
Abstract
Detection of pork meat adulteration in “halal” meat products is a crucial issue in the fields of modern food inspection according to implementation of very strict procedures for halal food labelling. Present study aims at detecting and quantifying pork adulteration in both raw and cooked manufactured sausages. This is by applying an optimized species-specific PCR procedure followed by QIAxcel capillary electrophoresis system. Manufacturing experiment was designed by incorporating pork with beef meat at 0.01 to 10 % substitution levels beside beef and pork sausages as negative and positive controls, respectively. Subsequently, sausages were divided into raw and cooked sausages then subjected to DNA extraction. Results indicated that PCR amplifications of mitochondrial D-loop and cytochrome b (cytb) genes by porcine-specific primers produced 185 and 117 bp pork-specific DNA fragments in sausages, respectively. No DNA fragments were detected when PCR was applied on beef sausage DNA confirming primers specificity. For internal control, a 141-bp DNA fragment of eukaryotic 18S ribosomal RNA (rRNA) gene was amplified from pork and beef DNA templates. Although PCR followed by either QIAxcel or agarose techniques were efficient for targeted DNA fragments differentiation even as low as 0.01 % (pork/meat: w/w). For proficiency, adequacy, and performance, PCR-QIA procedure is highly sensitive, a time-saver, electronically documented, mutagenic-reagent free, of little manual errors, accurate in measuring PCR fragments length, and quantitative data supplier. In conclusion, it can be suggested that optimized PCR-QAI is considered as a rapid and sensitive method for routine pork detection and quantification in raw or processed meat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species authentication, food safety, and food control are a growing concern in today’s marketplace worldwide. As minced meat is added in most of the processed meats (Tanabe et al. 2007a; Tanabe et al. 2007b), verification of food labelling should ensure food safety (i.e., unexpected occurrence of food allergies), gain consumer trust, and promote fair trades in local and international markets. Identification of meat source from different species of animals is considered important because of social, forensic, and public health reasons (Karabasanavar et al. 2014). Since almost one third of the human population do not eat pork meat, including Muslims and Jews, religious concerns are becoming facts. Authentically, this religious population knew the pork-free food as “halal” food. Religious belief is also another main factor that raises consumer concerns. Several religions impose some food restrictions. For instance, pork in addition to not ritually slaughtered meat is prohibited in Islam. The higher valued halal meat, such as beef and lamb, is easily adulterated by pork due to its similarity in color and texture. Thus, it is not only affecting food sanctity but also a fraud against the customer rights, religions, and beliefs (Bonne and Verbeke 2008). Pork is a potential source for adulteration of higher value meat such as beef and lamb due to its similarity in color and texture (Wissiack et al. 2003). Furthermore, mechanically recovered meats that are increasingly used in food industry are prone to various forms of pork adulteration (Skarpeid et al. 2001). Detection of pork in meat and meat products requires simple, specific, sensitive, and reliable analytical and authentication techniques (Ali et al. 2012; Che Man et al. 2012; Karabasanavar et al. 2014).
In some countries, pork DNA is detected in a number of halal meat products supplied to supermarkets despite being labeled as halal-certified foods (Calvo et al. 2002; Di Pinto et al. 2005; Karabasanavar et al. 2014; Montiel-Sosa et al. 2000; Tanabe et al. 2007a; Yusop et al. 2012). Veal is also substituted by pork due to its physical appearance (Toorop et al. 1997). Therefore, identification of pork adulteration in processed meat has become a necessity. Conventional methods of routine examination are not always able to detect species of meat present in processed, cooked, and adulterated mixtures. Hence, different analytical methods based on anatomical, histological, microscopic, organoleptic, chemical, electrophoretic, chromatographic, and immunological characteristics have been developed to differentiate meats. However, limitations of these techniques have led to apply the DNA-based molecular techniques for the purpose because of their sensitivity, repeatability, and reproducibility compared with other protein-based methods. Moreover, DNA is a relatively stable molecule allowing analysis of processed and heat-treated food products. Also, protein-based assays cannot differentiate closely related species due to cross-reactivity (Karabasanavar et al. 2014). Several DNA-based assays namely species-specific PCR (Karabasanavar et al. 2011; Kumar et al. 2011), restriction fragment length polymorphism (RFLP) (Ali et al. 2012; Girish et al. 2005), random amplified polymorphic DNA (RAPD) fingerprinting (Calvo et al. 2001), DNA hybridization (Chikuni et al. 1990), single-strand conformation polymorphism (PCR-SSCP) (Rehbein et al. 1997), mitochondrial D-loop based PCR (Che Man et al. 2012; Karabasanavar et al. 2014), and PCR product sequencing (Bartlett and Davidson 1992) have been employed for detection of meat authenticity. Species-specific PCR has the advantage over other DNA-based methods in terms of rapidity and specificity keeping in mind the need for development of a rapid and robust technique for the pork authentication.
