
Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains the leading cause of death due to a single bacterial agent, with approximately 10.6 million people developing active disease and 1.6 million deaths reported globally in 2021. After exposure, some, but not all individuals, will become infected with the bacillus. However, only a small fraction (approximately 5 to 15%) of these individuals will progress to clinical disease, while in the remainder, infection is seemingly contained, and no signs of clinical disease are shown. Numerous observations have advocated for the role of host genetics in the display of these inter-individual variabilities in infection and disease phenotypes. In this review, we will provide an overview of the approaches, findings and limitations of the very first studies investigating TB genetic susceptibility to more recent studies. Lastly, we highlight several approaches, namely, linkage analyses and association studies, proposed to discover genetic markers associated with TB susceptibility. This review also explored the concept of polygenic risk scores (PRS) for prediction of tuberculosis susceptibility. The identification of host genetic factors influencing TB susceptibility/resistance is paramount to not only better understand the physiopathology of the disease but also explore more effective approaches for the development of both optimal preventive measures (i.e. better vaccines) and treatments of TB disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is one of the oldest diseases to affect humans. Due to its devastating effect, and after the discovery of the causative bacterium, Mycobacterium tuberculosis (Mtb), exhaustive methods have been explored to not only detect, but also prevent infection and cure active disease (Gradmann 2001). This resulted in the development of the Pirquet and Mantoux tuberculin skin test (TST), a skin test to detect the presence of proteins of the TB bacillus in an individual, the first anti-tuberculous vaccine (i.e. the Bacille Calmette-Guérin (BCG) vaccine) (Calmette 1927) and the first anti-TB drug, streptomycin (Bogen 1947).
Undeniably, at the time, these findings better equipped us in the fight against TB, by tackling three aspects: (1) detection, (2) prevention through vaccination and (3) treatment in the event of active disease. However, despite these commendable efforts, TB remains the leading cause of death due to a single bacterial agent, with approximately 1.6 million deaths reported globally in 2021 (Geneva: World Health Organization 2022). This is a small, but significant increase from the global 1.4 million TB-related deaths reported in 2019 (WHO 2021; Geneva: World Health Organization 2022). This major death resurgence is a multifactorial setback in the World Health Organization’s (WHO) conscious effort to reach the “End of TB Strategy” aimed for 2035. In addition, approximately 10 million people developed active TB in 2020. The consequences are especially alarming for developing countries, which bear the brunt of the disease. Africa, alone, contributed 23% of new TB cases globally in 2021 (Geneva: World Health Organization 2022).
It has been reported that approximately 25% of the global population is latently infected with Mtb (Houben and Dodd 2016). However, only a small fraction (approximately 5 to 15%) of these individuals will progress to clinical disease, while in the remainder, infection is seemingly contained, and the individuals do not show any sign of clinical disease (El Baghdadi et al. 2013; Möller et al. 2018). These inter-individual variabilities in infection and disease phenotypes have been attributed to both extrinsic factors (e.g. diabetes, age, sex, alcohol consumption, smoking and HIV infection) and intrinsic factors (e.g. host genetic component) (Bellamy 2000; Bellamy et al. 2000; Brites and Gagneux 2015; Verhein et al. 2018). The identification of host genetic factors influencing TB susceptibility/resistance is paramount to not only better understand the physiopathology of the disease but also explore more effective approaches for the development of both optimal preventive measures (i.e. better vaccines) and treatments of TB disease.
In this review, we will discuss early and more recent genetic epidemiology studies on TB infection and progression to clinical disease while highlighting their limitations and bringing focus to future developments.
Tuberculosis susceptibility and resistance: evidence of genetic underlining
Several studies have suggested that host genetics contribute to TB susceptibility, regardless of the host immunocompetency. A good illustration of that evidence was brought forth, in 1930 at Lübeck, by the observed inter-individual variabilities in responses to accidental administration of virulent tubercule bacilli as opposed to attenuated ones (Fox et al. 2016). The tragic incident involved 251 children who had been inadvertently injected with BCG vaccines contaminated with varying amounts of Mtb. Of these children, 72 died from extensive TB infection within a year of inoculation, and 228 showed clinical evidence of TB. The affected infants displayed variation in disease severity (62 infants developed severe TB, 94 infants developed moderate TB and 17 infants did not present clinical signs of TB infection despite a positive TST test) (Fox et al. 2016). Contamination level was a critical factor determining outcome after exposure—children that received dosages with high Mtb levels were more likely to develop disease, while among those that received low dosages of Mtb, various clinical TB phenotypes were observed, indicative of the range of innate resistance in the human population (Fox et al. 2016). In addition, this accident showed that some individuals are more genetically susceptible to severe disease or death even at low infection levels—two children that received vaccines with the lowest Mtb contamination levels quickly progressed to disease and death (Fox et al. 2016).
A family clustering study by Puffer in 1943 which found that spouses with a family history of active TB developed disease more frequently than those without familial histories also contributed to the idea that host genetics contribute to TB susceptibility (Puffer 1944). Twin studies have also shown high levels of concordance when comparing TB incidence rate in affected and healthy twin siblings (Kallmann and Reisner 1943). Kallmann and Reisner found that monozygotic twins compared to dizygotic twins were more likely to progress to active disease (66.7% compared to 23%, respectively) (Kallmann and Reisner 1943). Interethnic disparities in the occurrence and prevalence of TB in different global populations have also been previously observed with Stead and colleagues seeking to understand the rate of TB infection in nursing homes in Arkansas, USA (Stead et al. 1990). Stead and colleagues measured the incidence of tuberculin conversion in over 25,000 nursing home residents. In this study, tuberculin conversion was said to have occurred when the initially TST-negative residents produced a positive TST result 60 days following the last negative test. It was found that individuals classified as African-American were twice as likely to become infected with TB compared to their European-American counterpart (relative risk, 1.9 [95% confidence interval (CI) = 1.2–2.1], P < 0.001). Therefore, hinting at the possibility for a genetic component that may render certain population groups relatively more susceptible to TB infection than others (see section Ancestry and TB susceptibility). However, the fact that participants were classified as African American or European-American by the colour of their skin, without considering genetic ancestry, is a major limitation of this study (Teteh et al. 2020).
Animal models to elucidate host genetic influence on TB infection
The findings presented above have sparked major efforts into understanding the mechanisms of action that govern a host’s genetic susceptibility to TB. This implies, in other words, finding the genes that are involved, how they are involved and to what extent they contribute to disease phenotype. To answer these questions, animal models have long been used as proxies to study the genetics and pathogenesis of disease susceptibility (Jouanguy et al. 1996; Bellamy et al. 1998; Fortin et al. 2007; Verhein et al. 2018; Ruiz‑Bedoya et al. 2022). For instance, mice are a good experimental model because when infected with Mtb, they exhibit pathological and immunological responses similar to those seen in humans (Fortin et al. 2007); which allows for an easier extrapolation of mouse model findings to human. Additionally, animal models offer easier ways to control for parameters such as stress and diet, route of infection, bacterial dose, strain and virulence of the mycobacteria (Vidal et al. 1993; Fortin et al. 2007; El Baghdadi et al. 2013). Furthermore, as opposed to other animal models, mice have low maintenance cost, require less space, are fast breeders and have been extensively used for research purposes; thus the availability of different well categorized strains (Fonseca et al. 2017; Singh and Gupta 2018).
The use of such models to study the underlying genetics of diseases mainly consists of two opposing types of studies: (1) reverse genetics and (2) a traditional/forward genetics approaches. In the former, the role of a gene in the disease is studied by infecting model organisms carrying loss-of-function mutations at a locus of interest. In the latter approach, the genetic basis of a disease is evaluated by linking an observed disease aetiology to a genetic mutation or locus of interest (Teare 2011). Both methods are hypothesis-based approaches relying either on our understanding and knowledge of genes known to influence the immune response against a pathogen or on results obtained from previous studies.
Using the forward genetics approach, Vidal et al. studied the association between Bcg (a gene known to control innate immune response to Mtb infection by enabling the early destruction of intracellular pathogens by macrophages during infection) and permissiveness of microbial replication in the spleen of mice after intravenous injection with M. bovis BCG strain (Vidal et al. 1993). First, Vidal et al. cloned the genomic region encompassing Bcg using a 400 kb bacteriophage and cosmid contig. Then, exon amplification was performed to search for transcription units within that genomic region, and 6 genes were subsequently identified. To characterise the role of these genes, Vidal and colleagues conducted RNA expression studies; however, only one of them, the Nramp1 (natural resistance associated macrophage protein 1) gene was found to be associated with resistance to Mtb infection. Nramp1 encodes the Nramp1 protein which is an integral membrane protein found to be exclusively expressed in lysosomes of macrophage populations and functions as a transporter of divalent metals across the phagosome membrane (Vidal et al. 1993; Fortin et al. 2007; Holder et al. 2020; Wahyuni et al. 2021; Agbayani et al. 2022). This is of particular interest as this function allows the Nramp1 protein to limit microbial access to micro-nutrients such as manganese and iron, thus affecting microbial replication within phagosomes and therefore containing Mtb infection (Canonne‑Hergaux et al. 1999; Cellier et al. 2007). Nramp1 was the first gene found to be associated with TB resistance in mice and has a human homolog commonly known as SLC11A1 (solute carrier family 11 member 1). Other animal model studies looking to elucidate mechanisms of susceptibility to Mtb infection have found results that were extrapolated to human settings. For instance, mice with defects in the production of Type 1 T helper (Th1) cells or cytokine interferon gamma (IFNγ) were found to be more susceptible to Mtb infection (Green et al. 2013), and a similar observation was made in humans where similar defects cause Mendelian susceptibility to mycobacterial disease (MSMD) (Bustamante et al. 2014).
