
Abstract
The mechanism of inactivation of mammalian voltage-gated Na+ channels involves transient interactions between intracellular domains resulting in direct pore occlusion by the IFM motif and concomitant extracellular interactions with the β1 subunit. Navβ1 subunits constitute single-pass transmembrane proteins that form protein–protein associations with pore-forming α subunits to allosterically modulate the Na+ influx into the cell during the action potential of every excitable cell in vertebrates. Here, we explored the role of the intracellular IFM motif of rNav1.4 (skeletal muscle isoform of the rat Na+ channel) on the α-β1 functional interaction and showed for the first time that the modulation of β1 is independent of the IFM motif. We found that: (1) Nav1.4 channels that lack the IFM inactivation particle can undergo a “C-type-like inactivation” albeit in an ultraslow gating mode; (2) β1 can significantly accelerate the inactivation of Nav1.4 channels in the absence of the IFM motif. Previously, we identified two residues (T109 and N110) on the β1 subunit that disrupt the α-β1 allosteric modulation. We further characterized the electrophysiological effects of the double alanine substitution of these residues demonstrating that it decelerates inactivation and recovery from inactivation, abolishes the modulation of steady-state inactivation and induces a current rundown upon repetitive stimulation, thus causing a general loss of function. Our results contribute to delineating the process of the mammalian Na+ channel inactivation. These findings may be relevant to the design of pharmacological strategies, targeting β subunits to treat pathologies associated to Na+ current dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fast and efficient transmission of information among excitable cells through action potentials is initiated by the transient activation of voltage-gated Na+ channels (Navs) upon membrane depolarization. The rapid inactivation (1–2 ms) of these channels is essential to preserve the transmembrane Na+ gradient and to ensure the availability of channels for reactivation and high frequency action potential firing (Catterall 2012). Na+ channel accessory β subunits are absent in invertebrates but are orthologs in teleost fish, amphibians, birds and mammals. They are believed to have originated before the divergence of teleosts and tetrapods and may have played a role in the electric signal specialization and diversification of the first vertebrates (Chopra et al. 2007). The β1 subunit modulates the Na+ current by accelerating the inactivation and recovery from inactivation and modifying the voltage dependence of gating in mammalian Navs, including the skeletal muscle isoform Nav1.4 (Patton et al. 1994; Wallner et al. 1993). This modulation occurs through an extracellular non-covalent interaction between the α and β1 subunits (Chen and Cannon 1995; McCormick et al. 1999; Qu et al. 1999; Zimmer et al. 2002). In vertebrates, an intracellular hydrophobic cluster of residues in between domains III and IV of the α subunit is highly implicated in fast inactivation; three key amino acids, IFM (isoleucine, phenylalanine and methionine), known as the inactivation particle, occlude the pore intracellularly, arresting the inward Na+ current within milliseconds after depolarization (Armstrong and Bezanilla 1973; Catterall 2012; Eaholtz et al. 1994). Here, we studied Na+ channel inactivation with a two-fold objective: (1) to describe the behavior of Nav1.4 in the absence of the IFM inactivation particle (via a triple deletion) and to determine the role of β1 on these channels; (2) to validate previous theoretical work by detailing the electrophysiological effects of double mutant β1-T109A/N110A, otherwise known as β1-TANA (Scior et al. 2015). We found that Nav1.4 channels can undergo inactivation despite the absence of the IFM motif (albeit in an ultraslow gating mode), previously thought to be essential for inactivation (West et al. 1992), displaying an independent but cooperative mechanism. In addition, we show that wild-type (WT) β1 can also significantly accelerate the time constant of inactivation of IFM-deleted Nav1.4 channels. With the use of molecular biology and electrophysiology methods, we describe the general loss of effect produced by β1-TANA, contributing to delineation of the molecular determinants of a drug-targetable interface between Nav1.4 α and β1 subunits.