Conventionally, gel electrophoresis has been used to separate PCR DNA fragments. However, this method is laborious, time-consuming, and hazardous due to the use of ethidium bromide or similar dyes that are mutagenic and dangerous for human being. In addition, gel data cannot be used directly for electronic documentation or data archiving (Armand et al. 2004; Marois et al. 2001). As an alternative method detecting pork adulteration in processed meat, the present study applied the QIAxcel capillary electrophoresis system, a computer-controlled system that provides electronic documentation which was innovatively used in different aspects (Matsumoto et al. 2013; McMurray et al. 2010; Melake et al. 2012; Mercimek-Mahmutoglu et al. 2012; Zhang et al. 2013). Using the QIAxcel system, at least 24 samples were analyzed in approximately 30 min. Detection using agarose gel electrophoresis, which involves more steps for handling and documentation, requires at least three times as long. Fragments shorter than 50 bp were detected with the QIAxcel system but might be not visible with agarose gels which greatly reduce the practical value of the agarose gel. Also, QIAxcel analysis system is more accurate in measuring the PCR fragments’ length. Identification of meat animal species as a source in meat products is an important subject in the field of modern food control and global market concerns (Di Pinto et al. 2005).
Therefore, the main objective of the present work is to optimize a species-specific PCR analysis followed by QIAxcel system targeting mitochondrial D-loop and cytb genes for identification of pork in raw and cooked sausages as processed meat product module. Also, 18S ribosomal RNA (rRNA) gene as an endogenous control was used. For inspection of the adulteration and test the procedure efficiency, manufacturing experiment of sausage incorporating pork at different substitution levels was designed. Conventional method and QIAxcel analysis were applied, compared, and then QIA-PCR was optimized.
Materials and methods
Meat samples
A fresh raw pork meat sample (2 kg) was purchased from especial slaughtering house in Qalama, Qaliuobia Governorate, while the fresh beef sample (5 kg) and mutton fat (3 kg) were purchased from a local supermarket “El-abed”, Qaliuobia Governorate, Egypt. In addition, the whole sausage ingredients were obtained from a local spices supermarket “Khedr El-Atar” in Cairo, Egypt. The meat and mutton fat samples were kept under frozen condition at −18 ± 1 °C until use to prevent enzymatic degradation of DNA in meat samples.
Preparation of beef and pork meat mixtures
Beef and pork meats were manually defatted and grounded separately by meat grinder (SIEMENS, type CNCM11ST, Germany). Subsequently, pork meat was taken and mixed with beef meat to be impregnated in sausage as 0, 0.01, 0.1, 0.5, 1, 2, 5, and 10 % pork in beef separately to avoid contamination. Pure beef for negative control (−C) and pure pork for positive control (+C) sausages were also prepared (Table 1). The grounded and mixed meat samples were packed in polyethylene bags and then kept under frozen conditions at −18 ± 1 °C until starting of sausage manufacturing.
Sausage manufacturing
The different raw sausage formulas were immediately prepared according to Table 1 as previously described by (Moghazy and El-Shaarawy 2001; Moghazy et al. 2004). Each prepared meat mixture was mixed with mutton fat tissues very well. Soybean flour was rehydrated by distilled water as 1:2 (w/v), then the other ingredients were added gradually to produce different adulterated beef sausage incorporated by pork meat. Finally, the ice flakes were added to the final mixture, and then the whole mixture was filed up into mutton natural sausage capillary intestine which prepared in the lab. Subsequently, each sausage batch was divided into two groups; one of them was kept as fresh, and the second was cooked in steam pot by wet live steam at 100 °C for 10 min then cooled down. Both sausage groups were stored under frozen conditions or subjected immediately to the DNA extraction.
DNA extraction
Total DNA extraction using 200 mg of each well grounded meat and sausage samples were performed using DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany) as per the instructions given by the manufacturer. The DNA concentration was estimated by spectrophotometric analysis using Thermo Scientific NanoDrop 1000 Spectrophotometer (Thermo Scientific, USA) at 260 nm/280 nm after appropriate dilution, and the DNA integrity was visualized by ethidium bromide staining of DNA on 1 % agarose gel. The extracted DNA solutions were stored at −20 °C for further applications.
