA succinate dehydrogenase, flavoprotein subunit; SdhB succinate dehydrogenase, iron-sulfur subunit; SdhC succinate dehydrogenase, cytochrome b558 subunit; FumA fumarate hydratase, class I; FumC fumarate hydratase, class II; CitH malate dehydrogenase; Mqo malate dehydrogenase (quinone); AspB aspartate transaminase; YclM aspartate kinase III; DapG aspartate kinase I; LysC aspartate kinase II; Asd aspartate-semialdehyde dehydrogenase; DapA 4-hydroxy-tetrahydrodipicolinate synthase; DapB 4-hydroxy-tetrahydrodipicolinate reductase; DapH tetrahydrodipicolinate N-acetyltransferase; PatA acetyl-diaminopimelate aminotransferase; DapL N-acetyldiaminopimelate deacetylase; DapF diaminopimelate epimerase; LysA diaminopimelate decarboxylase; GltA glutamate synthase (large subunit); GltB glutamate synthase (small subunit); GltA2 glutamate synthase (NADPH); YweB glutamate dehydrogenase; GlnA glutamine synthetase. [e − ] indicates production or consumption of NAD(P)H. ATP indicates production or consumption of adenosine triphosphate. Names and abbreviations are given according to the Uniprot database (http://www.uniprot.org). Metabolites involved: H-6-P 3-hexulose 6-phosphate; F-6-P fructose 6-phosphate; F-1,6-dP fructose 1,6-bisphosphate; GAP glyceraldehyde 3-phosphate; DHAP dihydroxyacetone phosphate; E-4-P erythrose 4-phosphate; S-1,7-dP sedoheptulose 1,7-bisphosphate; S-7-P sedoheptulose 7-phosphate; Ri-5-P ribose 5-phosphate; X-5-P xylulose 5-phosphate; Ru-5-P ribulose 5-phosphate; MTHF methylenetetrahydrofolate; MeTHF methenyltetrahydrofolate; FTHF formyltetrahydrofolate; G-6-P glucose 6-phosphate; 6-PG 6-phosphogluconate; 1,3-BPG 1,3-bisphosphoglycerate; 3-PG 3-phosphoglycerate; 2-PG 2-phosphoglycerate; PEP phosphoenolpyruvate; Thr threonine; Ile isoleucine; Met methionine; meso-dap meso-diaminopimelate
Assimilation of formaldehyde into biomass
It was proposed earlier that the RuMP cycle is used also for carbon assimilation (Dijkhuizen et al. 1988), but it was unclear which of the four potential variants (Anthony 1982) is used. Initially, missing activities of enzymes of the Entner-Doudoroff pathway and sedoheptulose-bisphosphatase (SBPase, GlpX) led to the suggestion that the fructose-bisphosphate aldolase (Fba)/transaldolase variant of the RuMP cycle is employed. Later, the discovery and sequencing of the pBM19 plasmid (Brautaset et al. 2004) and biochemical characterization of the encoded fructose-bisphosphatase (GlpX) (Stolzenberger et al. 2013a) revealed that this enzyme also displays sedoheptulose-bisphosphatase activity (Table 2). Thus, the Fba/SBPase variant of the RuMP cycle could be employed as well (Fig. 2). This assumption is supported by the observed upregulation of the plasmid-encoded GlpX on transcriptome and proteome level during methylotrophic growth (Brautaset et al. 2004; Heggeset et al. 2012; Müller et al. 2014) (Table 3).
In all variants of the RuMP cycle, carbon fixation is initiated by condensation of formaldehyde with ribulose 5-phosphate, leading to the formation of hexulose 6-phosphate which is subsequently isomerized to fructose 6-phosphate. These reactions are catalyzed by 3-hexulose-6-phosphate synthase (Hps) and 6-phospho-3-hexuloisomerase (Phi). Both enzymes have been purified from methanol-grown B. methanolicus C1 and shown to be active (Arfman et al. 1990). In contrast to many other key enzymes of the RuMP cycle (see below), they are encoded solely on the chromosome and are presumably transcribed from a single, formaldehyde inducible promoter (Brautaset et al. 2004; Jakobsen et al. 2006). Overexpression of the two genes increased formaldehyde tolerance in B. methanolicus MGA3, which is in line with their role in formaldehyde assimilation (Jakobsen et al. 2006). Interestingly, non-methylotrophic B. subtilis has an analogous hxlAB operon encoding active Hps and Phi enzymes presumably playing roles in formaldehyde detoxification, and this operon is under transcriptional control of the upstream hxlR gene (Yasueda et al. 1999; Yurimoto et al. 2005). No homologue to the hxlR gene was found in the B. methanolicus genome sequence. Fructose 6-phosphate is either used in the dissimilatory RuMP cycle starting with its conversion into glucose 6-phosphate by glucose-6-phosphate isomerase (Pgi) or phosphorylated by phosphofructokinase (Pfk) and cleaved into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate by fructose-bisphosphate aldolase (Fba). Transcriptome and proteome data showed that only the plasmid-encoded RuMP cycle homologues are upregulated during growth on methanol (Heggeset et al. 2012; Jakobsen et al. 2006; Müller et al. 2014) (Table 3). Recent characterization of the fructose-bisphosphate aldolases revealed that the two versions are biochemically different. The chromosomally encoded version preferentially catalyzes the aldol cleavage reaction of fructose 1,6-bisphosphate while the plasmid-encoded enzyme is working best in the reverse aldol condensation direction (Stolzenberger et al. 2013b) (Table 2). Glyceraldehyde 3-phosphate and fructose 6-phosphate are subsequently metabolized in a series of reactions known as the rearrangement phase, involving Pfk, Tkt, Fba, Rpe, and ribose 5-phosphate isomerase (RpiB) to regenerate ribulose 5-phosphate as an acceptor for formaldehyde. In the proposed SBPase variant of the rearrangement phase, sedoheptulose 1,7-bisphosphate is formed in an Fba-catalyzed reaction out of erythrose 4-phosphate and dihydroxyacetone phosphate (Anthony 1982; Stolzenberger et al. 2013b) (Fig. 2). Based on biochemical properties (Table 2), the plasmid-encoded GlpX is likely to be responsible for the production of sedoheptulose 7-phosphate by dephosphorylation of sedoheptulose 1,7-bisphosphate (see above). After three rounds of formaldehyde fixation and C5 precursor regeneration, a triose-phosphate unit in the form of glyceraldehyde or dihydroxyacetone phosphate remains available for biomass formation via the lower glycolysis. Contrary to the duplicate enzymes GlpX and Fba that have specific biochemical properties, the duplicate Tkt enzymes showed similar biochemical properties (Markert et al. 2014) (Table 2). Nonetheless, only the plasmid-encoded version is upregulated during growth on methanol (Table 3). A putative transaldolase (TA) gene (tal) was also identified on the B. methanolicus chromosome, and this gene is not transcriptionally upregulated on methanol. However, while tal of B. methanolicus PB1 could be shown to encode functional TA, the tal gene in strain MGA3 contained mutations (Brautaset and Wendisch, unpublished).