Materials and methods
Site-directed mutagenesis, deletion and heterologous expression
The alanine substitutions of T109 and N110 were introduced in the rNavβ1 construct (NCBI accession no. M26643), cloned in a pGEMHEnew vector with a single pair of mutagenic primers. Similarly, the deletion of the IFM motif on the rNav1.4 cDNA sequence (Uniprot accession no. P15390) cloned into the pGW1H vector was achieved through a single PCR hybridization using the QuikChange® II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The PCR product was transformed by heat shock into ultracompetent One Shot® bacteria for amplification and nick repair (see Supplementary Materials and methods). After antibiotic selection and culture, the mutated plasmids were obtained using the HiSpeed® Plasmid Purification Kit (Qiagen, Mexico City, Mexico). Successful mutagenesis was confirmed by DNA sequencing with the Applied Biosystems 3730 DNA Analyzer (UNAM, Cuernavaca, Mexico). Adult Xenopus laevis female frogs (Xenopus 1, Dexter, MI, USA) were anesthetized by immersion in 0.2% tricaine (Sigma Chemical, St Louis, MO, USA). Oocytes were surgically removed and placed in OR-2 buffer containing (in mM) 82.5 NaCl, 2.5 KCl, 1 MgCl2 and 5 4-2-hydroxyethylpi-97 perazine-1-ethanesulfonic acid (HEPES), pH 7.6. To remove the follicular membrane the cells were treated with 1.3 mg/ml collagenase. The nuclei of stage V and VI oocytes were injected using a nanoliter automatic injector (model A203XVY, WPI, Sarasota, FL, USA) with 25–30 ng of wild-type rNav1.4 or IFM-deleted rNav1.4 cDNA in a 1:5 ratio of WT or mutant β1. Eggs were then maintained for up to 3 days at 18 °C in ND-96 solution (in mM), 96 NaCl, 2 KCl, 1 MgCl2, 5 HEPES and 1 CaCl2, supplemented with 0.5 mM theophylline, 0.5 mM pyruvate and 50 μg/ml gentamicin. The pH was adjusted to 7.6 with 1 M NaOH. Oocytes in recording chambers were continuously superfused at a flow rate of 500 μl/min with ND-96 solution with 1 mM BaCl2 without CaCl2.
All surgical procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals from the Mexican Council for Animal Care (Norma Oficial Mexicana NOM-062-ZOO-1999) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. This study was approved by the internal ethics committee of the Instituto de Fisiología of the Universidad Autónoma de Puebla. All efforts were made to reduce the number of animals used and minimize animal suffering.
Electrophysiological recording and data analysis
Two electrode voltage-clamp recordings were performed at room temperature (20–22 °C) using an OC-725C amplifier (Warner, New Haven, CT). The electrodes were pulled on a horizontal P-97 puller (Sutter Instruments, Novato, CA) and filled with 3 M KCl with a resistance of 0.6–1.2 MΩ. The Na+ current signals were filtered at 2 kHz, digitized at a sampling rate of 10 kHz by a Digidata 1200 analog-to-digital converter (Axon Instruments, Foster City, CA) and stored on a computer for analysis with pClamp software, version 8.02 (Axon Instruments, Foster City, CA). Channel overexpression on oocytes can generate large currents of ~100 μA, and currents larger than 20 μA generate considerable voltage errors due to potential drop in the series resistance (R s) in the extracellular solution, which equals the sum of the pipette resistance and the access resistance resulting from the pieces of membrane and debris (Baumgartner et al. 1999). An R s value of 40 MΩ will introduce a steady-state error of 10% when clamping a cell of a membrane resistance (R m) of 400 MΩ. To control for quality and minimize voltage-clamping errors we: (1) only record oocytes displaying currents of ≤7 μA; (2) achieve a good R s:R m theoretical ratio by keeping the pipette resistance to a maximum of 1.2 MΩ. Given that X. laevis oocytes have a R m of 0.1–2 MΩ (Sobczak et al. 2010), our maximum steady-state error would be 0.8%, considering only the pipette resistance. (3) The access resistance is manually compensated using positive feedback circuits in the amplifier before each recording, and (4) only oocytes with minimal leak ≤0.1 μA (a maximum leak of less than 1.4%) are included in the study. Na+ currents (I Na) were generated by step depolarizations of 10-mV increments (30-ms durations) from a holding potential of −100 to +50 mV. Because of the abruptness of the I–V relationship at a short range of membrane potentials during channel activation and to confirm that the voltage was efficiently clamped, we applied the same protocol with 5-mV increments and observed a smoother Na+ channel activation (Supplementary Figure S4). Na+ conductances (g Na) were obtained from the peak currents generated by step