Primers
A pair of porcine-specific primers targeting a 185 and 117 bp fragments of swine D-loop and cytochrome b (cytb) genes, and for internal control, a 141-bp conserved fragment of eukaryotic 18S rRNA gene were amplified from both species (beef and pork) using the primers described by (Ali et al. 2012; Che Man et al. 2012). The specificity of those primers was checked by alignments with the original GenBank sequences using the standard nucleotide-nucleotide BLAST (blastn; provided online by National Center for Biotechnology Information (NCBI)). The nucleotide sequences of used primers are given in Table 2. The designed primers were ordered from Invitrogen™, Germany.
PCR amplification and QIAxcel procedures
Specific fragments (185, 117, and 141 bp) of D-loop, cytb, and eukaryotic 18S rRNA genes were selectively amplified in 25 μl reaction mixtures composed of 1× PCR reaction buffer, 2 mM MgCl2, 2.5 units Taq DNA polymerase, 400 μM of dNTP mix, 0.4 μM of each primer, and 1 μg DNA from each extracted sample. The amplification conditions on a Mastercycler (Eppendorf) for D-loop gene were as follows: initial denaturation step at 93 °C for 3 min, 35 cycles of amplification (30 s of denaturation at 93 °C, 30 s of annealing at 58.8 °C, 45 s of extension at 72 °C), and followed by final extension at 72 °C for 5 min. For cytb and eukaryotic 18S rRNA genes, the cycling conditions were preheating at 95 °C for 10 min, 35 cycles of amplification consisting of (20 s of denaturation at 95 °C, 30 s of annealing at 61 °C, and 20 s of extension at 72 °C), and 5 min of final extension at 72 °C. Negative template control of PCR reaction (PCR reaction mixture without template DNA and replaced with double sterilized deionized water) was carried out to ensure the purity of the PCR reaction mixture from contaminating DNA. Amplified products were analyzed by electrophoresis in 2 % agarose gel, then stained with ethidium bromide solution, and visualized under ultraviolet light (Highest Ultraviolet Intensity Spectroline (model TVC-312A) Variable Intensity Trans-Illuminator 312 nm Ultraviolet, USA). On the other hand, DNA analysis was performed on the QIAxcel system (Version: 9001421, QIAGEN, Germany) using the QIAxcel DNA high resolution kit (QIAGEN, Cat. No. 929002) as the method described in QIAxcel® DNA Handbook (OM400). The QX Alignment Marker 15 bp/1 kb was included in the analysis. Typically, 10 μl of the PCR products was added, and the instrument aspired 0.1 μl in each capillary tube applied with 0.1 μl of alignment marker into each of the 12 sample wells of QIAxcel capillary electrophoresis system. One microliter of DNA ladder 50 bp/800 bp was injected into the ladder well once at the first time of cartilage usage (1200 samples), while the alignment marker is injected with each sample. The samples were gently mixed for 1 min at 2500 rpm and were immediately run on the QIAxcel capillary electrophoresis system. The separation was achieved in 30 min by the application of high voltage (6 kV) in the sieving using the supplemented polymer and specialized buffer in the microfluidic channels through independent electrodes for each well. The results were displayed as gel image and electropherogram as obtained from QIAxcel advanced system software. Quantification analysis has been integrated using the QIAxcel software.
Results
Although QIAxcel analysis estimates the PCR fragments qualitatively and quantitatively (Graf et al. 2011), the conventional agarose method was compared with QIAxcel procedure. However, once pork adulteration was detected, it does not matter the quantity itself in the examined products as pork is avoided according to halal meat authorization (Bonne and Verbeke 2008; Che Man et al. 2012). But quantification analysis may expect the adulteration percentage in processed meat products.
Pork detection in raw and processed sausages
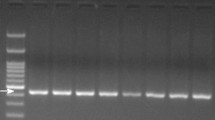
As the primary aim in the present study, detection of adulterated pork in processed meat products by species-specific PCR procedure is concerned. To achieve this, a lab sausage-manufacturing experiment has been carried out. The manufactured sausage was impregnated with pork in substitution levels ranged from 0.01 to 10 % pork in sausage beside pure beef meat and pure pork meat sausages as negative and positive control, respectively. Subsequently, sausages were subdivided similarly into two groups; one was cooked, whereas the second was handled as raw. Commonly, sausage is most distributed not only as raw in cold or frozen status but also as steam cooked or grilled. Cooking has been done to test the stability of the template DNA and to check whether if it affects pork meat detection in processed products. DNA was isolated from each individual sausage sample in both raw and cooked groups, quantified, and the purity was estimated then subjected to PCR amplification. The designed primer sets, D-loop, cytb, and eukaryotic 18S rRNA genes (Table 2) were tested with extracted DNA. Subsequently, the target DNA was analyzed using conventional agarose method. The gel image of the PCR products, obtained from pork–beef raw sausages, was displayed in Fig. 1. It was also necessary to test the performance of the PCR assay in raw and cooked sausages to distinguish pork contamination. Results found that cooking treatment and commercial meat additives such as spices, starch, fats, and curing agents from extraneous sources do not interfere the PCR amplifications (data not shown). In this case, no obvious difference has been found between the raw and cooked sausage samples which demonstrated similar results, and no effect of cooking conditions on the DNA has been found (raw sausage gel images were exemplary shown in Fig. 1). Our amplified products with ∼185, ∼117, and ∼141 bp are confirming the sensible stability of smaller size DNA templates. Likewise, our conventional PCR detection was not affected may be to confirm that the short heat treatments are not influencing the template DNA, but to exude successful detections (Fig. 1a–c). Obtained results reflected the sensitivity, specificity, stability, and reliability of the PCR assay in the screening of pork in processed meat products.