Summarized, it has been experimentally shown that methanol assimilation in B. methanolicus involves both plasmid and chromosomally encoded RuMP cycle genes and it presumably uses the fructose-bisphosphate aldolase for the cleavage phase and the SBPase for the rearrangement phase. Levels of enzymes involved in glycolysis and gluconeogenesis are mainly unaffected during growth with different substrates (Heggeset et al. 2012; Müller et al. 2014). Notably, the plasmid-encoded Fba, which is involved in both the RuMP cycle and glycolysis, is upregulated (Table 3). Since this enzyme preferentially catalyzes the formation of fructose 1,6-bisphosphate (Stolzenberger et al. 2013b), it was proposed that its increased expression counteracts the loss of molecules needed by the RuMP cycle for assimilation of methanol (i.e., ribulose 5-phosphate = RuMP) (Müller et al. 2014). One might speculate that the downregulation of pyruvate dehydrogenase (Pdh) on methanol (Table 3) serves the same purpose by reducing the carbon flux into the TCA cycle. Interestingly, during methylotrophic growth, five genes of the TCA cycle, icd, odhAB, korAB, sucCD, and sdhAB, encoding isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, 2-oxoglutarate synthase, succinyl-CoA ligase, and succinate dehydrogenase, respectively, are significantly downregulated on the proteome level. In contrast to growth on sugar substrates, B. methanolicus presumably does not require a functional TCA cycle to produce reduction equivalents during methylotrophic growth and it is likely that the remaining reactions of the TCA cycle mainly function to provide precursors for biosynthetic purposes under such conditions.
l-Glutamate and l-lysine biosynthesis (and degradation) pathways
The flavor enhancer l-glutamate and the essential amino acid l-lysine are industrially important compounds, and approximately 3 million tons of l-glutamate and 2 million tons of l-lysine are produced annually worldwide, primarily using Corynebacterium glutamicum and sugar-based raw materials (Anastassiadis 2007; Brautaset and Ellingsen 2011; Fernstrom 2009; Wendisch 2014). B. methanolicus MGA3 is a natural overproducer of l-glutamate, and in methanol fed-batch cultivation, it has been shown to produce up to 60 g/L l-glutamate at 50 °C (Brautaset et al. 2003; Heggeset et al. 2012). In comparison, B. methanolicus PB1 only produces around 1.6 g/L under such conditions, despite being genetically very similar (Heggeset et al. 2012). Classical B. methanolicus mutants overproducing l-lysine have been developed; for example mutant M168-20 was found to secrete 11 g/L lysine and 59 g/L l-glutamate, while mutant NOA2#13A52-8A66 secreted 65 g/L l-lysine and 28 g/L l-glutamate. So far, no B. methanolicus strains producing high l-lysine amounts without concurrently producing l-glutamate as considerable by-product have been described (Brautaset et al. 2010). Very recently, B. methanolicus strains for methanol-based production of the important platform chemical 1,5-diaminopentane (also denoted cadaverine) have been engineered by heterologous expression of a lysine decarboxylase gene from E. coli (Nærdal et al. 2014).
L-Glutamate and l-lysine are both produced from intermediates of the TCA cycle; glutamate is synthesized from 2-oxoglutarate, while l-lysine is a product of the aspartate pathway branching from oxaloacetate (Fig. 2). Withdrawal of TCA cycle intermediates for biosynthesis requires replenishment by anaplerotic reactions. Bacilli typically lack phosphoenolpyruvate carboxylase (PEPC) but possess pyruvate carboxylase (PC) (Brautaset et al. 2010; Heggeset et al. 2012; Kondo et al. 1997; Sauer and Eikmanns 2005). In B. methanolicus, the PC is encoded by pyc and the bacterium in addition possesses the key enzymes of the glyoxylate shunt isocitrate lyase and a malate synthase encoded by the aceA-aceB operon (Heggeset et al. 2012). Overexpression of the PC-encoding gene pyc in MGA3 surprisingly reduced l-glutamate production by nearly 50 % without affecting the l-lysine production (Brautaset et al. 2010), whereas PC activity and pyc messenger RNA (mRNA) levels were threefold enhanced in the classical mutant NOA2#13A52-8A66 (Brautaset et al. 2010, 2003). C. glutamicum has both PEPC and PC, and the latter enzyme is reported to be a key target for both l-lysine and l-glutamate production in different strains of this bacterium (Ohnishi et al. 2002; Peters-Wendisch et al. 2001; Yao et al. 2009). Clearly, more work is needed to characterize anaplerosis in B. methanolicus in order to improve l-lysine and l-glutamate production. In a classical citY mutant of B. methanolicus displaying reduced citrate synthase (CS) activity, the l-glutamate production was reduced sevenfold, and the ratio of l-lysine to l-glutamate was increased 4.5-fold (Brautaset et al. 2003). The CS activity was not completely abolished in this mutant, and it was proposed that an alternative enzyme with CS activity must be encoded by B. methanolicus, as in the case in B. subtilis (Brautaset et al. 2003). However, only one CS-encoding gene was identified in the B. methanolicus genome sequence, and the source for the CS activity measured in the citY knockout mutant is still unknown (Heggeset et al. 2012). In other bacteria, mutations leading to increased methylcitrate synthase levels are known to compensate for the lack of CS as shown in the amino acid-producing C. glutamicum (Radmacher and Eggeling 2007).
