
Abstract
Venous-related brain injury is a common form of cerebrovascular injury in children and encompasses a diverse group of cerebrovascular diagnoses. The purpose of this pictorial essay is to introduce the relevant anatomy, pathophysiology and various imaging patterns of venous-related cerebral injury in children. Unifying concepts to better understand the effects of venous hypertension in the developing brain will be emphasized. These unifying concepts will provide the imaging professional with a conceptual framework to better understand and confidently identify imaging patterns of venous-related cerebral injury.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The developmental and acquired causes of venous-related cerebral injury comprise a diverse, heterogeneous group of diagnoses. Examples include hemorrhagic venous infarction in premature neonates, congenital angiomatosis (Sturge-Weber syndrome), acute and chronic cerebral venous thrombosis, and congenital and acquired arteriovenous malformations. Despite the disparate origins of these diagnoses, injury to the brain occurs along three common overlapping pathways of venous hypertension, ischemia and hemorrhage. To illustrate the imaging spectrum of acute and chronic venous injury in children, we utilized case material from a tertiary care academic pediatric hospital (Cincinnati Children’s Hospital Medical Center).
Anatomical localization
Understanding the venous drainage pathways of the brain is important to allow accurate diagnosis of venous-related injury and to localize the source of venous disease. This is particularly true in the setting of focal venous disease such as venous thrombosis. Figure 1 demonstrates the major cerebral venous drainage territories. It is important to note that the venous drainage territories of the deep and superficial cerebral veins are variable, although the generalized drainage areas can be estimated [1]. The superior sagittal sinus drainage territory comprises the majority of the superior cerebrum. The sylvian veins drain the peri-insular region and drain into the basal dural sinuses (sphenoparietal and cavernous sinuses.) The transverse sinuses drain blood from the lateral surfaces of the temporal, parietal and occipital lobes. The vein of Labbe´, if dominant, can comprise much of the transverse sinus drainage territory. The deep medullary veins, which are tributaries of the thalamostriate and septal veins, drain the deep gray structures, deep white matter and deep central midline cortex. The thalamostriate and septal veins coalesce into the internal cerebral veins.

Supratentorial venous territories (a–d). Gray superior sagittal sinus drainage territory, white sylvian veins, black transverse sinus territory (vein of Labbe´), speckled white thalamostriate veins (drain to internal cerebral veins), black stripes septal veins (drain to internal cerebral veins) and inferior sagittal sinus
Pathophysiology of venous injury
Venous injury, depending upon its severity, manifests characteristic imaging patterns. The type of imaging manifestation depends on a number of pathological factors, including age, venous pressure, location (i.e. types of draining veins involved), acute versus chronic time frame and availability of collateral drainage pathway [2,3,4]. Figure 2 demonstrates the pathophysiology of acute venous occlusion and insufficiency and the expected imaging patterns. In acute venous occlusion, there is local increased venous pressure that leads to decreased cerebral perfusion pressure (i.e. mean arterial pressure minus venous pressure). The main determinant of whether pathological imaging changes become evident is the adequacy of collateral venous drainage pathways. If such drainage pathways are present, local cerebral perfusion pressure is maintained and no imaging changes become evident. If, however, collateral venous drainage pathways are not present, venous hypertension and decreased perfusion pressure lead to blood-brain barrier breakdown and a variety of imaging findings, including edema (signal abnormality without diffusion restriction), cytotoxic edema (signal abnormality with diffusion restriction) and hemorrhage. Figure 3 demonstrates the pathophysiology of delayed or chronic venous occlusion and insufficiency with the expected imaging patterns. Chronic venous occlusion is seen in different disease processes, but the common imaging findings of atrophy and calcification are the result of similar pathophysiology. As in acute venous occlusion, the most important determinant of whether parenchymal imaging findings become evident is adequacy of collateral venous pathways. However, unlike acute occlusion where collateral pathways must be immediately available, collateral pathways (or lack thereof) in chronic venous occlusion develop over time. If collateral venous pathways successfully develop, as is often seen in chronic venous thrombosis, venous pressures and cerebral perfusion pressures will be maintained and no parenchymal sequelae will be evident. If, however, collateral pathways are inadequate (as in Sturge-Weber syndrome) or venous outflow stenosis develops (as in high flow arteriovenous malformation/arteriovenous fistula), then venous pressures increase and cerebral perfusion pressures decrease leading to ischemia and poor maintenance of the blood brain barrier, which manifests as atrophy and mineralization over time.