Specificity test of the primers. The conventional gel images shows PCR products amplified by a D-loop-specific gene (185 bp), b swine (cytb) gene (117 bp), and c eukaryotic 18Sr RNA (141 bp). In the gel image, L DNA ladder, lane +C pork DNA and positive control, lane −C negative control, lanes 3–7 PCR products from raw sausage samples mixed with 0.01, 0.1, 0.5, 2, and 10 % pork, respectively
Subsequently, clear PCR product targeting D-loop gene was obtained from raw adulterated sausage with different pork levels produced ∼185-bp fragment (Fig. 1a, lanes 3, 4, 5, 6, and 7), while the analysis of similar types of products of negative control did not yield any PCR products (lane 2). Similarly, PCR amplification resulted to a ∼117-bp fragment when swine gene was targeted in raw adulterated sausage samples (Fig. 1b, lanes 3, 4, 5, 6, and 7), while nothing is detectable in the negative control samples (lane 2). Obviously, the assay was sensitive enough to detect as low as 0.01 % of contaminated sausage with pork in raw and cooked sausages (Fig. 1a, b). Furthermore, since the endogenous primers annealed with the eukaryotic template and the amount of eukaryotic DNA did not changed with the deliberate variation of pork adulteration, homogeneous amplification of endogenous control (∼141 bp) was observed from all specimens (Fig. 1c, lanes 1–7). Generally, the gel images (Fig. 1a–c) show the PCR products as well as the primers’ sensitivity of D-loop gene, cytb, and eukaryotic 18S rRNA genes to produce PCR fragments with ∼185, ∼117, and ∼141 bp, respectively.
Pork detection in raw and processed sausages applying PCR-QIAxcel procedure
Recently, PCR has become an essential and daily performed experimental technique in food analysis, bioanalytical, clinical, and research laboratories, but still not accepted yet as a definitive analytical method in routine tests (Yang et al. 2005). In current study, PCR-QIA procedure was optimized to detect pork meat in processed meat products based on D-loop, cytb, and 18S rRNA genes, data were illustrated in Figs. 2, 3, and 4. The amplification by targeting the D-loop gene exudes a fragment of 185 bp in all tested sausage samples analyzed using QIAxcel system (Fig. 2). No difference has been found between raw and cooked sausage samples. Interestingly, the applied PCR was efficient to detect as low as 0.01 % pork in both raw and cooked pork–beef sausages. However, no PCR fragment has been detected in the negative control as confirmed by the specificity of used primer for targeting only the mitochondrial D-loop gene from pork meat as mentioned previously. QIAxcel-computerized system provides also a fragment band signal for any detectable band which shows the relative fluorescent units response to band intensity as shown in Fig. 2a, b. On the other hand, the amplification by targeting the cytb gene exudes a fragment of 117 bp in all tested sausage samples which were analyzed by QIAxcel system as shown in Fig. 3. Interestingly, the applied PCR was efficient to detect as low as 0.01 % pork in both raw and cooked pork–beef sausages. Besides that, no PCR fragment has been found in the negative control which contains no pork meat. A signal appeared in parallel as a response of band intensity which shows the relative fluorescent units of raw and cooked samples which provided by QIAxcel system as shown in Fig. 3a, b.