Enzymes converting 2-oxoglutarate to l-glutamate
Two alternative l-glutamate biosynthetic pathways are known. The NADPH-dependent glutamate dehydrogenase (GDH) generates l-glutamate by reductive amination of 2-oxoglutarate, while the glutamate synthase (GOGAT) catalyzes the transfer of the amide group from l-glutamine to 2-oxoglutarate, generating two molecules of l-glutamate. l-Glutamine consumed in the GOGAT reaction is synthesized by the ATP-dependent glutamine synthetase (GS) that incorporates ammonium into l-glutamate.
Notably, B. methanolicus harbors only one GDH, encoded by yweB (Heggeset et al. 2012) (Fig. 2). YweB displays a high K m value for l-glutamate (250 mM) and much lower K m values for ammonium (10 mM) and 2-oxoglutarate (20 mM) (Krog et al. 2013a) (Table 4). This is in contrast to both B. subtilis GDHs where the K m for l-glutamate was reported to be 3 and 18 mM for RocG and GudB, while the K m values of the two proteins for ammonium were 18 and 41 mM, respectively (Gunka et al. 2010). The high K m value of YweB for l-glutamate indicates that it likely plays no major role in l-glutamate degradation in B. methanolicus but rather in l-glutamate synthesis since the V max value for the deamination reaction (l-glutamate degradation) was sevenfold lower than for the amination reaction (l-glutamate synthesis). Still, yweB expression was not sufficient to complement l-glutamate synthesis in a B. subtilis glutamate auxotroph, and the exact role of YweB in l-glutamate regulation in B. methanolicus therefore remains unclear (Krog et al. 2013a).
B. methanolicus possesses two different GOGATs encoded by the gltAB operon and by gltA2, in contrast to B. subtilis which has only one gltAB operon (Heggeset et al. 2012). The B. methanolicus gltAB operon encodes the large (GltA) and small (GltB) subunit of the first GOGAT while the gltA2 gene product constitutes the second GOGAT (Fig. 2). The amino acid identity between GltA and GltA2 is only 27 %, indicating that they have evolved independently from each other. In addition to B. methanolicus, only a few thermophilic Bacilli were found to encode both GOGAT variants. Bacteria utilizing GltA2 generally lack gltB genes. Interestingly, both GltA2 and GltA were found to be active and independent of GltB in vitro with similar V max and K m values for glutamine and 2-oxoglutarate (Table 4), while complementation experiments with E. coli and B. subtilis mutants suggest that GltA2 and GltAB are active in vivo (Krog et al. 2013a). This represented the first example of two different active GOGATs in a bacterium, and the biological impact of this for l-glutamate synthesis and degradation in B. methanolicus remains to be further investigated.
In C. glutamicum, synthesis and degradation of l-glutamate are controlled by GDH and the GS/GOGAT pathway, depending on ammonium availability. This organism has one GDH which displays much higher affinities for ammonium and 2-oxoglutarate than for l-glutamate, and it is considered to contribute to l-glutamate synthesis at high ammonium concentrations. Under such conditions, transcription of gltAB encoding GOGAT is repressed, while at low ammonium concentrations, GOGAT is important for l-glutamate synthesis (Beckers et al. 2001; Eggeling and Bott 2005; Tesch et al. 1999).
2-Oxoglutarate dehydrogenase plays a key role in regulating l-glutamate synthesis
2-Oxoglutarate is generated by isocitrate dehydrogenase and converted to succinyl-CoA by 2-oxoglutarate dehydrogenase (OGDH) (Brautaset et al. 2003) (Fig. 2). Controlling OGDH activity plays a key role in achieving l-glutamate overproduction in C. glutamicum (Kim et al. 2009; Niebisch et al. 2006; Schultz et al. 2007). OGDH is encoded by the odhAB operon and OGDH activity in MGA3 is low. When odhAB was overexpressed in a wild-type B. methanolicus MGA3 genetic background, l-glutamate secretion was eightfold reduced, indicating that odhAB represents a key target for control of l-glutamate production in B. methanolicus MGA3 (Krog et al. 2013a). Interestingly, proteome analyses showed that also the level of putative 2-oxoglutarate synthase (KorAB) level is higher on mannitol versus on methanol, suggesting an analogous biological role in the TCA cycle; however, the latter needs to be further investigated. Although the TCA cycle may play a minor role for energy generation (Brautaset et al. 2003), the finding that the B. methanolicus genome encodes a complete TCA cycle (Heggeset et al. 2012) and that all the enzymes are synthesized (Müller et al. 2014) implies that the TCA cycle is functional. As expected for growth on less-reduced C sources, several TCA proteins were found at elevated levels in cells growing on mannitol compared to methanol as sole C-source.