Schematic outlines the pathophysiology of acute venous occlusion and insufficiency and expected imaging patterns. AV arteriovenous, BBB blood-brain barrier

Schematic outlining pathophysiology of delayed or chronic venous occlusion and insufficiency with the expected imaging patterns. AVF arteriovenous fistula, AVM arteriovenous malformation, BBB blood-brain barrier
Imaging patterns of venous injury
This section will be divided into acute and chronic patterns of venous injury. To explore the acute imaging patterns (cerebral swelling, signal changes, venous infarction and hemorrhage), we will use cerebral venous sinus thrombosis and medullary venous thrombosis in premature infants as the primary examples. In our exploration of chronic imaging patterns of venous disease (volume loss and mineralization), we will use Sturge-Weber syndrome and arteriovenous malformations as our primary examples. Each of these disease processes can demonstrate both acute and chronic imaging patterns depending on the time frame of the disease.
Acute venous injury
Cerebral swelling and signal changes
Brain swelling occurs when sinus pressures reach 20–25 mmHg. Signal changes begin to occur when sinus pressures reach 32–42 mmHg [4]. The precise pathophysiological mechanisms of cerebral edema in the setting of venous hypertension are not well understood, but proposed models suggest that the primary mechanism is hydrostatic edema, which is supported by animal studies of acute venous occlusion. There is secondary contribution from cell dysfunction due to ischemia [5]. Imaging findings in edema should resolve over time if the primary cause is treated or adequate venous return pathways develop (Fig. 4).

A 10-year-old girl with a headache and papilledema. a Two-dimensional time-of-flight (TOF) MR venography images show thrombosis of the entire superior sagittal sinus (arrows). b Axial fluid attenuated inversion recovery (FLAIR) images show diffuse sulcal effacement from cerebral swelling. c Following anticoagulation, 2-D TOF MR venography images show the superior sagittal sinus (arrows) is recanalized. d Axial FLAIR images demonstrate resolution of previously seen cerebral swelling
Cerebral sinus venous thrombosis is a known cause of diffuse cerebral swelling. Evidence for venous thrombosis should be looked for in this setting. It is typically reversible with therapy, but can be lethal if not identified and treated appropriately. In the setting of acute cerebral sinus venous thrombosis, the amount of collateral flow potential determines the severity of parenchymal sequelae. On follow-up imaging, areas of encephalomalacia are usually much smaller than the initial signal abnormality, reflecting the capacity of collateral drainage pathways to restore adequate perfusion to affected cerebral venous drainage territories (Fig. 5).

A 10-year-old boy with a headache and altered mental status. a Axial CT with contrast shows lack of enhancement in the anterior aspect of the superior sagittal sinus (arrow) and hypoattenuation in the right frontal lobe. b Two-dimensional time-of-flight (TOF) MR venography shows lack of flow-related signal (arrow) in the anterior half of the superior sagittal sinus. c Axial T2-W image shows hyperintense signal, suggesting edema. d A CT performed 7 years later shows encephalomalacia affecting only a small portion of the frontal lobe, suggesting that most of the signal on the MRI represented reversible cerebral edema
Venous infarction
The term venous infarction has been used loosely to describe the entire spectrum of parenchymal manifestations of cerebral sinus venous thrombosis. The term venous infarction may be used when diffusion restriction is present, with the understanding that such imaging findings may be at least partially reversible. Diffusion restriction of brain parenchyma significantly increases the risk of permanent parenchymal sequelae, but many authors have shown that such changes are often reversible in the setting of venous thrombosis [6,7,8,9]. Figure 6 shows an example of a patient with superior sagittal sinus thrombosis and extensive cortical diffusion restriction, which resolved without parenchymal imaging sequelae. Signal changes on diffusion-weighted imaging in this setting are thought to represent a combination of vasogenic and cytotoxic edema [7]. When marked diffusion restriction occurs with associated parenchymal hemorrhage, the risk of permanent imaging changes is much higher. Figure 7 shows an example of infarction and resultant encephalomalacia related to cerebral sinus venous thrombosis.