depolarizations of 10-mV increments (30-ms durations) from a holding potential of −100 to 0 mV and calculated with the equation: g Na = I Na/(E m–E q), where E m was the membrane potential, I Na was the current amplitude, and E q was the equilibrium potential calculated for each cell. Steady-state inactivation data were obtained using a two-pulse protocol. First, a variable voltage conditioning pulse (from −120 to 0 mV, 1000-ms duration) was given from a holding potential of −100 mV and a gap of 2 ms. Then, a test pulse (25-ms duration) at −20 mV was given. The peak current from the second pulse was plotted as a function of the conditioning pulse potential. Activation and steady-state inactivation curves from every group were fitted using the Boltzmann distribution equation: I norm = 1/(1 + exp[(V−V 1/2)/dx] + A, where V was the potential of the voltage pre-pulse, V 1/2 was the half voltage of inactivation, dx was the slope, and A was a residual linear component. The time course of inactivation data from the peak current at −20 mV was fitted to a single exponential equation: y = A 1 exp(−x/τ) + y 0, where A 1 was the relative fraction of current inactivation, τ was the time constant, x was the time, and y 0 was the amplitude of the steady-state component. The recovery from inactivation was examined using a 500-ms conditioning pulse at −20 mV from a holding potential of −100 mV followed by a variable recovery interval (Δt = 1–10,000 ms) and a test pulse at −20 mV. Recovery data from each cell were fitted to a double exponential equation: y = A 1 exp[−x/τ 1] + A 2 exp[−x/τ 2] + y 0, where A 1 and A 2 were relative fractions of the recovery currents, τ 1 and τ 2 were time constants, and y 0 was the amplitude of the steady-state component. The frequency-dependent inhibition was determined by applying trains of 30 ms pulses from a holding potential of −100 mV to a test potential of −20 mV at 1, 2 and 5 Hz. Current amplitudes were normalized with respect to the first pulse. All the currents were analyzed using the pClamp version 10.2 software (Axon Instruments, Foster City, CA). Values were shown as mean ± SEM. Statistical comparisons between mutant and wild-type mean values were carried out using unpaired Student’s t test. A one-way analysis of variance (ANOVA) was used for comparisons of more than two mean values, followed by a pairwise multiple comparisons using the Holm-Sidak method. The graphs were built and fitted using Sigmaplot 11.0 (SPSS, Inc., Chicago, IL) and Origin 8.02 (OriginLab Corp., Northampton, MA, USA).
Homology molecular modeling
Several three-dimensional models of the α subunit of the rat Nav1.4 (Uniprot accession no. P15390) were generated with the SCWRL server (Canutescu et al. 2003) domain per domain excluding the interdomain loop regions and energy minimized under the CHARMM27 force field with MOE 2008.10 (Maier and Labute 2014) in every step. The template used was the 2.7 Å crystal structure of the bacterial sodium channel NavAb (PDB code: 3RVY). The stereochemical and energetic quality was assessed with MOE 2008.10. The 2.5 Å crystal structure of the Na+ channel hβ3 subunit (PDB code 4L1D) was used as a template to build rβ1 models using MODELER 9.12 (Webb and Sali 2014). The lead candidates were energy-minimized, and the stereochemical soundness was assessed with MOE 2008.10. The model's ensemble orientation was modeled with VEGA ZZ 2.3.2 (Pedretti et al. 2004). The prediction of local protein flexibility was performed with PredyFlexy (de Brevern et al. 2012). For more details on the mutant prediction and modeling, see (Scior et al. 2015).
Results
Nav1.4 channels lacking the IFM motif can undergo a “C-type-like” ultraslow inactivation, and this inactivation is significantly accelerated by the presence of the β1 subunit
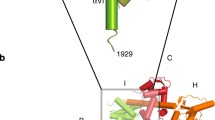
In Fig. 1, we show the mutated residues on the β1 subunit and the location of the IFM motif that was deleted. Co-expression of the Na+ channel α and β1 subunits on frog oocytes is sufficient to reconstitute cation channels that display typical fast-inactivating Na+ currents in native cells (Isom et al. 1992). The inactivation of Nav1.4 channels lacking the IFM motif (hereinafter called Nav1.4-DelIFM) was evidenced by applying depolarizations on the scale of seconds. The co-expression of WT β1 on these channels accelerated the ultraslow inactivation but failed to restore wild-type-like rapid inactivation within the millisecond time scale (Fig. 2). The values of the time constants of inactivation upon 1- and 5-s duration peak voltage depolarizations were significantly decreased when β1 was present (Table 1).