Despite the proven utility of animal models in TB susceptibility research, most results replicate poorly in humans (Pan et al. 2005; Fonseca et al. 2017; Singh and Gupta 2018). One of the reasons for this lack of replication could be attributed to inter-species differences. Although experimental infections in mice mimic several aspects of the pathophysiology and host response to Mtb seen in humans, other features are species-specific. For example, the long latency period typically observed in humans does not occur in the mouse model (Fortin et al. 2007). Additionally, because mice are not natural host of Mtb, they lack the prime characteristics of disease transmission observed in humans which represent additional limitations of the model. After infection in mice, the bacilli reside primarily intracellularly in the lungs which then develop inflammatory but non-necrotic lesions as opposed to humans who exhibit necrotic lesions with most of the mycobacteria located extracellularly within the lesion (Fonseca et al. 2017; Singh and Gupta 2018). Furthermore, mice do not possess the HLA (human leukocyte antigens) class II antigens which in humans is responsible for the regulation of the immune system (Nordquist and Jamil 2022). This issue has prompted the development of transgenic/humanized mice, which are generated by engrafting human haematopoietic stem cells into the mice to reproduce the critical features of human TB disease pathology absent in normal mice (Mangalam et al. 2008). However, transgenic mice also have important limitations pertaining to abnormal T-cell responses after Mtb infection and cost-effectiveness of the model (Fonseca et al. 2017). Therefore, when studying TB clinical manifestation using the mice model, results may not be readily transferable to humans. Moreover, animal studies are not suited to investigate single nucleotide polymorphisms (SNPs), the most common type of genetic variations among humans and environmental factors that may influence the genes associated with complex diseases, such as TB.
Linkage analysis and association studies using human models
To find SNPs that influence the outcomes of active TB, genetic epidemiological studies have mainly adopted two approaches: (1) family-based association analysis using linkage mapping and (2) case–control association studies. Linkage studies test whether a disease and an allele show correlated transmission within a pedigree, referred to as concordant inheritance, while association studies test whether an allele co-occurs with a disease in a population more frequently than expected by chance. Both offer the opportunity to better under the host genetic basis of disease.
Family-based association analysis using linkage mapping
Family studies are not as often cited as twin and adoption studies, but nonetheless, they are still a valid and important piece in the puzzle of heredity versus the environment. Family-based association studies are performed using linkage analysis, which aims to locate genomic regions or haplogroups on a chromosome that are co-inherited with a trait/disease under study. This is done by identifying a known gene on the chromosome which can serve as a marker. That marker’s location, and the location of the actual diseased gene, is very important. The closer the two are, the higher the likelihood that they will be passed on or linked together. To obtain said likelihood, a logarithm (base 10) of odds (LOD) score is used to assess the probability that a given pedigree, where the disease and the marker are co-segregating, is due to the existence of linkage or to chance (Lander and Kruglyak 1995). According to a widely used guideline for the interpretation of linkage results, LOD of 2.2 is classified as suggestive linkage, while LOD of 3.6 as significant linkage (Lander and Kruglyak 1995). However, linkage studies with insufficient statistical power are unable to discern true-positive signals from false-positive signals, resulting in inconsistent findings among studies (Baron 2001).
The first reported genome-wide linkage study of TB susceptibility was performed by Bellamy and colleagues who conducted a two-stage (discovery and replication) genome wide search using 169 sib-pair families from the Gambia and South Africa (Bellamy et al. 2000). The results suggested evidence for the presence of genomic regions on chromosome 15q and Xq that showed linkage to TB (LOD = 2.00 and 1.70, respectively). The genomic region of interest on chromosome 15 harbours the OCA2 gene and the HERC2 gene which encodes a protein that functions as a E3-ubiquitin ligase and is involved in DNA repair and regulatory cell-cycle (Morice‑Picard et al. 2016; Elpidorou et al. 2021). However, the HERC2 gene has mainly been found to be associated with skin-pigmentation, neurodegenerative disorders, as well as cancers and has not been replicated in other infectious diseases. Bellamy et al. presented the CD40LG gene (encoding the CD40 ligand) as a potential candidate gene located near the region of interest on chromosome Xq. The CD40 ligand is a member of the TNF-receptor superfamily and is believed to be involved in mediating a broad variety of inflammatory and immune responses (Elgueta et al. 2009; Françoise et al. 2022). Together, these results indicate that genome-wide linkage analysis can contribute to the mapping and identification of major genes for multifactorial infectious diseases.
Bellamy’s findings encouraged additional linkage studies to assess the genetic susceptibility to TB. Greenwood et al. identified a link between TB susceptibility and the D2S424 locus in a group of 81 Aboriginal Canadian individuals (LOD = 3.81). Interestingly, the D2S424 locus is located 235.92 Mb downstream to SLC11A1, the human homolog of the murine Nramp1 gene (Greenwood et al. 2000). Contrastingly, Jamieson et al. found evidence for a cluster of genes on chromosome 17q11-q21 to be associated with TB susceptibly in a family-based cohort of 627 Brazilians (LOD = 2.48) (Jamieson et al. 2004). Compared to Greenwood et al. who used a dominance mode of inheritance to evaluate allele sharing between affected family members (Greenwood et al. 2000), Jamieson and colleagues used a non-parametric approach to linkage analysis (Jamieson et al. 2004). Both approaches have the same basic functionalities, but the latter does not assume any particular model of inheritance to evaluate the sharing of alleles. Inconsistent results among these linkage studies may be attributed to the use of different study populations and statistical methods. However, it is also important to consider that TB susceptibility, as a multifactorial trait, is a product of environmental exposure and genetic risk factors (which are often population specific) and inconsistencies among study results reflects the complex nature of multifactorial traits.
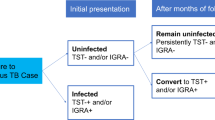
Stein et al. examined three outcomes after Mtb exposure (i.e. active TB, persistent TST negative and TST positive latently TB infection (LTBI)) in a household contact study of Ugandan individuals from 193 pedigrees (Stein et al. 2008). The aim of the study was to demonstrate the different genetic components underlining different stages of TB natural progression. For persistently TST negative individuals, loci at 2q21-2q24 and 5q13-5q22 were found to be associated with resistance to TB (P < 0.0003 and P < 0.0005, respectively), while the 7p22-7p21 locus, which contains the IL6 gene, was associated with increased susceptibility (P < 0.0002). IL6 gene encodes the interleukin 6 (IL-6) which is believed to play a role in host defence against infections and tissue injuries by stimulating immune reactions, the formation of blood cellular components as well as acute phase responses (Tanaka et al. 2014). The study also replicated the 20q13 TB-susceptibility locus (housing the SLC11A1 gene) (P < 0.002) that had been previously identified in linkage studies of South African and Malawian populations (Cooke et al. 2008). The results suggest three implications: (1) There are in fact genetic factors that influence the relationship between Mtb and humans, (2) these genetic factors differ based on the disease progression or outcome, and (3) some genetic loci associated with TB susceptibility/resistance could be population-specific. However, the sample size was too small to draw definitive conclusions.
Although linkage studies were able to find genomic regions linked with TB outcome, most were not replicated by any independent study. This lack of replication could be attributed to the overestimation of genetic effects on TB susceptibility as well as population-specific effects (Thuong et al. 2012). Moreover, linkage studies do not take into account the effect of shared environmental factors which have been shown to have large effect on TB outcome (van der Eijk et al. 2007). Due to their limited statistical power, linkage studies are not able to identify weak genetic effects of TB. However, their most useful feature resides in the ability to identify a large number of genes for which a role in infection may not have been suspected, albeit their functions cannot be addressed. Nevertheless, a few loci worthy of attention were reported, although besides the SLC11A1 gene, no other TB-associated genes had been identified using linkage analysis (Greenwood et al. 2000; Baghdadi et al. 2006; Stein et al. 2008; Cobat et al. 2009; Adams et al. 2011).