Biochemical characterization and manipulation of key enzymes of the aspartate pathway for overproduction of l-lysine
Aspartate kinase (AK) controls the carbon flow into the aspartate pathway (Fig. 2), which is branched into three main paths responsible for the generation of l-lysine and meso-diaminopimelate, of l-methionine and l-threonine, and of dipicolinate, respectively (Chen et al. 1993). meso-Diaminopimelate is a constituent of the bacterial cell wall peptidoglycan while dipicolinate is required for spore formation; thus, the common steps in the aspartate pathway need to be tightly controlled to allow for a balanced synthesis of the various end products (Chen et al. 1993). Oxaloacetate is aminated to l-aspartate by aspartate transaminase (AspB), and l-aspartate is then phosphorylated by one of three AK (DapG, LysC, YclM) generating 4-phospho-l-aspartate. Aspartate 4-phosphate is oxidized by aspartate-semialdehyde dehydrogenase (Asd) to aspartate semialdehyde, a branching metabolite and precursor of l-methionine, l-threonine, and l-isoleucine (Fig. 2). Aspartate semialdehyde is further converted to (2S,4S)-4-hydroxy-2,3,4,5-tetrahydrodipicolinate, a precursor of dipicolinic acid, by 4-hydroxy-tetrahydrodipicolinate synthase (DapA) followed by a reduction to 2,3,4,5-tetrahydrodipicolinate by 4-hydroxy-tetrahydrodipicolinate reductase (DapB). meso-2,6-Diaminopimelate (DAP) is then generated by tetrahydrodipicolinate N-acetyltransferase (DapH), acetyl-diaminopimelate aminotransferase (PatA), N-acetyldiaminopimelate deacetylase (DapL), and diaminopimelate epimerase (DapF). Finally, DAP is decarboxylated by DAP decarboxylase (LysA) to l-lysine (Fig. 2).
B. methanolicus possesses three AK isoenzymes; AKI is encoded by dapG, AKII by lysC, and AKIII by yclM (Jakobsen et al. 2009; Schendel and Flickinger 1992). The main role of AKI is linked to the biosynthesis of diaminopimelate for peptidoglycan synthesis where AKI is providing a constant level of 4-phospho-l-aspartate (Graves and Switzer 1990; Roten et al. 1991). AKII is regulating l-lysine biosynthesis, while the function of AKIII is primarily to regulate threonine biosynthesis (Graves and Switzer 1990). The AK of B. methanolicus MGA3 has similar V max values and K m values for aspartate (Table 4). AKI and AKII are allosterically inhibited by meso-diaminopimelate (IC50, 0.1 mM) and l-lysine (IC50, 0.3 mM), respectively, while AKIII is inhibited by l-threonine (IC50, 4 mM) and l-lysine (IC50, 5 mM) and synergistically inhibited by both amino acids at low concentrations (Jakobsen et al. 2009). In the classical mutant strain NOA2#13A52-8A66, a single base change C1125A in dapG results in a mutated gene product AKI-D375E, which is not feedback inhibited by meso-diaminopimelate in vitro (Nærdal et al. 2011). Overexpression of dapG, lysC, and yclM increased the l-lysine production in wild-type B. methanolicus MGA3 2-, 10-, and 60-fold (corresponding to 11 g/L) in fed-batch methanol fermentations, respectively, without negatively affecting the specific growth rate (Jakobsen et al. 2009). In shake-flask methanol cultivation, the overexpression of lysC and yclM increased the l-lysine production by 8- and 20-fold (up to 140 mg/L). In contrast, while dapG overexpression had no effect on l-lysine production, the recombinant strain MGA3 (pTH1mp-dapG_D375E) overexpressing AKI-D375E secreted 120 mg/L under these conditions, showing that all the three AKs can play important roles in l-lysine-overproducing B. methanolicus strains (Nærdal et al. 2011). The presence of three different AKs that are regulated in a distinct manner is presumably common among Bacilli, while C. glutamicum has only one AK enzyme that is feedback inhibited by lysine as well as by threonine (Eggeling and Bott 2005) and its importance for controlling l-lysine biosynthesis in this organism is well documented (see below).
Like in B. subtilis, synthesis of the AKs in B. methanolicus MGA3 is controlled by amino acids. qPCR studies have shown that dapG transcription is essentially unaffected by the presence of l-lysine, l-threonine, dl-methionine, or the combination of l-lysine and l-threonine in the growth medium (Nærdal et al. 2011). The transcription of lysC was fivefold repressed in the presence of l-lysine, slightly reduced in the presence of l-threonine, and twofold induced in the presence of dl-methionine. The yclM-transcription on the other hand was fivefold repressed in the presence of dl-methionine, while l-lysine and l-threonine either alone or together had a minor effect on the transcription level of this gene (Nærdal et al. 2011). In addition, a lysine riboswitch in the lysC leader region with two regions of dyad symmetry and a second region containing a series of T-residues after the hairpin loop, typical of a rho-independent terminator, has been found in B. methanolicus MGA3 (Schendel and Flickinger 1992). In analogy to B. subtilis, lysC may be regulated by premature transcriptional termination in the presence of high l-lysine concentrations, whereas the full-length transcript is produced due to transcriptional anti-termination with low l-lysine concentrations (Grundy et al. 2003; Phan and Schumann 2009). In C. glutamicum, expression of an allosterically deregulated AK has proven to be the major step for increased l-lysine production (Eggeling and Bott 2005).