Cartoon illustrating the topology of the sodium channel subunits α and β1, showing the location of the sites that we mutated or deleted. A 3D homology model of β1 is shown in yellow, and the 3D structure of inactivation particle IFM motif is shown in blue (PDB code: 1BYY)

Wild-type β1 accelerates the slow inactivation of IFM-deleted Nav1.4 channels upon long-duration depolarizations. Representative traces evoked by peak voltage depolarizations of 5-s duration of wild-type Nav1.4 channels co-expressed with β1 (in black), Nav1.4 with the deletion of the inactivation gate, the IFM motif in the presence (purple) and absence of the β1 subunit (gray). The inset shows the difference in the inactivation of DelIFM mutant channels (purple), wild-type channels in the presence (black) and absence of β1 (in green) on a shorter time scale
Figure 3a and b illustrates how the currents recorded from oocytes transfected only with the α subunit of Nav1.4 inactivate significantly slower than the currents obtained from the co-transfection of both subunits WT Nav1.4 + β1 (control group). The deletion of the IFM motif in the α subunit co-transfected with β1 produces currents that do not inactivate upon short-duration depolarizations (Fig. 3c). To investigate whether β1 modulation of the inactivation process is unique or cooperative, we decided to focus on the mutant β1-TANA (a double alanine substitution of residues T109 and N110) that we previously identified as a strong candidate to disrupt Na+ inactivation. In Fig. 3d we show that the co-expression of WT α with β1-TANA critically reduced the typical acceleration of fast inactivation that this protein exerts on the Na+ current. Note that this double mutant also induced a significant increase of the sustained current component (arrows in Fig. 2) compared to the control group, measured as the remaining current at the end of the pulse, normalized with respect to the peak current (Table 2).

Co-expression of β1 speeds the inactivation of Nav1.4 currents. Mutant β1-TANA disrupts this effect, and the deletion of the IFM on Nav1.4 channels abolishes short-pulse inactivation. Representative Na+ inward currents of oocytes transfected with a wild-type Nav1.4 and β1, b Nav1.4, c DelIFM Nav1.4 and β1 wild-type, and d wild-type Nav1.4 co-expressed with mutant T109A/N110A β1. The cell membrane was stepped by a standard two-electrode voltage clamp (TVC). Currents were elicited by depolarizing pulses of 30-ms duration, from a holding potential of −100 mV to +50 mV in 10-mV increments. Note that the remaining current components (or sustained components) before the capacitive discharge in b and d are similar (arrows) while the sustained component upon this short depolarizations of wild type a and DelIFM Nav1.4 + β1 c is practically 0 and 100%, respectively
Double mutant Navβ1-TANA causes loss of function on Nav1.4 kinetics
The time constant of inactivation was markedly increased over a range of voltages when Nav1.4 channels were co-expressed with β1-TANA compared to WT channels Nav1.4 + β1 (Fig. 4a), and a delay in the recovery from inactivation was also observed (Fig. 4b). Efficient action potential conduction requires fast Na+ channel recovery from inactivation and channel availability upon high frequency stimulation. Under control conditions, ~50% of Nav1.4 channels co-expressed with wild-type β1 recovered from peak voltage depolarization within the first millisecond, while Nav1.4 + β1-TANA channels took ~4 times longer, severely impairing fast inactivation. The mean time constants of peak voltage inactivation and recovery from fast inactivation (τ fast) of Nav1.4 + β1-TANA channels were significantly different from those of the control group Nav1.4 + β1 (Table 2).