Population-based case–control studies
Candidate gene association studies
To overcome the limitations of linkage studies, genetic studies of TB susceptibility have primarily turned their focused to hypothesis-based association studies using candidate gene association studies (CGAS). While linkage analysis looks at actual physical segments of the genome that are associated with given traits, association analysis compares allele frequency differences in unrelated cases and population-matched controls. It is important to note that discovered alleles could either be directly influencing the disease or be in linkage disequilibrium (LD) with the actual disease-predisposing gene. With higher resolution than linkage in family studies, association studies provide the opportunity to identify SNPs with smaller effect that could be involved with gene expression levels and subsequent disease response (Bellamy 2000; Möller et al. 2009; Velez et al. 2009; Baker et al. 2011; Bahari et al. 2012; Möller and Kinnear 2020).
Several CGAS have identified genetic markers associated with TB susceptibility across diverse ancestral groups (reviews of CGAS have been provided by Möller et al. (2010) and Möller and Kinnear (2020)). Among the most interesting CGAS, we note the association of SLC11A1 polymorphisms with TB susceptibility in a Gambian population (Bellamy et al. 1998). Polymorphisms in the HLA-DR and HLA-DQ genes have sparked multiple studies, with results involving the HLA-DR2 and HLA-DQB1 genes class II antigen subtypes (Sriram et al. 2001; Kettaneh et al. 2006). Genes implicated in immunity such as those coding for IFNG, TLR1 and TLR2 have been previously identified and reviewed, but the interpretation remains difficult as results are often contradictory (Möller et al. 2010). The role of the VDR gene, encoding the vitamin D receptor (VDR) protein, has largely been studied as well, since vitamin D deficiency has been associated with increase susceptibility to TB (Wilkinson et al. 2000; Mohammadi et al. 2020). However, although many associations have been found with TB susceptibility, replication across studies have been limited (Möller and Kinnear 2020).
Genome-wide association studies: genotype–phenotype associations
Similar to candidate genes association studies, genome-wide association studies (GWAS) compare the frequency of alleles present in cases and controls for a given trait or disease. However, unlike CGAS, GWAS involve testing thousands or millions of genetic markers and are not limited by the current understanding of biological pathways involved in the phenotypic expression of said trait or disease.
GWAS have allowed for the discovery of common SNPs that influence a trait/disease outcome. Due to its success, GWAS have seen an exponential growth (~40-fold) in almost two decades of its implementation, with currently more than 5000 published studies from which hundreds of thousands of SNP-trait associations have been found (www.ebi.ac.uk/gwas (Buniello et al. 2019)). However, only a few published studies (from diverse populations) on TB susceptibility have emerged in the last 10 years (2010 to 2021) with minimal overlap between results (Uren et al. 2017a; Möller et al. 2018; Möller and Kinnear 2020). This lack of replication has been attributed to differences in allele frequencies and LD patterns between and across population groups; allelic and phenotypic heterogeneity associated with TB; and, more importantly, small study sample sizes which strongly affect the statistical power required to detect or replicate associations. However, where replication has been successful, it was mostly in populations that were more genetically similar. A summary of previous TB GWAS results is provided in Table 1.
Thye et al. (2010) conducted a combined GWAS of two African populations from Ghana (921 TB cases and 1740 healthy controls) and the Gambia (1316 cases and 1382 controls) to investigate host susceptibility to TB (Thye et al. 2010). The G allele of SNP rs4331426, located in a gene desert region at 18q11.2, was found to be significantly associated with increase TB susceptibility (odds ratio (OR) = 1.19, 95% CI [1.13–1.27]; P = 6.8 × 10−9). This was replicated in an independent cohort from Malawi with comparable OR estimates, suggesting that this variant is common in African populations and therefore suggesting population-specific effects.
Oki et al. (2011) conducted a pilot GWAS study to investigate genetic susceptibility to extrapulmonary TB (ETB) (Oki et al. 2011) using 24 ETB patients as cases and, as controls, 56 patients with Mtb infection and 24 with pulmonary TB (PTB) (from the USA). Oki and colleagues argued that since Mtb infection, PTB and ETB have different clinical manifestations and therefore different pathophysiology, they must exhibit different genetic factors influencing patient’s outcome. Four SNPs (rs4893980, rs10488286, rs2026414, rs10487416; OR = 0.13, 11.15, 3.11, 5.56 at suggestive P = 0.0007, 0.0009, 0.0009, 0.0007 respectively) showed an association with ETB compared to participants with Mtb infection and 2 SNPs (rs340708 [OR = 5.44, P = 0.0008] and rs1886870 [OR = 6.00, P = 0.0009]) showing suggestive, but not significant, association with ETB compared with PTB. Although the sample size was small and P value of association suggestive, this study supports the idea of TB susceptibility phenotypic heterogeneity (Oki et al. 2011; Mahasirimongkol et al. 2012). In agreement with these findings, Luo et al. (2019) using a Peruvian population (2175 cases and 1827 controls) found a significant association between rs73226617*A in the ATP1B3 gene region at 3q23 and early progression to active TB following Mtb infection (OR = 1.80; P = 3.93 × 10−8) (Luo et al. 2019). This study also detected a previously reported TB locus at rs9272785*A (Sveinbjornsson et al. 2016; Luo et al. 2019). However, this locus does not have a strong association with TB progression following Mtb. infection (OR = 1.04, P = 4.49 × 10−3 in the study performed by Luo et al.; OR = 1.14, P = 9.3 × 10−9 in the study performed by Sveinbjornsson et al.). These studies encourage the use of detailed phenotyping in GWAS of infectious disease to investigate the pathogenic mechanisms of different TB infection stages, which should aid in the identification of novel variants in diverse populations.
Thye et al. (2012) reported SNP rs2057178*A in an intergenic region of 11p13 located 45 kb downstream of WT1 gene to be associated with resistance to TB in the Ghanaian population (1329 cases, 1847 controls) (OR = 0.77, 95% CI [0.71–0.84]; P = 2.63 × 10−9) (Thye et al. 2012). This was successfully replicated in populations from The Gambia (1207 cases, 1349 controls), Russia (4441 cases, 5874 controls) and Indonesia (1025 cases, 983 controls) with consistent effect sizes (OR = 0.77, 95% CI [0.71–0.84]) across the study cohorts. The rs2057178*A variant was also later replicated by Chimusa et al. using 642 PTB cases and 91 controls from the admixed SAC population of South Africa, thus suggesting its association with TB resistance in diverse populations (Chimusa et al. 2014). This provides evidence for the relevance of WT1 in TB pathogenesis. WT1 encodes a transcription factor WT1, which induces expression of VDR gene and suppresses IL-10 gene expression (Maurer et al. 2001; Thye et al. 2012). Both VDR and IL-10 play important roles in TB pathophysiology, and variation in their respective genes has previously been associated with TB susceptibility (Ottenhoff et al. 2005). In contrast, some of these findings failed to be successfully replicated from one population to another, as was the case in a case–control study in a Russian population who failed to replicate the locus at 18q11.2 albeit with a bigger sample size (5530 cases, 5607 controls) (Curtis et al. 2015). This locus was previously identified in West and East African populations (Thye et al. 2010). In a relatively recent study, Quistrebert et al. discovered a cluster of variants, in intronic regions and upstream of C10orf90 at chromosome 10q26.2 to be associated with resistance to Mtb infection in three distinct populations, namely, Vietnam, France and South Africa (Quistrebert et al. 2021). In the Vietnamese cohort of 353 cases and 185 controls, rs17155120 (with T as protective allele) was found to display the strongest association with resistance to Mtb (OR = 0.42, 95% CI [0.45 – 0.55]; P = 3.71 × 10−8). This association was replicated with similar effect in cohorts from France (157 cases and 30 controls) and South Africa (136 cases and 118 controls) with overall OR = 0.50, 95% CI [0.45–0.55]; P = 1.26 × 10−9. Using in silico analysis, Quistrebert and colleagues assessed the functionality of the T protective allele and found that it was associated with a decrease in monocytes expression which could then lead to an enhance Th17 lymphocytes response (Quistrebert et al. 2021).
As opposed to the studies presented above which only looked at autosomal chromosomes due to past GWAS statistical analyses constraints, Schurz et al. (2019) proposed a sex-stratified and X-linked GWAS of TB susceptibility (Schurz et al. 2019). Schurz and colleagues hypothesized that as the X chromosome houses several immunity-related genes, the removal of this chromosome would likely hinder the discovery of novel associations. Using an admixed SAC population of 410 TB cases and 405 controls, the rs17410035*T variant in the DROSHA gene region was associated with disease (OR = 0.40, 95% CI [0.28–0.58]; P = 1.50 × 10−6), but did not reach the GWAS significance level of P value < 5 × 10−8 (Schurz et al. 2019). DROSHA initiates maturation of miRNA molecules, which regulate many biological processes (including the innate immune response), and it is thought that dysfunction of the miRNA pathway could be involved in the pathogenesis of human diseases, including TB (Leal et al. 2022). However, the role of miRNA expression in TB requires further research.