AKI is expressed from the dap operon, which includes dpaA and dpaB (named spoVFA and spoVFB in B. subtilis) encoding the two subunits of dipicolinate synthase, asd, dapG, and dapA (Chen et al. 1993; Heggeset et al. 2012; Jakobsen et al. 2009). A translational attenuator as present in the B. subtilis asd-dapG mRNA was not found in the asd-dapG intergenic region in B. methanolicus MGA3 (Jakobsen et al. 2009). Like for dapG, no significant change was seen in the transcription level of asd and dapA in response to amino acids in the growth medium, similar to the results reported for B. subtilis (Belitsky 2002; Nærdal et al. 2011).
LysA is allosterically inhibited by l-lysine, with a K i of 0.93 mM and displays a K m for DAP of 0.8 mM (Mills and Flickinger 1993). Unlike in B. subtilis where lysA expression is repressed by l-lysine (Belitsky 2002), no significant change was seen in the lysA expression in B. methanolicus MGA3 in response to amino acids (Nærdal et al. 2011). Overexpression of lysA in a wild-type MGA3 background increased l-lysine production 20-fold, while overexpression of dapA or asd did not. In contrast, dapA overexpression caused increased l-lysine production in C. glutamicum (Eggeling and Bott 2005). Interestingly, B. methanolicus mutant NOA2#8A52-8A66 displayed threefold increased lysA transcription compared to the wild-type strain, which was likely due to a point mutation in the lysA promoter region and likely contributed to l-lysine overproduction of this strain (Nærdal et al. 2011). Both the LysC and LysA gene products are inhibited by l-lysine (Nærdal et al. 2011).
l-Methionine and l-threonine are synthesized from the common precursor l-homoserine, which is generated from l-aspartate semialdehyde by homoserine dehydrogenase. B. methanolicus MGA3 encodes two homoserine dehydrogenases, hom1 and hom2, while most Bacilli, including B. subtilis, encode only one (Brautaset et al. 2010). hom1 forms an operon with thrC and thrB, encoding threonine synthase and homoserine kinase, respectively, while hom2 is encoded elsewhere (Heggeset et al. 2012). Surprisingly, B. methanolicus PB1 has no hom2 gene, although the biological impact remains unknown. hom1 transcription was repressed 33-fold by addition of l-threonine to cells grown with methanol and 2-fold by l-methionine, while repression of hom2 was 2-fold by l-threonine and 14-fold by l-methionine (Brautaset et al. 2010). The classical mutants M168-20 and NOA2#A52-8A66 had mutations in the hom1-gene. Recombinant strains of M168-20 overexpressing unmutated hom1 displayed l-lysine production close to wild-type MGA3 levels, while overexpressing hom2 only slightly reduced l-lysine production, indicating that hom1 is the major homoserine dehydrogenase in B. methanolicus (Brautaset et al. 2010). C. glutamicum has one homoserine dehydrogenase gene, and mutations abolishing or reducing HD activity cause increased l-lysine production (Ohnishi et al. 2002). A series of recombinant B. methanolicus MGA3 strains were made overexpressing up to three different l-lysine biosynthetic enzymes resulting in additive (above 80-fold) l-lysine production levels (Nærdal et al. 2011). We previously made a stoichiometric comparison showing that the yield of l-lysine from methanol by B. methanolicus using the acetylase variant of the l-lysine biosynthesis pathway is similar (0.71 and 0.81 g l-lysine-HCl per gram methanol depending on whether there is NAD(P)H formation upon formaldehyde oxidation or not) to the yield from glucose by C. glutamicum using the succinylase or the dehydrogenase variants (0.68 and 0.82 g l-lysine-HCl per gram glucose, respectively) (Brautaset et al. 2007). This, together with a high methanol consumption rate, should make B. methanolicus a potentially efficient candidate for production of l-lysine from methanol.
Concluding remarks
Methanol represents an alternative and highly attractive non-food raw material for biotechnological processes. The thermophilic and methylotrophic bacterium B. methanolicus can grow rapidly on low-cost methanol medium at 50 °C and is a natural l-glutamate producer, and classical mutagenesis and metabolic engineering approaches have demonstrated a high capacity to overproduce l-lysine. Recent genome sequencing of two B. methanolicus isolates has contributed to complete insight into genes and pathways involved in methylotrophy and amino acid biosynthetic pathways. This, accompanied with transcriptome and proteome analyses, has unraveled several unique traits of this organism that make it an interesting model strain for basic studies aiming at understanding bacterial methylotrophy. Multiple key enzymes involved in methanol oxidation and assimilation pathways, as well as in l-lysine and l-glutamate biosynthesis pathways, have been biochemically characterized, and such knowledge has been used to construct amino acid overproducers by metabolic engineering. In parallel, new and better genetic tools are being established, and together, this knowledge and new technologies should serve as a valuable basis for future systems-level metabolic engineering of B. methanolicus for production of commodity chemicals from methanol.