Mutant β1-TANA significantly reduces the allosteric effect of β1 on Nav1.4 channel kinetics. a Time constants of inactivation (peak decay) from short-duration depolarizations, as a function of the membrane voltage step. Statistically significant differences between Nav1.4 + β1 and Nav1.4 + β1-TANA are shown. b Time course of recovery from inactivation obtained by a two-pulse protocol (inset). The relative fraction of channels recovered is plotted as a function of the time elapsed between the first and second pulses and reflects the availability of channels to open, inactivate and re-open at peak voltage (−20 mV). Data were expressed as mean ± SE (n = 4–6, **P < 0.01, ***P < 0.001). Data from each cell were individually fitted to a double exponential equation, and statistically significant differences were found between Nav1.4 + β1 and Nav1.4 + β1-TANA (Table 2)
The functional interaction between α and β1 subunits also allows Nav1.4 channels to recover after short repetitive depolarizations, abolishing the frequency-dependent inhibition that is observed when Nav1.4 channels are expressed in the absence of β1. A rundown of 60, 66 and 79% was observed after repetitive peak voltage stimulation at 1, 2 and 5 Hz, respectively, in recordings from oocytes expressing the α subunit alone. Unlike the control group (Nav1.4-β1), Nav1.4 channels co-expressed with mutant β1-TANA exhibited frequency-dependent inhibition; at these three escalating frequencies the rundowns induced by the mutant were 34, 37 and 41%, respectively (Fig. 5). Note that the rundown is not as significant as when β1 is absent, suggesting that other residues in this auxiliary subunit may interact with the α subunit.

Mutant β1-TANA induces a rundown upon repetitive depolarizations on Nav1.4 channels. Frequency-dependent inhibition was determined by applying repetitive peak depolarizing pulses (−20 mV, 30-ms duration) from a holding potential of −100 mV at a frequency of 1 Hz (a), 2 Hz (b) and 5 Hz (c). Peak currents were normalized to the first pulse, and data were expressed as mean ± SE (n = 4–6). Data from Nav1.4 + β1-TANA were statistically different from Nav1.4 + β1 at 1 Hz (P = 0.001), 2 Hz (P = 0.005) and 5 Hz (P ≤ 0.001)
Double mutant Na+ channel β1-TANA causes loss of function on Nav1.4 voltage dependence
Loss-of-function effects caused by this β1 double mutant were observed on the modulation of voltage dependence that β1 exerts on Nav1.4 channels. The presence of β1 typically shifts the conductance and steady-state inactivation curve to more hyperpolarizing potentials. The β1-TANA mutant significantly impaired both these features (Fig. 6, Table 3). Interestingly, the half voltage of activation significantly increased when these channels were co-expressed with the β1-TANA mutant in a manner comparable to the disruption caused by the deletion of the inactivation particle IFM on Nav1.4 channels. While the half voltages of activation and inactivation of Nav1.4 + β1-TANA and Nav1.4-DelIFM + β1 were significantly different from wild-type Nav1.4 + β1, the slope factors were not (Table 3), which indicates that the channels' sensitivity to voltage has been altered but the ratio of change has not.

Mutant β1-TANA significantly reduces the allosteric effect of β1 on Nav1.4 voltage dependence of activation and steady-state inactivation. The activation curves show the normalized conductance in the right axis as a function of the voltage potential and the steady-state inactivation curves of the same groups in the left axis. Mean ± SEM data were fitted with a Boltzman equation, and the parameters are reported in Table 3
On the other hand, the IFM deletion caused a prominent shift to the depolarizing potentials beyond the behavior of Nav1.4 channels alone and reduced the channel unavailability (~50%) at any prepulse within the activation voltage (diamonds in Fig. 5), which indicates that the deletion prevents channel inactivation during this stimulation protocol.
Discussion
Given the role of β1 in accelerating the inactivation kinetics of most isoforms of Na+ channels, we wondered whether the presence of this integral protein would still be able to accelerate the kinetics of channels lacking the IFM motif, which is necessary to maintain the closure of the fast inactivation gate (West et al. 1992), and this seemed to be the case. β1 was able to speed the process of inactivation whether or not the inactivation particle was present (Fig. 2, Table 1). Balser et al. observed that upon long-duration depolarizations, Nav1.4 channels in which the IFM motif had been substituted by glutamines underwent slow inactivation upon a 5-s peak depolarization (Balser et al. 1996). However, this substitution (QQQ) may generate an electrostatic interaction at the internal mouth of the channel with the polar amide side chain of glutamines. Hence, the issue of whether this glutamine triad could still occlude the intracellular channel pore remains. On the one hand, the slower inactivation of homotetrameric prokaryotic Navs (that inherently lack the IFM motif) is proposed to involve conformational rearrangements of the pore and the pore vestibule on the extracellular side, i.e., C-type inactivation (Irie et al. 2010; Pavlov et al. 2005). On the other hand, it is well established that β1 binds eukaryotic heterotetrameric channels on the extracellular side exerting an allosteric effect on N-type fast inactivation that involves intracellular pore occlusion. Hence, it is plausible that β1 may also play a role in a hypothetical IFM-independent “C-like-type” inactivation in eukaryotic channels, as the results of Balser et al. and our group suggest. Here, the deletion of the IFM motif on this α subunit isoform produced a comparably slow inactivation upon depolarizations of 1- and 5-s duration and the co-expression of β1 accelerated this IFM-independent ultraslow inactivation (see Fig. 2, Table 1). To our knowledge, this is the first study that demonstrates that Nav1.4 channels lacking the IFM motif still undergo inactivation.