GWAS are confounded by hidden population substructure. To account for this issue, global ancestry proportions, which refer to the average ancestry across the whole genome, are usually inferred and the resultant used as covariates in the association analysis. However, global ancestry does not account for the role of both ancestry and allelic effects as well as the interplay between the two on the phenotypic display of a disease (Wang et al. 2011; Duan et al. 2018). This is particularly hindering for GWAS of admixed individuals where the frequency of certain alleles does not always correlate with observed disease phenotypic expression but with subtle variations of ancestry at chromosomal level (i.e. local ancestry proportions) (Duan et al. 2018). To this end, Duan et al., using a two-way admixed African American cohort, proposed a local ancestry allelic adjustment (LAAA) model. This approach leverages information on local ancestry at specific risk locus and includes this as a covariate in the logistic regression. The LAAA model captures ancestry-specific allelic effects that would have otherwise been missed by standard population stratification methods (Duan et al. 2018).
For more complex admixed population group, the task of using such an approach can be daunting owing to the fact that an LAAA model would have to be developed for every chosen SNP and every global ancestry present in the admixed cohort. Despite the obvious statistical burden of the approach, a recent study by Swart et al. utilized LAAA models in a TB-GWAS of 820 multi-way admixed individuals from South Africa (Swart et al. 2021). Swart and colleagues were able to identify a specific SNP, located on chromosome 4q22, in a Bantu-speaking African population (rs28647531*G) that showed significant association with TB susceptibility (OR = 3.07; P = 5.52 × 10−7) which had previously been missed when correcting for global ancestry proportions only (Swart et al. 2021). This highlights the increase in statistical power that can be gained by using the LAAA model in GWAS of complex admixed individuals.
Genome-wide association studies: host–pathogen associations
GWAS have also shown that the level of association between TB and a SNP can vary based on different Mtb lineages. A first study in Vietnam has revealed that individuals with the C allele of TLR2 gene had an increased susceptibility to TB meningitis when infected with Mtb strains of Beijing lineage (OR = 1.57, 95% CI [1.15–2.15]; P = 0.004) (Caws et al. 2008). Another study using a Ghanaian cohort has indicated that an autophagy gene variant IRGM -261 T contributed to protection from PTB in patients infected with Mtb (OR = 0.63, 95% CI [0.49–0.81]; P < 0.0019) but not in those with M. africanum or M. bovis (Intemann et al. 2009). A study in South Africa conducted pathogen lineage-based association analysis and identified HLA types to be associated with disease caused by the Euro-American or East Asian lineages (Salie et al. 2014). It was also been reported that the Beijing strains occurred more frequently in individuals with multiple disease episodes (Salie et al. 2014). To provide further evidence for host–pathogen interactions on TB clinical presentation, Omae et al. conducted a pathogen lineage-based GWAS in a Thai cohort (n = 1 457) by stratifying the study cohort based on lineage and age of onset of disease (Omae et al. 2017). The results revealed that SNP rs1418425 was associated with non-Beijing lineage-infected old age onset cases (OR = 1.74, 95% CI [1.43–2.12], P = 2.54 × 10−8) when compared to healthy controls. In a more recent study, Müller et al. used South African (n = 853) and Ghanaian (n = 1 359) cohorts to identify genetic variations associated with members of clades causing TB (Müller et al. 2021). The results show that loci on chromosomes 5, 6 and 17 was associated with strains of different Mycobacteria tuberculosis complex (MTBC) superclades, albeit with suggestive significance level of association (P < 1 × 10−7). In the South African cohort, 11 SNPs were found to be associated with strains of different MTBC superclades. Individuals carrying the risk allele for associated SNPs on chromosome 5 were twice as likely to get infected with a member of the Quebec superclade when compared with BeijingCAS1 or HaarlemsLCC or other superclades. For those carrying the risk allele A for SNP rs9389610 on chromosome 6, the likelihood of infection was twice as much with a member of the BeijingCAS1 superclade (OR = 2.19, 95% CI [1.35–3.55]; P = 1.0 × 10−7) as opposed to members of HaarlemsLCC or Quebec superclades. Risk allele of SNPs on chromosome 17 renders study participants highly susceptible infection with members of the Quebec superclade (OR ~ 5) as opposed to members of other clades. A similar pattern of differential infection risk due to members of different superclades was also observed for the Ghanaian cohort. For instance, the risk allele G of the intergenic variant rs529920 (chromosome 6) was found to double the risk of infection with members of the Ghana2 superclade (OR = 1.04, 95% CI [0.60–1.80]; P = 1.86 × 10−7) and halve the risk with BeijingCAS (OR = 0.40, 95% CI [0.22 – 0.70]; P = 1.86 × 10−7) and East African Indian (EAI)_afri superclades (OR = 0.69, 95% CI [0.57–0.84]; P = 1.86 × 10−7). Similarly, rs41472447*G (intron variant on chromosome 12) was found to nearly triple the risk of infection with a member of the BeijingCAS superclade (OR = 2.56, 95% CI [1.48–4.41]; P = 5.70 × 10−9) or the Ghana2 superclade (OR = 2.94, 95% CI [1.71–5.08]; P = 5.70 × 10−9), whereas the risk is reduced with members of the EAI_afri superclade (OR = 0.97, 95% CI [0.78–1.21]; P = 5.70 × 10−9) and HaarlemX (OR = 0.92, 95% CI [0.68 – 1.26]; P = 5.70 × 10−9). The relationship between ancestry and strains of Mtb should be highlighted. This relationship could be explained by the high prevalence of some TB strains in certain geographical areas as opposed to others, which then naturally led to the co-evolution of the host and the pathogen in those regions (Brites and Gagneux 2015; Dou et al. 2015; Nebenzahl‑Guimaraes et al. 2015; Duarte et al. 2017; Ejo et al. 2020; Müller et al. 2021). For instance, M. africanum is prevalent in Western Africa and is the main cause of TB in that region while for other parts of the world. TB is caused by Mtb (Gagneux and Small 2007). Similarly, the Haarlem (Dutch) strain was found to be dominant in the aborigines of Eastern and Central Taiwan, while the EAI strain was found at a higher frequency in Southern Taiwan (Dou et al. 2015). This is indicative of the historical migration in Taiwan. Taken together, these studies reveal the potential role of the interaction between host genetic and pathogen lineages in the heterogeneity of association analyses of TB susceptibility. Therefore, future studies should consider sample stratification by clades to avoid confounding effect due to differences in TB strain lineages.
Meta-analysis of TB GWAS
The International TB Host Genetics Consortium (ITHGC) performed a multi-ancestry meta-analysis of TB GWAS using 12 case–control data sets with a total of 14153 TB cases and 19536 controls (Schurz et al. 2022). Study participants were from China (two unpublished datasets and (Qi et al. 2017)), Thailand (Mahasirimongkol et al. 2012), Japan (Mahasirimongkol et al. 2012), Russia (Curtis et al. 2015), Estonia (unpublished data), Germany (unpublished data), Gambia (Wellcome Trust Case Control Consortium 2007), Ghana (Thye et al. 2010) and South Africa (two datasets) (Daya et al. 2014a; Schurz et al. 2018). Only three of these (South Africa, China and Thailand) are included in the WHO’s global list (Geneva: World Health Organization 2021) of high burden countries for TB (Fig. 1). Polygenic heritability estimates performed by Schurz et al. ranged from 5 to 36% (average of 26%) suggesting that besides host genetic factors, environmental, socio-economic factors and genetic ancestry contribute significantly to TB susceptibility. This has been demonstrated by the near elimination of TB in several countries that experienced economic development and improved public health action. Interestingly, only one association, rs28383206 in the HLA region, was identified at genome-wide significance, albeit with a high level of heterogeneity. Upon further investigation of the HLA locus, multiple epitopes significantly associated with TB were identified, and these associations had less heterogeneity and consistent directions of effects. As was evident from the heritability analysis, this consistent direction of effects once again suggests a degree of shared TB susceptibility at a global scale. Even so, dominant epitopes, which refers to the part of the antigen molecule that is easily recognized by the immune system and therefore influences the specificity of the induced antibody, varied between the input studies. Taken together, the ITHGC results confirm that there is a complex shared genetic architecture to TB susceptibility and reaffirms that additional large-scale GWAS, including cohorts from multiple ancestries and regions with different infection pressures, are required.

World map indicating countries involved in the International TB Host Genetics consortium and the 30 high burden countries for TB as identified by the World Health Organization in 2021. Only three of these (South Africa, China and Thailand) are included in both.
Ancestry and TB susceptibility
Differential demographic histories, geographic dispersal and pathogen exposures have led to distinct capacities in innate immunity and infection control among human populations (Quach and Quintana‑Murci 2017). For instance, centuries of exposure to TB in densely populated Europe has likely resulted in adaptive selection for TB resistance, whereas groups that were otherwise previously geographically isolated were only relatively recently exposed to virulent European Mtb strains, thus fostering the spread and increased TB mortality in those regions (Comas et al. 2015; Uren et al. 2017b). There are both evolutionary and epidemiological evidence that support the notion that genetic ancestry influences risk disparity across populations (Barreiro and Quintana‑Murci 2010; Manry et al. 2011; Deschamps et al. 2016; Nédélec et al. 2016; Nahid et al. 2018).