References
Anastassiadis S (2007) L-lysine fermentation. Recent Pat Biotechnol 1:11–24
Anthony C (1982) The biochemistry of methylotrophs. Academic, London
Arfman N, Watling EM, Clement W, van Oosterwijk RJ, de Vries GE, Harder W, Attwood MM, Dijkhuizen L (1989) Methanol metabolism in thermotolerant methylotrophic Bacillus strains involving a novel catabolic NAD-dependent methanol dehydrogenase as a key enzyme. Arch Microbiol 152:280–288
Arfman N, Bystrykh L, Govorukhina NI, Dijkhuizen L (1990) 3-Hexulose-6-phosphate synthase from thermotolerant methylotroph Bacillus C1. Methods Enzymol 188:391–397
Arfman N, Van Beeumen J, De Vries GE, Harder W, Dijkhuizen L (1991) Purification and characterization of an activator protein for methanol dehydrogenase from thermotolerant Bacillus spp. J Biol Chem 266:3955–3960
Arfman N, de Vries KJ, Moezelaar HR, Attwood MM, Robinson GK, van Geel M, Dijkhuizen L (1992a) Environmental regulation of alcohol metabolism in thermotolerant methylotrophic Bacillus strains. Arch Microbiol 157:272–278
Arfman N, Dijkhuizen L, Kirchhof G, Ludwig W, Schleifer KH, Bulygina ES, Chumakov KM, Govorukhina NI, Trotsenko YA, White D, Sharp RJ (1992b) Bacillus methanolicus sp. nov., a new species of thermotolerant, methanol-utilizing, endospore-forming bacteria. Int J Syst Bacteriol 42:439–445
Arfman N, Hektor HJ, Bystrykh LV, Govorukhina NI, Dijkhuizen L, Frank J (1997) Properties of an NAD(H)-containing methanol dehydrogenase and its activator protein from Bacillus methanolicus. Eur J Biochem 244:426–433
Beckers G, Nolden L, Burkovski A (2001) Glutamate synthase of Corynebacterium glutamicum is not essential for glutamate synthesis and is regulated by the nitrogen status. Microbiology 147:2961–2970
Belitsky BR (ed) (2002) Bacillus subtilis and its closest relatives: from genes to cells. Biosynthesis of amino acids of the glutamate and aspartate families, alanine, and polyamines. American Society of Microbiology, Washington DC
Brautaset T, Ellingsen TE (2011) Food ingredients | lysine: industrial uses and production. In: Moo-Young M (ed) Comprehensive biotechnology, vol 3, 2nd edn. Elsevier, Amsterdam, The Netherlands, pp 541–554
Brautaset T, Williams MD, Dillingham RD, Kaufmann C, Bennaars A, Crabbe E, Flickinger MC (2003) Role of the Bacillus methanolicus citrate synthase II gene, citY, in regulating the secretion of glutamate in L-lysine-secreting mutants. Appl Environ Microbiol 69:3986–3995
Brautaset T, Jakobsen ØM, Flickinger MC, Valla S, Ellingsen TE (2004) Plasmid-dependent methylotrophy in thermotolerant Bacillus methanolicus. J Bacteriol 186:1229–1238
Brautaset T, Jakobsen ØM, Josefsen KD, Flickinger MC, Ellingsen TE (2007) Bacillus methanolicus: a candidate for industrial production of amino acids from methanol at 50 °C. Appl Microbiol Biotechnol 74:22–34. doi:10.1007/s00253-006-0757-z
Brautaset T, Jakobsen ØM, Degnes KF, Netzer R, Nærdal I, Krog A, Dillingham R, Flickinger MC, Ellingsen TE (2010) Bacillus methanolicus pyruvate carboxylase and homoserine dehydrogenase I and II and their roles for L-lysine production from methanol at 50 °C. Appl Microbiol Biotechnol 87:951–964. doi:10.1007/s00253-010-2559-6
Chen NY, Jiang SQ, Klein DA, Paulus H (1993) Organization and nucleotide sequence of the Bacillus subtilis diaminopimelate operon, a cluster of genes encoding the first three enzymes of diaminopimelate synthesis and dipicolinate synthase. J Biol Chem 268:9448–9465
Chistoserdova L (2011) Modularity of methylotrophy, revisited. Environ Microbiol 13:2603–2622. doi:10.1111/j.1462-2920.2011.02464.x
Chistoserdova L, Lidstrom ME (2013) Aerobic methylotrophic prokaryotes. In: Rosenberg E, DeLong EF, Thompson F, Lory S, Stackebrandt E (eds) The prokaryotes. Springer, New York, pp 267–285
Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME (2009) The expanding world of methylotrophic metabolism. Annu Rev Microbiol 63:477–499
Cue D, Lam H, Hanson RS, Flickinger MC (1996) Characterization of a restriction-modification system of the thermotolerant methylotroph Bacillus methanolicus. Appl Environ Microbiol 62:1107–1111
Cue D, Lam H, Dillingham RL, Hanson RS, Flickinger MC (1997) Genetic manipulation of Bacillus methanolicus, a gram-positive, thermotolerant methylotroph. Appl Environ Microbiol 63:1406–1420
Dijkhuizen L, Arfman N, Attwood MM, Brooke AG, Harder W, Watling EM (1988) Isolation and initial characterization of thermotolerant methylotrophic Bacillus strains. Fems Microbiol Lett 52:209–214
Eggeling L, Bott M (eds) (2005) Handbook of Corynebacterium glutamicum. CRC Press Boca Raton, USA
Fernstrom JD (2009) Symposium summary. The roles of glutamate in taste, gastrointestinal function, metabolism, and physiology. Am J Clin Nutr 90:881S–885S. doi:10.3945/ajcn.2009.27462DD
Graves LM, Switzer RL (1990) Aspartokinase III, a new isozyme in Bacillus subtilis 168. J Bacteriol 172:218–223
Grundy FJ, Lehman SC, Henkin TM (2003) The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc Natl Acad Sci U S A 100:12057–12062. doi:10.1073/pnas.2133705100