Specificity test of the primers to D-loop gene. The electropherograms of D-loop-specific gene (185 bp) PCR products for raw and cooked sausages are shown. In the image, L DNA ladder, lane 1 pork DNA positive control, lane 2 negative control, and lanes 3–7 PCR products from raw and cooked sausage samples mixed with 0.01, 0.1, 0.5, 2, and 10 % pork, respectively. (See also relative singles below)

Specificity test of the primers to cytb gene. The electropherograms of cytb-specific gene (117 bp) PCR products for raw and cooked sausages are shown. In the image, L DNA ladder, lane 1 pork DNA positive control, lane 2 negative control, and lanes 3–7 PCR products from raw and cooked sausage samples mixed with 0.01, 0.1, 0.5, 2, and 10 % pork, respectively. (See also relative singles below)

Specificity test of the primers to 18S rRNA gene. The electropherograms of endogenous control eukaryotic 18S rRNA gene (141 bp) PCR products for raw and cooked sausages are shown. In the image, L DNA ladder, lane 1 pork DNA positive control, lane 2 beef DNA negative control, lanes 3–7 PCR products from raw and cooked sausage samples mixed with 0.01, 0.1, 0.5, 2, and 10 % pork, respectively. (See also relative singles below)
As a positive control, a 141-bp fragment of eukaryotic 18S rRNA gene was amplified from all species, demonstrating the presence of good quality DNA templates in all specimens. Obviously, the alignment analysis of the endogenous primers demonstrated 100 % matching with animal 18S rRNA gene of the animal species. As expected, the amount of eukaryotic DNA did not changed with the deliberate variation of pork adulteration and homogeneous amplification of endogenous control was recorded in all specimens. This result could reflect the primer annealing potential level that significantly shows the efficiency in PCR amplification. Our results indicated that optimized PCR followed by QIAxcel analysis was rapid, efficient, and practical and provides automatic documentation and quantification data as compared to analysis of PCR products by conventional agarose gel.
The quantitative data (ng μl−1) of all PCR products, as integrated by QIAxcel software, is displayed in Table 3. The obtained PCR products of D-loop and cytb from 0.01 to 10 % pork adulteration level in both raw and processed sausages were detectable as shown in electropherogram (Figs. 2a, b and 3a, b), and quantification data are given in (Table 3). The mean of D-loop and cytb PCR products from 0.01 to 10 % increased exponentially with gradual increase of pork substitution level. The quantity of detected pork fragment DNA based on D-loop gene was higher than detected pork fragment DNA based on cytb gene (Table 3). The likelihood of detecting pork adulteration—as low as 0.1 to 10 % in beef sausage—reached 83 %, since a little variation was shown in the PCR products. Thus, 0.1 to 10 % of pork adulteration in a background of beef sausage was robustly detected as little variation was found in the amount of PCR products. A plot of the PCR products against the adulterated beef sausage with pork (%) showed exponential fit for D-loop (185 bp) and cytb (117 bp) PCR products with R 2 = 0.97, Fig. 5. Successful annealing of endogenous primers 18S rRNA with the eukaryotic template exudes homogeneous amplification in all specimens. The DNA averaged 1.03 ng μl−1 in all pork adulteration levels of both raw and processed sausages. However, a liner plot of eukaryotic 18S rRNA gene PCR products against the adulterated beef sausages with pork (%) was strongly supporting the theoretical expectation (Fig. 5).

Quantification profiles of D-loop (185 bp) as indicated in black squares, cytb (117 bp) in black triangles, and endogenous control eukaryotic 18S rRNA (141 bp) in black circles. PCR products were performed under the same conditions (n = 6)
Discussion
Rapid urbanization and industrialization have led to promote ready-to-eat food products including meat and their products. However, many authors proved that the PCR assays are sufficient to trace out pork in mixed and commercial meat products under various processing conditions using species-specific genes (Ali et al. 2012; Che Man et al. 2012; Karabasanavar et al. 2014; Kesmen et al. 2007; Mane et al. 2013;; Tanabe et al. 2007a; Tanabe et al. 2007b; Yusop et al. 2012). Obtained results clearly indicated that cooking treatment of sausage do not interfere the PCR amplification of swine D-loop, cytb-specific, or eukoryotic18S rRNA genes. As mentioned before, the fragment size of current genes are 185, 117, and 141 bp, respectively, which are seemed to be small in size templates. Our results were in agreement with Hird et al. (2006), who observed a little or no effect of autoclaving or other cooking methods on 81 and 116 bp templates of turkey’s cytb gene. On the contrary, Ali et al. (2011) showed that a 411-bp fragment of swine 12S rRNA gene did not amplify when the pork was extensively autoclaved for 2.5 h. These studies clearly demonstrated that the smaller size templates are more stable than the longer ones. However, the same authors observed a tremendous jump of Cross point (CT) values from less than 20 to more than 30 in autoclaved and canned turkey’s meat by changing the template size from 351 to 565 bp. As a higher CT value indicates lower PCR efficiency (Rojas et al. 2010; Yusop et al. 2012), their studies clearly reflect the higher rate of longer template degradation than the shorter ones by processing treatments.