Several studies of admixed individuals have investigated and attested to genetic ancestry as a factor that affects susceptibility to TB. Using a cohort of 733 South African multi-way admixed individuals, Chimusa et al. observed a significant positive correlation between African Khoe-San ancestry and TB susceptibility (OR = 0.16, 95% CI [0.0.9–0.23]; P = 1.58 × 10−5) and negative correlations with non-African ancestry proportions (Chimusa et al. 2014) with no differences in socioeconomic status between the cases and controls. Leal and colleagues also noted an increased TB susceptibility corresponding to an increase in the range of Amerindian ancestry proportion in an Amazonian population (n = 418) (Leal et al. 2020). This is in line with results from Asgari et al. who observed a 25% increase in TB progression risk for a 10% increase in Peruvian ancestry proportion in their cohort understudy (Asgari et al. 2022). These disparities could, in part, be explained by the fact that some populations have been exposed to virulent strains of Mtb longer than others (Hoal 2002; Comas et al. 2015; Hoal et al. 2018). In these populations, the genetic variants that had previously contributed to disease susceptibility may have been removed from the gene pool by natural selection (Hoal 2002; Daya et al. 2014b). Together, these studies lend credence to the role played by genetic ancestry variations in TB susceptibility and, more importantly, to the importance of including diverse populations in genetic investigations (Petersen et al. 2022).
Although the results of these studies are in alignment with previous epidemiological observations, they should be interpreted with caution in order to avoid the stigmatization and social exclusion of certain population groups. It is important to note that genetic ancestry alone does not explain all the differences in TB-incidence rate observed between population groups. Other aspects are likely contributors of the phenomenon. Firstly, TB is common in resource-constrained countries which means that in (most part of) those regions, access to adequate health care is limited, thus undeniably contributing to high disease burden in the populations. Secondly, in the event that health care is accessible, cultural, language, health knowledge and stigma associated with TB may discourage some to seek medical attention. Thirdly, should those individuals get the medical care they need, TB treatment regimen is lengthy and can last at least 6 months for drug sensitive TB treatment (given 3 times/day for 7 days/week) and could be twice as long for multi drug-resistant TB treatment (Winston and Mitruka 2012; Chirehwa et al. 2019). Coupled with the issue of adverse drug reactions which present a wild range of symptoms, patients are often reluctant or unable to complete treatment which can lead to the surge of drug resistance TB-strains, thus adding on to the burden of disease in the region. Lastly, current genetic study design may skew the results due to both inadequate sample sizes used and poor phenotypic characterization (Möller and Kinnear 2020). Moreover, good genetic ancestry markers are lacking for populations that are underrepresented in genomic studies (i.e. African populations and other non-European population groups) which could lead to misunderstanding of ancestry population structure (Campbell and Tishkoff 2008; Daya et al. 2013; Bentley et al. 2020). All of the factors mentioned above could easily contribute to the disparities observed in TB incidence rate across global populations.
Polygenic risk scores
Advances in the field of precision medicine have allowed for the exploration of polygenic risk scores (PRS) to assess the relative risk of developing a disease, which can in turn provide more specific patients genetic risk information as well as tailored approaches to healthcare. Multiple studies have shown evidence for the ability of PRS to stratify individuals based on their genetic risk of developing a disease (Abraham et al. 2014; Mavaddat et al. 2019; Sharp et al. 2019; Willoughby et al. 2019; Cánovas et al. 2020). However, the clinical utility of PRS have disproportionally been assessed in European populations and for non-communicable diseases (e.g. breast cancers, cardiovascular diseases) and shown limited portability to GWAS-underrepresented populations, such as African and admixed individuals (De La Vega and Bustamante 2018; Grinde et al. 2019; Martin et al. 2019; Cavazos and Witte 2021). In addition, methodological approaches to PRS calculation have proven to be challenging, particularly for admixed individuals (Márquez‑Luna et al. 2017; Bitarello and Mathieson 2020). To bridge the gap between risk prediction for non-communicable and infectious diseases, to our knowledge, only one study that used GWAS-derived PRSs for prediction of TB susceptibility has been published, to date (Hong et al. 2017). Hong and colleagues achieved an in-sample predictive performance AUC of 0.69 (69%) using 10 SNPs from GWAS of 679 individuals of Korean descent. However, the model was unable to successfully stratify between cases and controls. Combining the PRS-only model with factors that influence the progression of active TB (e.g. sex, body mass index (BMI), cigarette smoking, systemic blood pressure and haemoglobin) increased the accuracy obtained (AUC = 0.80 versus 0.69 from PRS-only model). This substantial increase allowed for the stratification of individuals based on relative risks of developing active TB from a latent stage and observed a 3.7-fold increased risk of TB outcome in the top quartile of the distribution compared to the bottom quartile. This is in agreement with previous reported studies on PRS that indicated an improvement in predictive power by combining the effects of SNPs with modifiable/environmental risk factors and family history (Mavaddat et al. 2019; Kachuri et al. 2020). Although these results have shown an insightful trajectory for genetic risk prediction of TB, they cannot be extrapolated to other more genetically diverse populations. Lack of portability is a key challenge of PRS. This have been credited to genetic drift as a result of the bottle neck effect during the “Out-of-Africa” expansion, variant frequency and LD pattern differences (which are known to be population specific) and the effect of gene–gene and gene-environment interactions (Martin et al. 2017; Duncan et al. 2019). Additionally, PRS is constrained by the statistical power of GWAS; therefore, as GWAS sample sizes increase, the predictive power of PRS will also be expected to increase until it reaches the limit determined by the disease/trait heritability (Choi et al. 2020). For the implementation of PRS in clinical settings that ensure a truly representative and equitable PRS clinical utility, both methods development and recruitments of more diverse populations are absolute prerequisite (Martin et al. 2017, 2019; Khera et al. 2018; Cavazos and Witte 2021).
Conclusion
Given the considerable and continuing progress in genomic techniques, it is clear that human genetics is an effective strategy for identifying the molecules and circuits that are really important in the response to Mtb infection and that contribute to progression to TB. As noted in this review, much has been done in the field of host susceptibility to TB and identifying the genetic risk factors associated with this phenotype. However, much still needs to be done especially with regard to replication of study findings and discovery of novel associations. A step towards reducing these setbacks and improving genetic risk prediction should include further exploring the effects of sex-, age- and population-specific on genetic susceptibility to TB. Future studies should also consider sample stratification by clades to avoid confounding effect due to differences in infection with TB strain lineages. Additional emphasis should also be placed on the use of high throughput sequencing methods such as whole exome sequencing and whole genome sequencing.
The search for genetic factors predisposing to TB is fundamental from both an immunological and medical point of view. This will allow a better understanding of the biological mechanisms involved in the immune response to mycobacteria and in the clinical expression of the disease under natural conditions of infection. From a medical point of view, there are direct implications in terms of diagnosis and prognosis and more generally, the ability to distinguish between TB susceptible and resistant individuals will be particularly important for optimal evaluation of trials of new vaccines or treatments against TB.