Gunka K, Newman JA, Commichau FM, Herzberg C, Rodrigues C, Hewitt L, Lewis RJ, Stülke J (2010) Functional dissection of a trigger enzyme: mutations of the Bacillus subtilis glutamate dehydrogenase RocG that affect differentially its catalytic activity and regulatory properties. J Mol Biol 400:815–827. doi:10.1016/j.jmb.2010.05.055
Haima P, Bron S, Venema G (1987) The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol Gen Genet 209:335–342
Heggeset TM, Krog A, Balzer S, Wentzel A, Ellingsen TE, Brautaset T (2012) Genome sequence of thermotolerant Bacillus methanolicus: features and regulation related to methylotrophy and production of L-lysine and L-glutamate from methanol. Appl Environ Microbiol 78:5170–5181. doi:10.1128/AEM. 00703-12
Hektor HJ, Kloosterman H, Dijkhuizen L (2002) Identification of a magnesium-dependent NAD(P)(H)-binding domain in the nicotinoprotein methanol dehydrogenase from Bacillus methanolicus. J Biol Chem 277:46966–46973. doi:10.1074/jbc.M207547200
Irla M, Neshat A, Winkler A, Albersmeier A, Heggeset TM, Brautaset T, Kalinowski J, Wendisch VF, Ruckert C (2014) Complete genome sequence of Bacillus methanolicus MGA3, a thermotolerant amino acid producing methylotroph. J Biotechnol 188C:110–111. doi:10.1016/j.jbiotec.2014.08.013
Jakobsen ØM, Benichou A, Flickinger MC, Valla S, Ellingsen TE, Brautaset T (2006) Upregulated transcription of plasmid and chromosomal ribulose monophosphate pathway genes is critical for methanol assimilation rate and methanol tolerance in the methylotrophic bacterium Bacillus methanolicus. J Bacteriol 188:3063–3072
Jakobsen ØM, Brautaset T, Degnes KF, Heggeset TM, Balzer S, Flickinger MC, Valla S, Ellingsen TE (2009) Overexpression of wild-type aspartokinase increases L-lysine production in the thermotolerant methylotrophic bacterium Bacillus methanolicus. Appl Environ Microbiol 75:652–661
Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H (2009) Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol 81:1097–1106. doi:10.1007/s00253-008-1743-4
Komives CF, Cheung LY, Pluschkell SB, Flickinger MC (2005) Growth of Bacillus methanolicus in seawater-based media. J Ind Microbiol Biotechnol 32:61–66
Kondo H, Kazuta Y, Saito A, Fuji K (1997) Cloning and nucleotide sequence of Bacillus stearothermophilus pyruvate carboxylase. Gene 191:47–50
Kovacs AT, van Hartskamp M, Kuipers OP, van Kranenburg R (2010) Genetic tool development for a new host for biotechnology, the thermotolerant bacterium Bacillus coagulans. Appl Environ Microbiol 76:4085–4088. doi:10.1128/AEM. 03060-09
Krog A, Heggeset TM, Ellingsen TE, Brautaset T (2013a) Functional characterization of key enzymes involved in L-glutamate synthesis and degradation in the thermotolerant and methylotrophic bacterium Bacillus methanolicus. Appl Environ Microbiol 79:5321–5328. doi:10.1128/AEM. 01382-13
Krog A, Heggeset TM, Muller JE, Kupper CE, Schneider O, Vorholt JA, Ellingsen TE, Brautaset T (2013b) Methylotrophic Bacillus methanolicus encodes two chromosomal and one plasmid born NAD+ dependent methanol dehydrogenase paralogs with different catalytic and biochemical properties. PLoS One 8:e59188. doi:10.1371/journal.pone.0059188
Markert B, Stolzenberger J, Brautaset T, Wendisch VF (2014) Characterization of two transketolases encoded on the chromosome and the plasmid pBM19 of the facultative ribulose monophosphate cycle methylotroph Bacillus methanolicus. BMC Microbiol 14:7. doi:10.1186/1471-2180-14-7
Mills DA, Flickinger MC (1993) Cloning and sequence analysis of the meso-diaminopimelate decarboxylase gene from Bacillus methanolicus MGA3 and comparison to other decarboxylase genes. Appl Environ Microbiol 59:2927–2937
Müller JE, Litsanov B, Bortfeld-Miller M, Trachsel C, Grossmann J, Brautaset T, Vorholt JA (2014) Proteomic analysis of the thermophilic methylotroph Bacillus methanolicus MGA3. Proteomics 14:725–737. doi:10.1002/pmic.201300515
Nærdal I, Netzer R, Ellingsen TE, Brautaset T (2011) Analysis and manipulation of aspartate pathway genes for L-lysine overproduction from methanol by Bacillus methanolicus. Appl Environ Microbiol 77:6020–6026. doi:10.1128/AEM. 05093-11
Nærdal I, Pfeifenschneider J, Brautaset T, Wendisch VF (2014) Methanol-based cadaverine production by genetically engineered Bacillus methanolicus strains. submitted
Nguyen HD, Nguyen QA, Ferreira RC, Ferreira LC, Tran LT, Schumann W (2005) Construction of plasmid-based expression vectors for Bacillus subtilis exhibiting full structural stability. Plasmid 54:241–248. doi:10.1016/j.plasmid.2005.05.001
Niebisch A, Kabus A, Schultz C, Weil B, Bott M (2006) Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem 281:12300–12307. doi:10.1074/jbc.M512515200
Nilasari D, Dover N, Rech S, Komives C (2012) Expression of recombinant green fluorescent protein in Bacillus methanolicus. Biotechnol Prog 28:662–668. doi:10.1002/btpr.1522
Ochsner AM, Muller JE, Mora CA, Vorholt JA (2014a) In vitro activation of NAD-dependent alcohol dehydrogenases by Nudix hydrolases is more widespread than assumed. FEBS Lett 588:2993–2999. doi:10.1016/j.febslet.2014.06.008