Previously, we elaborated on the usefulness of distinguishing analogical structural features in protein–protein interfaces by predicting two functional hotspots (i.e., key residues) in the Nav1.4-β1 functional association. We provided evidence that the designed mutant β1-TANA expressed in a mammalian cell line disrupted β1-like typical channel electrophysiological modulation (Scior et al. 2015). Here we detailed its effects on a different heterologous expression system (haploid amphibian cells) because it is known that some mammalian cells can express endogenous β1 subunits (Islas et al. 2013; Moran et al. 2000).
A great deal of progress has been made in demonstrating the many roles of Navβ1 subunits in health and disease (Patino and Isom 2010). By regulating Na+ channel gating, voltage dependence and kinetics, these subunits decisively contribute to the excitability of cells in vivo (Brackenbury and Isom 2011). This argues in favor of the design of agents that specifically target the α-β1 physical interaction, but in order to do so it is essential to locate the underlying critical structural elements and characterize their biophysical role in channel modulation. This study provides experimental quantitative confirmation of general loss-of-function effects upon mutation of T109 and N110 (β1-TANA) and the deletion of the IFM inactivation particle, dissecting its electrophysiology characteristics and thereby contributing to delineating the functional Nav α-β1 interface (for a discussion of a putative extracellular interface at the α subunit, see Supplementary Figure S1).
β1-TANA was able to significantly disrupt the acceleration of inactivation and recovery from inactivation that the β1 wild type induces on Nav1.4 (Fig. 2b, c) without modifying the activation, peak current membrane potential (−20 mV) or equilibrium potential (~40 mV) (Supplementary Figure S4; Supplementary Table 1). These effects along with the rundown generated upon repetitive stimulations constitute state-dependent reductions in Na+ permeability. State-dependent inhibition is a hallmark of local anesthetics that accounts for their therapeutic efficacy (Starmer et al. 1984); unfortunately, these drugs often produce cardiac side effects (Wolfe and Butterworth 2011). Their binding site is located at a transmembrane cavity at the Nav α subunit, thereby affecting cardiac isoform Nav1.5. This isoform is the least sensitive to β1 (Fozzard and Hanck 1996; Qu et al. 1999), and thus the design of compounds targeting β1 instead of α subunits, which produce use-dependent Na+ current reduction, may circumvent the problem of heart iatrogenesis that local anesthetics possess, given that the presence of β1 affects the potency of Na+ channel inhibitors such as phenytoin (Lucas et al. 2005), carbamazepine (Uebachs et al. 2010) and mefloquine (Paiz-Candia et al. 2016).
Furthermore, β1-TANA completely prevented the effect of β1 voltage-modulation of steady-state inactivation (Fig. 6) and increased the sustained current component, resembling the behavior of Nav1.4 alone. Taken together, these results support the idea that T109 and N110 constitute critical allosteric residues involved in the conformational and dynamical transitions of the Nav1.4 channels. This is in keeping with our prior computational and experimental work on β1 (Islas et al. 2013) (Supplementary figure S3); see the supplementary discussion for structural details and further theoretical discussion in relation to previous studies.
Site-directed mutational studies have advanced the concept of intramolecular communication between the extracellular and intracellular domains, highlighting the importance of residues on the DIV domain of the Nav α subunits that modulate inactivation kinetics on Na+ channels (Vedantham and Cannon 2000; Zarrabi et al. 2010). In the former study, the authors identified two conformational changes near the V1583 residue at transmembrane segment S6 of the DIV domain in Nav1.4; accordingly, they proposed an IFM-dependent and an IFM-independent inactivation mode (Vedantham and Cannon 2000). Our results upon short- and long-duration pulses may reflect the allosteric participation of β1 in both inactivation processes.