References
Abraham G, Tye-Din JA, Bhalala OG et al (2014) Accurate and robust genomic prediction of celiac disease using statistical learning. PLoS Genet 10:e1004137. https://doi.org/10.1371/journal.pgen.1004137
Adams LA, Möller M, Nebel A et al (2011) Polymorphisms in MC3R promoter and CTSZ 3’UTR are associated with tuberculosis susceptibility. Eur J Hum Genet 19:676–681. https://doi.org/10.1038/ejhg.2011.1
Agbayani G, Clark K, Sad S et al (2022) Roles of natural resistance-associated macrophage protein-1 in modulating bacterial distribution and immune responses during Salmonella enterica serovar Typhimurium infection in murine pregnancy. Am J Reprod Immunol. https://doi.org/10.1111/aji.13599
Asgari S, Luo Y, Huang CC et al (2022) Higher native Peruvian genetic ancestry proportion is associated with tuberculosis progression risk. Cell Genomics 2. https://doi.org/10.1016/j.xgen.2022.100151
Baghdadi JE, Orlova M, Alter A et al (2006) An autosomal dominant major gene confers predisposition to pulmonary tuberculosis in adults. J Exp Med 203:1679–1684. https://doi.org/10.1084/jem.20060269
Bahari G, Hashemi M, Taheri M et al (2012) Association of IRGM polymorphisms and susceptibility to pulmonary tuberculosis in Zahedan, Southeast Iran. Sci World J 950801. https://doi.org/10.1100/2012/950801
Baker AR, Zalwango S, Malone LL et al (2011) Genetic susceptibility to tuberculosis associated with cathepsin Z haplotype in a Ugandan household contact study. Hum Immunol 72:426–430. https://doi.org/10.1016/j.humimm.2011.02.016
Baron M (2001) The search for complex disease genes: fault by linkage or fault by association? Mol Psychiatry 6:143–149. https://doi.org/10.1038/sj.mp.4000845
Barreiro LB, Quintana-Murci L (2010) From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11:17–30. https://doi.org/10.1038/nrg2698
Bellamy R (2000) Identifying genetic susceptibility factors for tuberculosis in Africans: a combined approach using a candidate gene study and a genome-wide screen. Clin Sci 98:245–250
Bellamy R, Beyers N, McAdam KP et al (2000) Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc Natl Acad Sci USA 97:8005–8009. https://doi.org/10.1073/pnas.140201897
Bellamy R, Ruwende C, Corrah T et al (1998) Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med 338:640–644. https://doi.org/10.1056/NEJM199803053381002
Bentley AR, Callier SL, Rotimi CN (2020) Evaluating the promise of inclusion of African ancestry populations in genomics. NPJ Genom Med 5:5. https://doi.org/10.1038/s41525-019-0111-x
Bitarello BD, Mathieson I (2020) Polygenic scores for height in admixed populations. G3 (Bethesda) 10:4027–4036. https://doi.org/10.1534/g3.120.401658
Bogen E (1947) Streptomycin treatment of tuberculosis. Am Rev Tuberc 56:442–444. https://doi.org/10.1164/art.1947.56.5.442
Brites D, Gagneux S (2015) Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev 264:6–24. https://doi.org/10.1111/imr.12264
Buniello A, MacArthur JAL, Cerezo M et al (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47:D1005–D1012. https://doi.org/10.1093/nar/gky1120
Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L (2014) Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol 26:454–470. https://doi.org/10.1016/j.smim.2014.09.008
Calmette A (1927) Sur la vaccination préventive des enfants nouveau-nés contre la tuberculose par le BCG. Ann. de l'Inst. Pasteur.
Campbell MC, Tishkoff SA (2008) African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 9:403–433. https://doi.org/10.1146/annurev.genom.9.081307.164258
Canonne-Hergaux F, Gruenheid S, Govoni G, Gros P (1999) The Nramp1 protein and its role in resistance to infection and macrophage function. Proc Assoc Am Physicians 111:283–289. https://doi.org/10.1046/j.1525-1381.1999.99236.x
Cánovas R, Cobb J, Brozynska M et al (2020) Genomic risk scores for juvenile idiopathic arthritis and its subtypes. Ann Rheum Dis 79:1572–1579. https://doi.org/10.1136/annrheumdis-2020-217421
Cavazos TB, Witte JS (2021) Inclusion of variants discovered from diverse populations improves polygenic risk score transferability. HGG Adv 2. https://doi.org/10.1016/j.xhgg.2020.100017
Caws M, Thwaites G, Dunstan S et al (2008) The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog 4:e1000034. https://doi.org/10.1371/journal.ppat.1000034
Cellier MF, Courville P, Campion C (2007) Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect 9:1662–1670. https://doi.org/10.1016/j.micinf.2007.09.006
Chimusa ER, Zaitlen N, Daya M et al (2014) Genome-wide association study of ancestry-specific TB risk in the South African Coloured population. Hum Mol Genet 23:796–809. https://doi.org/10.1093/hmg/ddt462
Chirehwa MT, McIlleron H, Wiesner L et al (2019) Effect of efavirenz-based antiretroviral therapy and high-dose rifampicin on the pharmacokinetics of isoniazid and acetyl-isoniazid. J Antimicrob Chemother 74:139–148. https://doi.org/10.1093/jac/dky378
Choi SW, Mak TS-H, O’Reilly PF (2020) Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 15:2759–2772. https://doi.org/10.1038/s41596-020-0353-1
Cobat A, Gallant CJ, Simkin L et al (2009) Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med 206:2583–2591. https://doi.org/10.1084/jem.20090892
Comas I, Hailu E, Kiros T et al (2015) Population genomics of Mycobacterium tuberculosis in Ethiopia contradicts the virgin soil hypothesis for human tuberculosis in Sub-Saharan Africa. Curr Biol 25:3260–3266. https://doi.org/10.1016/j.cub.2015.10.061
Cooke GS, Campbell SJ, Bennett S et al (2008) Mapping of a novel susceptibility locus suggests a role for MC3R and CTSZ in human tuberculosis. Am J Respir Crit Care Med 178:203–207. https://doi.org/10.1164/rccm.200710-1554OC
Curtis J, Luo Y, Zenner HL et al (2015) Susceptibility to tuberculosis is associated with variants in the ASAP1 gene encoding a regulator of dendritic cell migration. Nat Genet 47:523–527. https://doi.org/10.1038/ng.3248
Daya M, van der Merwe L, Galal U et al (2013) A panel of ancestry informative markers for the complex five-way admixed South African coloured population. PLoS ONE 8:e82224. https://doi.org/10.1371/journal.pone.0082224
Daya M, van der Merwe L, Gignoux CR et al (2014a) Using multi-way admixture mapping to elucidate TB susceptibility in the South African Coloured population. BMC Genomics 15:1021. https://doi.org/10.1186/1471-2164-15-1021
Daya M, van der Merwe L, van Helden PD et al (2014b) The role of ancestry in TB susceptibility of an admixed South African population. Tuberculosis (Edinb) 94:413–420. https://doi.org/10.1016/j.tube.2014.03.012
De La Vega FM, Bustamante CD (2018) Polygenic risk scores: a biased prediction? Genome Med 10:100. https://doi.org/10.1186/s13073-018-0610-x
Deschamps M, Laval G, Fagny M et al (2016) Genomic signatures of selective pressures and introgression from Archaic Hominins at human innate immunity genes. Am J Hum Genet 98:5–21. https://doi.org/10.1016/j.ajhg.2015.11.014
Dou HY, Chen YY, Kou SC, Su IJ (2015) Prevalence of Mycobacterium tuberculosis strain genotypes in Taiwan reveals a close link to ethnic and population migration. J Formos Med Assoc 114:484–488. https://doi.org/10.1016/j.jfma.2014.07.006
Duan Q, Xu Z, Raffield LM et al (2018) A robust and powerful two-step testing procedure for local ancestry adjusted allelic association analysis in admixed populations. Genet Epidemiol 42:288–302. https://doi.org/10.1002/gepi.22104
Duarte TA, Nery JS, Boechat N et al (2017) A systematic review of East African-Indian family of Mycobacterium tuberculosis in Brazil. Braz J Infect Dis 21:317–324. https://doi.org/10.1016/j.bjid.2017.01.005
Duncan L, Shen H, Gelaye B et al (2019) Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 10:3328. https://doi.org/10.1038/s41467-019-11112-0
Ejo M, Hassane-Harouna S, Souleymane MB et al (2020) Multidrug-resistant patients receiving treatment in Niger who are infected with M. tuberculosis Cameroon family convert faster in smear and culture than those with M. tuberculosis Ghana family. Tuberculosis (Edinb) 122:101922. https://doi.org/10.1016/j.tube.2020.101922
El Baghdadi J, Grant AV, Sabri A et al (2013) Human genetics of tuberculosis. Pathol Biol 61:11–16. https://doi.org/10.1016/j.patbio.2013.01.004
Elgueta R, Benson MJ, de Vries VC et al (2009) Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229:152–172. https://doi.org/10.1111/j.1600-065X.2009.00782.x
Elpidorou M, Best S, Poulter JA et al (2021) Novel loss-of-function mutation in HERC2 is associated with severe developmental delay and paediatric lethality. J Med Genet 58:334–341. https://doi.org/10.1136/jmedgenet-2020-106873