Ochsner AM, Sonntag F, Buchhaupt M, Schrader J, Vorholt JA (2014b) Methylobacterium extorquens: methylotrophy and biotechnological applications. Appl Microbiol Biotechnol (In Press)
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) A novel methodology employing Corynebacterium glutamicum genome information to generate a new L-lysine-producing mutant. Appl Microbiol Biotechnol 58:217–223
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3:295–300
Phan TT, Schumann W (2009) Transcriptional analysis of the lysine-responsive and riboswitch-regulated lysC gene of Bacillus subtilis. Curr Microbiol 59:463–468. doi:10.1007/s00284-009-9461-4
Pluschkell SB, Flickinger MC (2002) Dissimilation of [13C]methanol by continuous cultures of Bacillus methanolicus MGA3 at 50 °C studied by 13C NMR and isotope-ratio mass spectrometry. Microbiology 148:3223–3233
Radmacher E, Eggeling L (2007) The three tricarboxylate synthase activities of Corynebacterium glutamicum and increase of L-lysine synthesis. Appl Microbiol Biotechnol 76:587–595. doi:10.1007/s00253-007-1105-7
Roten CA, Brandt C, Karamata D (1991) Genes involved in meso-diaminopimelate synthesis in Bacillus subtilis: identification of the gene encoding aspartokinase I. J Gen Microbiol 137:951–962
Sauer U, Eikmanns BJ (2005) The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol Rev 29:765–794. doi:10.1016/j.femsre.2004.11.002
Schendel FJ, Flickinger MC (1992) Cloning and nucleotide sequence of the gene coding for aspartokinase II from a thermophilic methylotrophic Bacillus sp. Appl Environ Microbiol 58:2806–2814
Schendel FJ, Bremmon CE, Flickinger MC, Guettler M, Hanson RS (1990) L-lysine production at 50 °C by mutants of a newly isolated and characterized methylotrophic Bacillus sp. Appl Environ Microbiol 56:963–970
Schrader J, Schilling M, Holtmann D, Sell D, Filho MV, Marx A, Vorholt JA (2009) Methanol-based industrial biotechnology: current status and future perspectives of methylotrophic bacteria. Trends Biotechnol 27:107–115. doi:10.1016/j.tibtech.2008.10.009
Schultz C, Niebisch A, Gebel L, Bott M (2007) Glutamate production by Corynebacterium glutamicum: dependence on the oxoglutarate dehydrogenase inhibitor protein OdhI and protein kinase PknG. Appl Microbiol Biotechnol 76:691–700. doi:10.1007/s00253-007-0933-9
Stolzenberger J, Lindner SN, Persicke M, Brautaset T, Wendisch VF (2013a) Characterization of fructose 1,6-bisphosphatase and sedoheptulose 1,7-bisphosphatase from the facultative ribulose monophosphate cycle methylotroph Bacillus methanolicus. J Bacteriol 195:5112–5122. doi:10.1128/JB.00672-13
Stolzenberger J, Lindner SN, Wendisch VF (2013b) The methylotrophic Bacillus methanolicus MGA3 possesses two distinct fructose 1,6-bisphosphate aldolases. Microbiology 159:1770–1781. doi:10.1099/mic. 0.067314-0
Tannenbaum SR, Wang DIC (1975) Single-cell protein. MIT Press, Cambridge
Tesch M, de Graaf AA, Sahm H (1999) In vivo fluxes in the ammonium-assimilatory pathways in Corynebacterium glutamicum studied by 15 N nuclear magnetic resonance. Appl Environ Microbiol 65:1099–1109
Vonck J, Arfman N, De Vries GE, Van Beeumen J, Van Bruggen EF, Dijkhuizen L (1991) Electron microscopic analysis and biochemical characterization of a novel methanol dehydrogenase from the thermotolerant Bacillus sp. C1. J Biol Chem 266:3949–3954
Vorholt JA (2002) Cofactor-dependent pathways of formaldehyde oxidation in methylotrophic bacteria. Arch Microbiol 178:239–249
Wendisch VF (2014) Microbial production of amino acids and derived chemicals: synthetic biology approaches to strain development. Curr Opin Biotechnol 30C:51–58. doi:10.1016/j.copbio.2014.05.004
Yao W, Deng X, Zhong H, Liu M, Zheng P, Sun Z, Zhang Y (2009) Double deletion of dtsR1 and pyc induce efficient L: -glutamate overproduction in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 36:911–921. doi:10.1007/s10295-009-0569-0
Yasueda H, Kawahara Y, Sugimoto S (1999) Bacillus subtilis yckG and yckF encode two key enzymes of the ribulose monophosphate pathway used by methylotrophs, and yckH is required for their expression. J Bacteriol 181:7154–7160
Yurimoto H, Hirai R, Matsuno N, Yasueda H, Kato N, Sakai Y (2005) HxlR, a member of the DUF24 protein family, is a DNA-binding protein that acts as a positive regulator of the formaldehyde-inducible hxlAB operon in Bacillus subtilis. Mol Microbiol 57:511–519. doi:10.1111/j.1365-2958.2005.04702.x
Acknowledgments
Work related to methylotrophy in the authors’ laboratories is supported by FP7 project Promyse and the Swiss SystemsX.ch, Norwegian RCN and the German initiatives within the framework of the ERA-Net ERASysAPP.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jonas E. N. Müller and Tonje M. B. Heggeset contributed equally to this work.
Rights and permissions
About this article

Cite this article
Müller, J.E.N., Heggeset, T.M.B., Wendisch, V.F. et al. Methylotrophy in the thermophilic Bacillus methanolicus, basic insights and application for commodity production from methanol. Appl Microbiol Biotechnol 99, 535–551 (2015). https://doi.org/10.1007/s00253-014-6224-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6224-3