Recent crystallographic and in vitro assay studies have boosted the interest in the structure of Na+ channel β subunits (Gilchrist et al. 2013; Namadurai et al. 2014; Kubota et al. 2014; Yereddi et al. 2013), in contrast to the former apparent stagnation in regard to the structural elements underlying the electrophysiological role of β1 (Islas et al. 2013; McCormick et al. 1999). The present results illustrate distinct and critical structure-activity determinants that contribute to the ongoing study of Na+ channels and may be particularly relevant in light of the evidence of β1 as a modifier of the response of some antiepileptic and anesthetic drugs (Uebachs et al. 2010; Wright et al. 1997) as well as for the future prospects of this subunit as a drug target (Brackenbury and Isom 2011).
References
Armstrong CM, Bezanilla F (1973) Currents related to movement of the gating particles of the sodium channels. Nature 242:459–461
Balser JR, Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Marban E, Tomaselli GF (1996) External pore residue mediates slow inactivation in mu 1 rat skeletal muscle sodium channels. J Physiol 494(Pt 2):431–442
Baumgartner W, Islas L, Sigworth FJ (1999) Two-microelectrode voltage clamp of Xenopus oocytes: voltage errors and compensation for local current flow. Biophys J 77:1980–1991
Brackenbury WJ, Isom LL (2011) Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol 2:53
Canutescu AA, Shelenkov AA, Dunbrack RL Jr (2003) A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci Publ Protein Soc 12:2001–2014
Catterall WA (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590:2577–2589
Chen C, Cannon SC (1995) Modulation of Na+ channel inactivation by the beta 1 subunit: a deletion analysis. Pflug Arch 431:186–195
Chopra SS, Watanabe H, Zhong TP, Roden DM (2007) Molecular cloning and analysis of zebrafish voltage-gated sodium channel beta subunit genes: implications for the evolution of electrical signaling in vertebrates. BMC Evol Biol 7:113
de Brevern AG, Bornot A, Craveur P, Etchebest C, Gelly JC (2012) PredyFlexy: flexibility and local structure prediction from sequence. Nucl Acids Res 40:W317–322
Eaholtz G, Scheuer T, Catterall WA (1994) Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron 12:1041–1048
Fozzard HA, Hanck DA (1996) Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev 76:887–926
Gilchrist J, Das S, Van Petegem F, Bosmans F (2013) Crystallographic insights into sodium-channel modulation by the beta4 subunit. Proc Natl Acad Sci USA 110:E5016–5024
Irie K, Kitagawa K, Nagura H, Imai T, Shimomura T, Fujiyoshi Y (2010) Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J Biol Chem 285:3685–3694
Islas AA, Sanchez-Solano A, Scior T, Millan-PerezPena L, Salinas-Stefanon EM (2013) Identification of Navbeta1 residues involved in the modulation of the sodium channel Nav1.4. PLoS One 8:e81995
Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256:839–842
Kubota T, Dang B, Finol-Urdaneta RK, Lacroix JJ, Frezza L, French RJ, Kent SBH, Correa AM, Bezanilla F (2014) Searching for the interaction sites of the beta1 subunit with the voltage-sensing domains of sodium channels using LRET. Biophys J 106(2):133a
Lucas PT, Meadows LS, Nicholls J, Ragsdale DS (2005) An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res 64:77–84
Maier JK, Labute P (2014) Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 82:1599–1610
McCormick KA, Srinivasan J, White K, Scheuer T, Catterall WA (1999) The extracellular domain of the beta1 subunit is both necessary and sufficient for beta1-like modulation of sodium channel gating. J Biol Chem 274:32638–32646
Moran O, Nizzari M, Conti F (2000) Endogenous expression of the beta1A sodium channel subunit in HEK-293 cells. FEBS Lett 473:132–134
Namadurai S, Balasuriya D, Rajappa R, Wiemhofer M, Stott K, Klingauf J, Edwardson JM, Chirgadze DY, Jackson AP (2014) Crystal structure and molecular imaging of the Nav channel beta3 subunit indicates a trimeric assembly. J Biol Chem 289:10797–10811