Interestingly, the optimized PCR was able to detect the pork in processed meat products as exemplarily shown in current study. Really, as low as 0.01 % pork in beef meat was detected by optimized PCR using both D-loop and cytb gene primers followed by conventional agarose gel procedure. Our results are in agreement with Ali et al. (2012) who detected a low pork contamination at levels of 0.1 and 0.01 % by targeting swine gene produce PCR products (109 bp) and found an equal level of amplification of endogenous control (141 bp). Moreover, Che Man et al. (2012) confirmed that porcine-specific primer designed based on a porcine-specific sequence of mitochondrial D-loop gene (174 bp) was used to detect pork in processed meat products. The assay was able to detect as low as 0.1 % (v/v) porcine DNA spiked on DNA of cattle, sheep, goat, chicken, and deer. Duplex polymerase chain reaction for detection of pork meat in horse meat fresh sausages from Italian retail sources had been established (Di Pinto et al. 2005). Specific primers and TaqMan probes were designed based on the mitochondrial ND2, ND5, and ATP 6–8 genes for donkey, pork, and horse, respectively. A convenient, sensitive, and specific RT-PCR assay was optimized for the species identification and their quantification in raw and cooked meat products (Kesmen et al. 2009). The mitochondrial D-loop gene was used to detect pork adulteration (up to 0.1 %) in raw and cooked meat samples along with acceptable sensitivity of 10 pg (Che Man et al. 2012; Karabasanavar et al. 2014; Mane et al. 2013).
Regarding DNA separation and visualization using conventional agarose procedure, several additional factors could affect the mobility of DNA fragments in agarose gels as mentioned before (Olive et al. 1992). Among these factors are (1) gel concentration, (2) voltage used, (3) electrophoresis buffer, and (4) effects of ethidium bromide. Those mentioned factors affect the results of PCR which may cause varied results that different laboratories demonstrated. Therefore, sustainable procedure is needed to obtain PCR results with ignorable factors.
The QIAxcel capillary electrophoresis system would be the perfect facility to analyze the PCR products in a systematic way which provides automatic documentation, rapid, sensitive, and reproducible results as well as quantification data. In addition, applying PCR-QAI procedure could provide an option to quantify the detectable DNA as optimized in present study. It was efficient to trace out as low as 0.01 % pork in detectable range of both applied species-specific genes. This study confirmed that detection of pork adulteration starting by 0.1 % pork was perfectly likely. However, detection of 0.01 % pork was only 83 % likely. The relative DNA quantification in PCR products by comparing the obtained fragment bands has been previously done by Barakat et al. (2010). It was a relative method to follow some pathogens during malting process where accuracy of quantification was not highlighted. Quantification facility presented in current study could be helpful to expect the pork adulteration levels.
Similarly, Ali et al. (2012) used PCR assay to trace out pork in mixed and commercial meat products using species-specific primers for endogenous control and cytb genes followed by a simple restriction fragment length polymorphism analysis, RFLP. The substitution of gel electrophoresis by automated and sensitive chip-based CE was practical enough to be sensitive to detect 0.0001 ng of swine DNA and 0.01 % pork in ternary mixture of pork, beef, and flour. In addition, Graf et al. (2011) differentiated between 14 different exotic species and compared between analysis results from the QIAxcel system and from agarose gel electrophoresis. Based on the results from both Graf et al. (2011) studies and ours, the assay procedure has a good applicability to be used by quality control labs, and at the same time, there is no need to use hazardous chemicals in tracing. The QIAxcel analysis enables significantly shorter running time, eliminates sample preparation and exposure to mutagenic reagents, and requires fewer analysis and handling steps, saving time for more demanding laboratory work and reducing manual errors as more or less mentioned by Xiao et al. (2012). Moreover, PCR-QAI procedure provides qualitative and quantitative data which could be applicable for archiving and handling. Finally, concluded results of combining PCR with QIAxcel capillary electrophoresis system named as “PCR-QAI” was optimized to be used instead of using PCR followed by conventional agarose gel electrophoresis in routine analysis of pork detection and quantification in processed halal meat products.
In conclusion, a reliable, accurate, sensitive, and easily performable PCR-QAI assay was successfully optimized for the routine analysis of pork adulteration in processed meat. The assay utilized species-specific primers for D-loop, cytb, and eukaryotic 18S rRNA genes. The performance of PCR-QAI was tested through manufacturing experiment model simulating meat products, e.g., raw and cooked beef adulterated sausages with pork. Interestingly, the optimized PCR protocol was sufficient and sensitive enough to detect 0.01 % pork in meat. In addition, it was less time-consuming, providing electronic documentation, eliminating exposure to mutagenic reagents, reducing manual errors, accurate in measuring PCR fragments length, and providing quantification data. Therefore, it can be suggested that optimized PCR-QAI is consider a rapid and sensitive method for routine pork meat detection and quantification in raw or processed meat products. Applying this technique by official and quality control laboratories for halal authentication upon religious reasons is highly recommended.