Fonseca KL, Rodrigues PNS, Olsson IAS, Saraiva M (2017) Experimental study of tuberculosis: From animal models to complex cell systems and organoids. PLoS Pathog 13:e1006421. https://doi.org/10.1371/journal.ppat.1006421
Fortin A, Abel L, Casanova JL, Gros P (2007) Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genomics Hum Genet 8:163–192. https://doi.org/10.1146/annurev.genom.8.080706.092315
Fox GJ, Orlova M, Schurr E (2016) Tuberculosis in newborns: the lessons of the “Lübeck Disaster” (1929–1933). PLoS Pathog 12:e1005271. https://doi.org/10.1371/journal.ppat.1005271
Françoise U, Lafont E, Suarez F et al (2022) Disseminated Cryptococcosis in a patient with CD40 ligand deficiency. J Clin Immunol. https://doi.org/10.1007/s10875-022-01329-y
Gagneux S, Small PM (2007) Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 7:328–337. https://doi.org/10.1016/S1473-3099(07)70108-1
Geneva: World Health Organization (2021) Global Tuberculosis Report 2021. WHO
Geneva: World Health Organization (2022) Global tuberculosis report 2022. WHO
Gradmann C (2001) Robert Koch and the pressures of scientific research: tuberculosis and tuberculin. Med Hist 45:1–32
Grant AV, Sabri A, Abid A et al (2016) A genome-wide association study of pulmonary tuberculosis in Morocco. Hum Genet 135(3):299–307. https://doi.org/10.1007/s00439-016-1633-2. PMID: 26767831; PMCID: PMC5042142
Green AM, Difazio R, Flynn JL (2013) IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol 190:270–277. https://doi.org/10.4049/jimmunol.1200061
Greenwood CM, Fujiwara TM, Boothroyd LJ et al (2000) Linkage of tuberculosis to chromosome 2q35 loci, including NRAMP1, in a large aboriginal Canadian family. Am J Hum Genet 67:405–416. https://doi.org/10.1086/303012
Grinde KE, Qi Q, Thornton TA et al (2019) Generalizing polygenic risk scores from Europeans to Hispanics/Latinos. Genet Epidemiol 43:50–62. https://doi.org/10.1002/gepi.22166
Hoal EG (2002) Human genetic susceptibility to tuberculosis and other mycobacterial diseases. IUBMB Life 53:225–229. https://doi.org/10.1080/15216540212644
Hoal EG, Dippenaar A, Kinnear C et al (2018) The arms race between man and Mycobacterium tuberculosis: time to regroup. Infect Genet Evol 66:361–375. https://doi.org/10.1016/j.meegid.2017.08.021
Holder A, Garty R, Elder C et al (2020) Analysis of genetic variation in the bovine SLC11A1 gene, its influence on the expression of NRAMP1 and potential association with resistance to bovine tuberculosis. Front Microbiol 11:1420. https://doi.org/10.3389/fmicb.2020.01420
Hong EP, Go MJ, Kim H-L, Park JW (2017) Risk prediction of pulmonary tuberculosis using genetic and conventional risk factors in adult Korean population. PLoS ONE 12:e0174642. https://doi.org/10.1371/journal.pone.0174642
Houben RMGJ, Dodd PJ (2016) The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med 13:e1002152. https://doi.org/10.1371/journal.pmed.1002152
Intemann CD, Thye T, Niemann S et al (2009) Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5:e1000577. https://doi.org/10.1371/journal.ppat.1000577
Jamieson SE, Miller EN, Black GF et al (2004) Evidence for a cluster of genes on chromosome 17q11-q21 controlling susceptibility to tuberculosis and leprosy in Brazilians. Genes Immun 5:46–57. https://doi.org/10.1038/sj.gene.6364029
Jouanguy E, Altare F, Lamhamedi S et al (1996) Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med 335:1956–1961. https://doi.org/10.1056/NEJM199612263352604
Kachuri L, Graff RE, Smith-Byrne K et al (2020) Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat Commun 11:6084. https://doi.org/10.1038/s41467-020-19600-4
Kallmann FJ, Reisner D (1943) Twin studies on the significance of genetic factors in tuberculosis1,2. Am Rev Tuberc 47:549–574
Kettaneh A, Seng L, Tiev KP et al (2006) Human leukocyte antigens and susceptibility to tuberculosis: a meta-analysis of case-control studies. Int J Tuberc Lung Dis 10:717–725
Khera AV, Chaffin M, Aragam KG et al (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50:1219–1224. https://doi.org/10.1038/s41588-018-0183-z
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247. https://doi.org/10.1038/ng1195-241
Leal DF da VB, Santana da Silva MN, Fernandes DCR de O et al (2020) Amerindian genetic ancestry as a risk factor for tuberculosis in an amazonian population. PLoS ONE 15:e0236033. https://doi.org/10.1371/journal.pone.0236033
Leal DF da VB, Santana da Silva MN, Pastana LF et al (2022) Genetic variants of microrna and DROSHA genes in association with the risk of tuberculosis in the amazon population. Front Genet 13:850058. https://doi.org/10.3389/fgene.2022.850058
Luo Y, Suliman S, Asgari S et al (2019) Early progression to active tuberculosis is a highly heritable trait driven by 3q23 in Peruvians. Nat Commun 10:3765. https://doi.org/10.1038/s41467-019-11664-1
Mahasirimongkol S, Yanai H, Mushiroda T et al (2012) Genome-wide association studies of tuberculosis in Asians identify distinct at-risk locus for young tuberculosis. J Hum Genet 57:363–367. https://doi.org/10.1038/jhg.2012.35
Mangalam AK, Rajagopalan G, Taneja V, David CS (2008) HLA class II transgenic mice mimic human inflammatory diseases. Adv Immunol 97:65–147. https://doi.org/10.1016/S0065-2776(08)00002-3
Manry J, Laval G, Patin E et al (2011) Evolutionary genetic dissection of human interferons. J Exp Med 208:2747–2759. https://doi.org/10.1084/jem.20111680
Márquez-Luna C, Loh PR (2017) South Asian Type 2 Diabetes (SAT2D) Consortium, Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol 41:811–823. https://doi.org/10.1002/gepi.22083
Martin AR, Gignoux CR, Walters RK et al (2017) Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet 100:635–649. https://doi.org/10.1016/j.ajhg.2017.03.004
Martin AR, Kanai M, Kamatani Y et al (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51:584–591. https://doi.org/10.1038/s41588-019-0379-x
Maurer U, Jehan F, Englert C et al (2001) The Wilms’ tumor gene product (WT1) modulates the response to 1,25-dihydroxyvitamin D3 by induction of the vitamin D receptor. J Biol Chem 276:3727–3732. https://doi.org/10.1074/jbc.M005292200
Mavaddat N, Michailidou K, Dennis J et al (2019) Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet 104:21–34. https://doi.org/10.1016/j.ajhg.2018.11.002
Mohammadi A, Khanbabaei H, Nasiri-Kalmarzi R et al (2020) Vitamin D receptor ApaI (rs7975232), BsmI (rs1544410), Fok1 (rs2228570), and TaqI (rs731236) gene polymorphisms and susceptibility to pulmonary tuberculosis in an Iranian population: a systematic review and meta-analysis. J Microbiol Immunol Infect 53:827–835. https://doi.org/10.1016/j.jmii.2019.08.011
Möller M, de Wit E, Hoal EG (2010) Past, present and future directions in human genetic susceptibility to tuberculosis. FEMS Immunol Med Microbiol 58:3–26. https://doi.org/10.1111/j.1574-695X.2009.00600.x
Möller M, Kinnear CJ, Orlova M et al (2018) Genetic resistance to Mycobacterium tuberculosis infection and disease. Front Immunol 9:2219. https://doi.org/10.3389/fimmu.2018.02219
Möller M, Kinnear CJ (2020) Human global and population-specific genetic susceptibility to Mycobacterium tuberculosis infection and disease. Curr Opin Pulm Med 26:302–310. https://doi.org/10.1097/MCP.0000000000000672
Möller M, Nebel A, Valentonyte R et al (2009) Investigation of chromosome 17 candidate genes in susceptibility to TB in a South African population. Tuberculosis (edinb) 89:189–194. https://doi.org/10.1016/j.tube.2008.10.001
Morice-Picard F, Benard G, Rezvani HR et al (2016) Complete loss of function of the ubiquitin ligase HERC2 causes a severe neurodevelopmental phenotype. Eur J Hum Genet 25:52–58. https://doi.org/10.1038/ejhg.2016.139
Müller SJ, Schurz H, Tromp G et al (2021) A multi-phenotype genome-wide association study of clades causing tuberculosis in a Ghanaian- and South African cohort. Genomics 113:1802–1815. https://doi.org/10.1016/j.ygeno.2021.04.024
Nahid P, Jarlsberg LG, Kato-Maeda M et al (2018) Interplay of strain and race/ethnicity in the innate immune response to M. tuberculosis. PLoS ONE 13:e0195392. https://doi.org/10.1371/journal.pone.0195392
Nebenzahl-Guimaraes H, Verhagen LM, Borgdorff MW, van Soolingen D (2015) Transmission and progression to disease of Mycobacterium tuberculosis phylogenetic lineages in the Netherlands. J Clin Microbiol 53:3264–3271. https://doi.org/10.1128/JCM.01370-15
Nédélec Y, Sanz J, Baharian G et al (2016) Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167:657-669.e21. https://doi.org/10.1016/j.cell.2016.09.025