Paiz-Candia B, Islas AA, Sánchez-Solano A, Mancilla-Simbro C, Scior T, Millan-PerezPeña M, Salinas-Stefanon EM (2016) Mefloquine inhibits voltage dependent Nav1.4 channel by overlapping the local anaesthetic binding site. Euro J Pharmacol (Submitted)
Patino GA, Isom LL (2010) Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett 486:53–59
Patton DE, Isom LL, Catterall WA, Goldin AL (1994) The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J Biol Chem 269:17649–17655
Pavlov E, Bladen C, Winkfein R, Diao C, Dhaliwal P, French RJ (2005) The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys J 89:232–242
Pedretti A, Villa L, Vistoli G (2004) VEGA—an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des 18:167–173
Qu Y, Rogers JC, Chen SF, McCormick KA, Scheuer T, Catterall WA (1999) Functional roles of the extracellular segments of the sodium channel alpha subunit in voltage-dependent gating and modulation by beta1 subunits. J Biol Chem 274:32647–32654
Scior T, Paiz-Candia B, Islas AA, Sanchez-Solano A, Millan-Perez Pena L, Mancilla-Simbro C, Salinas-Stefanon EM (2015) Predicting a double mutant in the twilight zone of low homology modeling for the skeletal muscle voltage-gated sodium channel subunit beta-1 (Nav1.4 beta1). Comput Struct Biotechnol J 13:229–240
Sobczak K, Bangel-Ruland N, Leier G, Weber WM (2010) Endogenous transport systems in the Xenopus laevis oocyte plasma membrane. Methods 51:183–189
Starmer CF, Grant AO, Strauss HC (1984) Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys J 46:15–27
Uebachs M, Opitz T, Royeck M, Dickhof G, Horstmann MT, Isom LL, Beck H (2010) Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J Neurosci Off J Soc Neurosci 30:8489–8501
Vedantham V, Cannon SC (2000) Rapid and slow voltage-dependent conformational changes in segment IVS6 of voltage-gated Na(+) channels. Biophys J 78:2943–2958
Wallner M, Weigl L, Meera P, Lotan I (1993) Modulation of the skeletal muscle sodium channel alpha-subunit by the beta 1-subunit. FEBS Lett 336:535–539
Webb B, Sali A (2014) Comparative protein structure modeling using MODELLER. Curr protoc Bioinform 47:561–563
West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA (1992) A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci USA 89:10910–10914
Wolfe JW, Butterworth JF (2011) Local anesthetic systemic toxicity: update on mechanisms and treatment. Curr Opin Anaesthesiol 24:561–566
Wright SN, Wang SY, Kallen RG, Wang GK (1997) Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophys J 73:779–788
Yereddi NR, Cusdin FS, Namadurai S, Packman LC, Monie TP, Slavny P, Clare JJ, Powell AJ, Jackson AP (2013) The immunoglobulin domain of the sodium channel beta3 subunit contains a surface-localized disulfide bond that is required for homophilic binding. FASEB J 27:568–580
Zarrabi T, Cervenka R, Sandtner W, Lukacs P, Koenig X, Hilber K, Mille M, Lipkind GM, Fozzard HA, Todt H (2010) A molecular switch between the outer and the inner vestibules of the voltage-gated Na+ channel. J Biol Chem 285:39458–39470
Zimmer T, Biskup C, Dugarmaa S, Vogel F, Steinbis M, Bohle T, Wu YS, Dumaine R, Benndorf K (2002) Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J Membr Biol 186:1–12
Acknowledgements
This work was supported by VIEP-BUAP (grant: SCJT-NAT-14-6) to E.S.S and TS and the National Council of Science and Technology of Mexico (CONACyT). In partial fulfillment of the PhD of Alfredo Sánchez-Solano (CONACyT #226507, grant no. 329859). We would like to thank Prof. Thomas Zimmer for kindly providing the β1 clone and the LNS (Laboratorio Nacional de Supercomputo del Sureste de Mexico) for the resources provided.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. Sanchez-Solano and A. A. Islas contribute equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sánchez-Solano, A., Islas, A.A., Scior, T. et al. Characterization of specific allosteric effects of the Na+ channel β1 subunit on the Nav1.4 isoform. Eur Biophys J 46, 485–494 (2017). https://doi.org/10.1007/s00249-016-1193-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-016-1193-3