References
Ali ME, Hashim U, Mustafa S, Che Man YB (2012) Swine-specific PCR-RFLP assay targeting mitochondrial cytochrome B gene for semiquantitative detection of pork in commercial meat products. Food Anal Methods 5:613–623
Ali ME, Hashim U, Mustafa S, Che Man YB, Yusop MHM, Kashif M, Dhahi TS, Bari MF, Hakim MA, Latif MA (2011) Nanobiosensor for detection and quantification of DNA sequences in degraded mixed meats. J Nanomater 2011:1–11
Armand R, Channon JY, Kintner J, White KA, Miselis KA, Perez RP, Lewis LD (2004) The effects of ethidium bromide induced loss of mitochondrial DNA on mitochondrial phenotype and ultrastructure in a human leukemia T-cell line (MOLT-4 cells). Toxicol Appl Pharmacol 196:68–79
Barakat H, Spielvogel A, Hassan M, El-Desouky A, El-Mansy H, Rath F, Meyer V, Stahl U (2010) The antifungal protein AFP from Aspergillus giganteus prevents secondary growth of different Fusarium species on barley. Appl Microbiol Biotechnol 87:617–624
Bartlett SE, Davidson WS (1992) FINS (forensically informative nucleotide sequencing): a procedure for identifying the animal origin of biological specimens. Biotechniques 12:408–411
Bonne K, Verbeke W (2008) Muslim consumer trust in halal meat status and control in Belgium. Meat Sci 79:113–123
Calvo JH, Osta R, Zaragoza P (2002) Quantitative PCR detection of pork in raw and heated ground beef and pâté. J Agric Food Chem 50:5265–5267
Calvo JH, Zaragoza P, Osta R (2001) Random amplified polymorphic DNA fingerprints for identification of species in poultry pate. Poult Sci 80:522–524
Che Man YB, Mustafa S, Khairil-Mokhtar NF, Nordin R, Sazili AQ (2012) Porcine-specific polymerase chain reaction assay based on mitochondrial D-loop gene for identification of pork in raw meat. Int J Food Prop 15:134–144
Chikuni K, Ozutsumi K, Koishikawa T, Kato S (1990) Species identification of cooked meats by DNA hybridization assay. Meat Sci 27:119–128
Di Pinto A, Forte VT, Conversano MC, Tantillo GM (2005) Duplex polymerase chain reaction for detection of pork meat in horse meat fresh sausages from Italian retail sources. Food Cont 16:391–394
Girish PS, Anjaneyulu ASR, Viswas KN, Shivakumar BM, Anand M, Patel M, Sharma B (2005) Meat species identification by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) of mitochondrial 12S rRNA gene. Meat Sci 70:107–112
Graf C, Rüegg-Kuslyte A, Kuhn R (2011) Species determination for meat using PCR-RFLP analysis on the QIAxcel® system. Application Note
Hird H, Chisholm A, Sanchez M, Hernandez R, Goodier K, Schneede C, Boltz B, Popping B (2006) Effect of heat and pressure processing on DNA fragmentation and implications for the detection of meat using a real-time polymerase chain reaction. Food Addit Contam 23:645–650
Karabasanavar NS, Singh SP, Kumar D, Shebannavar SN (2014) Detection of pork adulteration by highly-specific PCR assay of mitochondrial D-loop. Food Chem 145:530–534
Karabasanavar NS, Singh SP, Umapathi V, Girish PS, Shebannavar SN, Kumar D (2011) Authentication of carabeef (water buffalo, Bubalus bubalis) using highly specific polymerase chain reaction. Eur Food Res Technol 233:985–989
Kesmen Z, Gulluce A, Sahin F, Yetim H (2009) Identification of meat species by TaqMan-based real-time PCR assay. Meat Sci 82:444–449
Kesmen Z, Sahin F, Yetim H (2007) PCR assay for the identification of animal species in cooked sausages. Meat Sci 77:649–653
Kumar D, Singh SP, Singh R, Karabasanavar NS (2011) A highly specific PCR assay for identification of goat (Capra hircus) meat. Small Rumin Res 97:76–78
Mane BG, Mendiratta SK, Tiwari AK (2013) Pork specific polymerase chain reaction assay for authentication of meat and meat products. J Meat Sci Technol 1:21–27
Marois C, Dufour-Gesbert F, Kempf I (2001) Comparison of pulsed-field gel electrophoresis with random amplified polymorphic DNA for typing of Mycoplasma synoviae. Vet Microbiol 79:1–9
Matsumoto T, Koshii Y, Sakane K, Murakawa T, Hirayama Y, Yoshida H, Kurokawa M, Tamura Y, Nagai T, Kawase I (2013) A novel approach to automated genotyping of Mycobacterium tuberculosis using a panel of 15 MIRU VNTRs. J Microbiol Methods 93:239–241