Nordquist H, Jamil RT (2022) Biochemistry, HLA Antigens. In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Oki NO, Motsinger-Reif AA, Antas PR et al (2011) Novel human genetic variants associated with extrapulmonary tuberculosis: a pilot genome wide association study. BMC Res Notes 4:28. https://doi.org/10.1186/1756-0500-4-28
Omae Y, Toyo-Oka L, Yanai H et al (2017) Pathogen lineage-based genome-wide association study identified CD53 as susceptible locus in tuberculosis. J Hum Genet 62:1015–1022. https://doi.org/10.1038/jhg.2017.82
Ottenhoff THM, Verreck FAW, Hoeve MA, van de Vosse E (2005) Control of human host immunity to mycobacteria. Tuberculosis (edinb) 85:53–64. https://doi.org/10.1016/j.tube.2004.09.011
Pan H, Yan B-S, Rojas M et al (2005) Ipr1 gene mediates innate immunity to tuberculosis. Nature 434:767–772. https://doi.org/10.1038/nature03419
Petersen DC, Steyl C, Scholtz D et al (2022) African genetic representation in the context of SARS-CoV-2 infection and COVID-19 severity. Front Genet 13:909117. https://doi.org/10.3389/fgene.2022.909117
Png E, Alisjahbana B, Sahiratmadja E et al (2012) A genome wide association study of pulmonary tuberculosis susceptibility in Indonesians. BMC Med Genet 13:5. https://doi.org/10.1186/1471-2350-13-5. PMID: 22239941; PMCID: PMC3287960
Puffer RR (1944) Familial susceptibility to tuberculosis: its importance as a public health problem. Cambridge, MA: Harvard Univ. Press 106pp. https://doi.org/10.4159/harvard.9780674428720
Qi H, Zhang Y-B, Sun L et al (2017) Discovery of susceptibility loci associated with tuberculosis in Han Chinese. Hum Mol Genet 26:4752–4763. https://doi.org/10.1093/hmg/ddx365
Quach H, Quintana-Murci L (2017) Living in an adaptive world: genomic dissection of the genus Homo and its immune response. J Exp Med 214:877–894. https://doi.org/10.1084/jem.20161942
Quistrebert J, Orlova M, Kerner G et al (2021) Genome-wide association study of resistance to Mycobacterium tuberculosis infection identifies a locus at 10q26.2 in three distinct populations. PLoS Genet 17:e1009392. https://doi.org/10.1371/journal.pgen.1009392
Ruiz-Bedoya CA, Mota F, Tucker EW et al (2022) High-dose rifampin improves bactericidal activity without increased intracerebral inflammation in animal models of tuberculous meningitis. J Clin Invest 132. https://doi.org/10.1172/JCI155851
Salie M, van der Merwe L, Möller M et al (2014) Associations between human leukocyte antigen class I variants and the Mycobacterium tuberculosis subtypes causing disease. J Infect Dis 209:216–223. https://doi.org/10.1093/infdis/jit443
Schurz H, Kinnear CJ, Gignoux C et al (2018) A sex-stratified genome-wide association study of tuberculosis using a multi-ethnic genotyping array. Front Genet 9:678. https://doi.org/10.3389/fgene.2018.00678
Schurz H, Naranbhai V, Yates T et al (2022) Multi-ancestry meta-analysis of host genetic susceptibility to tuberculosis identifies shared genetic architecture. medRxiv 2022.08.26.22279009. https://doi.org/10.1101/2022.08.26.22279009
Schurz H, Salie M, Tromp G et al (2019) The X chromosome and sex-specific effects in infectious disease susceptibility. Hum Genomics 13:2. https://doi.org/10.1186/s40246-018-0185-z
Sharp SA, Rich SS, Wood AR et al (2019) Development and standardization of an improved type 1 diabetes genetic risk score for use in newborn screening and incident diagnosis. Diabetes Care 42:200–207. https://doi.org/10.2337/dc18-1785
Singh AK, Gupta UD (2018) Animal models of tuberculosis: lesson learnt. Indian J Med Res 147:456–463. https://doi.org/10.4103/ijmr.IJMR_554_18
Sobota RS, Stein CM, Kodaman N et al (2016) A locus at 5q33.3 confers resistance to tuberculosis in highly susceptible individuals. Am J Hum Genet 98(3):514–524. https://doi.org/10.1016/j.ajhg.2016.01.015. PMID: 26942285; PMCID: PMC4800052
Sriram U, Selvaraj P, Kurian SM et al (2001) HLA-DR2 subtypes & immune responses in pulmonary tuberculosis. Indian J Med Res 113:117–124
Stead WW, Senner JW, Reddick WT, Lofgren JP (1990) Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N Engl J Med 322:422–427. https://doi.org/10.1056/NEJM199002153220702
Stein CM, Zalwango S, Malone LL et al (2008) Genome scan of M. tuberculosis infection and disease in Ugandans. PLoS ONE 3:e4094. https://doi.org/10.1371/journal.pone.0004094
Sveinbjornsson G, Gudbjartsson DF, Halldorsson BV et al (2016) HLA class II sequence variants influence tuberculosis risk in populations of European ancestry. Nat Genet 48:318–322. https://doi.org/10.1038/ng.3498
Swart Y, Uren C, van Helden PD et al (2021) Local ancestry adjusted allelic association analysis robustly captures tuberculosis susceptibility loci. Front Genet 12:716558. https://doi.org/10.3389/fgene.2021.716558
Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295. https://doi.org/10.1101/cshperspect.a016295
Teare MD (2011) Approaches to genetic linkage analysis. In: Teare M (eds) Genetic epidemiology. Methods in molecular biology, vol 713. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60327-416-6_5
Teteh DK, Dawkins-Moultin L, Hooker S et al (2020) Genetic ancestry, skin color and social attainment: the four cities study. PLoS ONE 15:e0237041. https://doi.org/10.1371/journal.pone.0237041
Thuong NTT, Hawn TR, Chau TTH et al (2012) Epiregulin (EREG) variation is associated with susceptibility to tuberculosis. Genes Immun 13:275–281. https://doi.org/10.1038/gene.2011.83
Thye T, Owusu-Dabo E, Vannberg FO et al (2012) Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat Genet 44:257–259. https://doi.org/10.1038/ng.1080
Thye T, Vannberg FO, Wong SH et al (2010) Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat Genet 42:739–741. https://doi.org/10.1038/ng.639
Uren C, Henn BM, Franke A et al (2017a) A post-GWAS analysis of predicted regulatory variants and tuberculosis susceptibility. PLoS ONE 12:e0174738. https://doi.org/10.1371/journal.pone.0174738
Uren C, Möller M, van Helden PD et al (2017b) Population structure and infectious disease risk in southern Africa. Mol Genet Genomics 292:499–509. https://doi.org/10.1007/s00438-017-1296-2
van der Eijk EA, van de Vosse E, Vandenbroucke JP, van Dissel JT (2007) Heredity versus environment in tuberculosis in twins: the 1950s United Kingdom Prophit Survey Simonds and Comstock revisited. Am J Respir Crit Care Med 176:1281–1288. https://doi.org/10.1164/rccm.200703-435OC
Velez DR, Hulme WF, Myers JL et al (2009) Association of SLC11A1 with tuberculosis and interactions with NOS2A and TLR2 in African-Americans and Caucasians. Int J Tuberc Lung Dis 13:1068–1076
Verhein KC, Vellers HL, Kleeberger SR (2018) Inter-individual variation in health and disease associated with pulmonary infectious agents. Mamm Genome 29:38–47. https://doi.org/10.1007/s00335-018-9733-z
Vidal SM, Malo D, Vogan K et al (1993) Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73:469–485. https://doi.org/10.1016/0092-8674(93)90135-d
Wahyuni TD, Hatta M, Bukhari A et al (2021) Increasing natural resistance associated macrophage protein 1 serum level after Miana treatment in BALB/c induced Klebsiella pneumoniae experimental research. Ann Med Surg (Lond) 65:102262. https://doi.org/10.1016/j.amsu.2021.102262
Wang X, Zhu X, Qin H et al (2011) Adjustment for local ancestry in genetic association analysis of admixed populations. Bioinformatics 27:670–677. https://doi.org/10.1093/bioinformatics/btq709
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678. https://doi.org/10.1038/nature05911
WHO (2021) Impact of the COVID-19 pandemic on TB detection and mortality in 2020. WHO
Wilkinson RJ, Llewelyn M, Toossi Z et al (2000) Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet 355:618–621. https://doi.org/10.1016/S0140-6736(99)02301-6
Willoughby A, Andreassen PR, Toland AE (2019) Genetic testing to guide risk-stratified screens for breast cancer. J Pers Med 9. https://doi.org/10.3390/jpm9010015
Winston CA, Mitruka K (2012) Treatment duration for patients with drug-resistant tuberculosis, United States. Emerging Infect Dis 18:1201–1202. https://doi.org/10.3201/eid1807.120261
Zheng R, Li Z, He F et al (2018) Genome-wide association study identifies two risk loci for tuberculosis in Han Chinese. Nat Commun 9(1):4072. https://doi.org/10.1038/s41467-018-06539-w. PMID: 30287856; PMCID: PMC6172286.
Funding
We acknowledge the support of the DSI-NRF Centre of Excellence for Biomedical Tuberculosis Research, South African Medical Research Council Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ndong Sima, C.A.A., Smith, D., Petersen, D.C. et al. The immunogenetics of tuberculosis (TB) susceptibility. Immunogenetics 75, 215–230 (2023). https://doi.org/10.1007/s00251-022-01290-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-022-01290-5