McMurray CL, Hardy KJ, Hawkey PM (2010) Rapid, automated epidemiological typing of methicillin-resistant Staphylococcus aureus. J Microbiol Methods 80:109–111
Melake NA, Shaker GH, Salama MA (2012) Incidence of Helicobacter pylori infection and their clarithromycin-resistant strains in otitis media with effusion regarding phenotypic and genotypic studies. Saudi Pharm J 20:345–353
Mercimek-Mahmutoglu S, Sinclair G, van Dooren SJM, Kanhai W, Ashcraft P, Michel OJ, Nelson J, Betsalel OT, Sweetman L, Jakobs C, Salomons GS (2012) Guanidinoacetate methyltransferase deficiency: first steps to newborn screening for a treatable neurometabolic disease. Mol Genet Metab 107:433–437
Moghazy EA, El-Shaarawy MOA (2001) Quality attributes of beef burger as affected by using proplies and frozen storage. Egypt J Agric Res 79:1499–1512
Moghazy EA, Sharaf SM, El-Seesy TA (2004) Effect of substitutines mutton tailfat with vegetable oil on quality attributes of healthy beef sausage. Egypt J Appl Sci 19:191–205
Montiel-Sosa JF, Ruiz-Pesini E, Montoya J, Roncalés P, López-Pérez MJ, Pérez-Martos A (2000) Direct and highly species-specific detection of pork meat and fat in meat products by PCR amplification of mitochondrial DNA. J Agric Food Chem 48:2829–2832
Olive PL, Wlodek D, Durand RE, Banáth JP (1992) Factors influencing DNA migration from individual cells subjected to gel electrophoresis. Exp Cell Res 198:259–267
Rehbein H, Kress G, Schmidt T (1997) Application of PCR-SSCP to species identification of fishery products. J Sci Food Agric 74:35–41
Rojas M, González I, Pavón MA, Pegels N, Lago A, Hernández PE, García T, Martin R (2010) Novel TaqMan real-time polymerase chain reaction assay for verifying the authenticity of meat and commercial meat products from game birds. Food Addit Contam A 27:749–763
Skarpeid H, Moe RE, Indahl UG (2001) Detection of mechanically recovered meat and head meat from cattle in ground beef mixtures by multivariate analysis of isoelectric focusing protein profiles. Meat Sci 57:227–234
Tanabe S, Hase M, Yano T, Sato M, Fujimura T, Akiyama H (2007a) A real-time quantitative PCR detection method for pork, chicken, beef, mutton, and horseflesh in foods. Biosci Biotechnol Biochem 71:3131–3135
Tanabe S, Miyauchi E, Muneshige A, Mio K, Sato C, Sato M (2007b) PCR method of detecting pork in foods for verifying allergen labeling and for identifying hidden pork ingredients in processed foods. Biosci Biotechnol Biochem 71:1663–1667
Toorop R, Murch SJ, Ball RO (1997) Methodology and development of prediction equations for the determination of pork substitution in veal. Food Res Int 30:629–636
Wissiack R, de la Calle B, Bordin G, Rodriguez AR (2003) Screening test to detect meat adulteration through the determination of hemoglobin by cation exchange chromatography with diode array detection. Meat Sci 64:427–432
Xiao M, Kong P, Jin Q, Wang K, Xiao N, Jeoffreys G, James GL, Gilbert GL (2012) Comparison of two capillary gel electrophoresis systems for Clostridium difficile ribotyping, using a panel of ribotype 027 isolates and whole-genome sequences as a reference standard. J Clin Microbiol 50:2755–2760
Yang I, Kim Y, Byun J, Park S (2005) Use of multiplex polymerase chain reactions to indicate the accuracy of the annealing temperature of thermal cycling. Anal Biochem 338:192–200
Yusop MHM, Mustafa S, Che Man YB, Omar A, Mokhtar N (2012) Detection of raw pork targeting porcine-specific mitochondrial cytochrome B gene by molecular beacon probe real-time polymerase chain reaction. Food Anal Methods 5:422–429
Zhang S, Zhao S, Wang Z, Li C (2013) Investigation of parent-of-origin SNPs in 5 imprinted genes for forensic purpose. For Sci Int Genet Suppl Ser 4:e304–e305
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Barakat, H., El-Garhy, H.A.S. & Moustafa, M.M.A. Detection of pork adulteration in processed meat by species-specific PCR-QIAxcel procedure based on D-loop and cytb genes. Appl Microbiol Biotechnol 98, 9805–9816 (2014). https://doi.org/10.1007/s00253-014-6084-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6084-x