A 22-year-old man with leukemia and a headache. a Sagittal T1 FSPGR (fast spoiled gradient recalled acquisition in the steady state) with contrast shows a filling defect (arrows) throughout most of the superior sagittal sinus, consistent with thrombosis. b Axial diffusion-weighted image shows bilateral areas of increased cortical signal intensity (arrows). c Axial apparent diffusion coefficient (ADC) image shows corresponding decreased cortical signal, confirming true diffusion restriction (arrows). d Axial fluid attenuated inversion recovery (FLAIR) image 1 week later shows no abnormal signal or volume loss to suggest permanent parenchymal sequelae

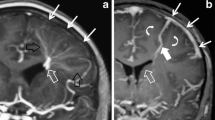
A 3-year-old girl with unequal pupils. a Sagittal CT demonstrates increased attenuation (arrows) in the right internal cerebral veins, vein of Galen and straight sinus, consistent with acute thrombus. Note the normal attenuation in the basilar artery (open arrow), excluding the possibility of these findings being related to increased hematocrit. b Axial T2-W image shows hyperintense signal and enlargement (arrow) of the right thalamus and internal capsule as well as mild hyperintense signal in the left thalamus (arrowhead). c Axial diffusion-weighted image shows diffusion restriction (arrow) in the right thalamus, suggesting ischemia. Note the lack of diffusion restriction in the left thalamus. d Axial T2-W image obtained 4 months later shows encephalomalacia (chevron) in the right thalamus in the region of diffusion restriction, consistent with true “venous infarct.” Note resolution of edema in the left thalamus
Parenchymal hemorrhage
Venous infarction commonly leads to hemorrhagic complications, much more so than arterial infarction [10, 11]. In the setting of cerebral sinus venous thrombosis, hemorrhagic infarction (microhemorrhage) occurs in approximately 37% of venous infarcts and frank intracerebral hemorrhage (hemorrhage >1 cm) occurring in 21% of venous infarcts [11]. Parenchymal hemorrhage correlates with high venous pressure (>42 mmHg) and is hypothesized to arise from venule rupture and infarction [4]. Gradient echo and susceptibility-weighted imaging allow for sensitive, early detection of hemorrhage [3]. Figure 8 shows hemorrhage in the left temporal lobe of a neonate, suggesting vein of Labbe´ thrombosis.

A 2-week-old boy with congenital heart disease and new apnea with a vein of Labbé hemorrhagic infarct. a Axial CT image shows mixed attenuation hemorrhage in the left temporal lobe with adjacent edema. b A 3-D MR venography image shows absence of the left vein of Labbé (black arrow). Note the normal right vein of Labbé (white arrow)
Grade IV germinal matrix/intraventricular hemorrhage in the premature infant is a prominent example of the pathophysiology of hemorrhagic venous infarction (Figs. 9 and 10). Once thought to represent direct extension of germinal matrix or intraventricular hemorrhage into the brain parenchyma, parenchymal hemorrhage in the setting of germinal matrix/intraventricular hemorrhage is now understood to represent hemorrhagic venous infarction. The periventricular hemorrhagic infarction can be closely associated with germinal matrix hemorrhage suggesting that the presence of germinal matrix hemorrhage causes obstruction of deep venous drainage pathways, leading to venous hypertension in the drainage territory. If the venous hypertension is significant enough, this leads to ischemia and, in many cases, hemorrhage [12, 13]. On imaging, hemorrhagic venous infarction typically is wedge-shaped or fan-shaped, conforming to the radiating medullary veins of the cerebral hemispheres. Deep medullary venous occlusion with parenchymal sequelae can also occur in isolation in this population, with typical fan-shaped signal abnormalities and hemorrhage in the deep white matter [14].

Schematic of proposed pathophysiology of grade IV germinal matrix/intraventricular hemorrhage. a Deep medullary veins drain to a deep central terminal vein in the region of the germinal matrix. b Germinal matrix hemorrhage causes mass effect on the terminal vein resulting in venous insufficiency of medullary veins. c Venous insufficiency leads to a venous infarct. d Venous infarct becomes hemorrhagic

A 9-day-old premature infant (29 weeks’ gestational age) status post tracheoesophageal fistula repair. a Coronal ultrasound shows echogenic germinal matrix hemorrhages (white arrows) in the bilateral caudothalamic grooves with a fan-shaped region of increased echogenicity (arrow) in the periventricular white matter, consistent with hemorrhagic venous infarction. b Coronal T2-W image shows hypointense fan-shaped hemorrhage (arrow)
Chronic venous injury
Parenchymal volume loss
Parenchymal volume loss may occur secondary to the effects of chronic ischemia in venous hypertension/insufficiency. Examples include chronic venous insufficiency in Sturge-Weber syndrome [15, 16] and venous hypertension in high flow arteriovenous malformations [17].
Sturge-Weber is a neurocutaneous disorder typically characterized by facial port-wine stain, leptomeningeal angiomatous changes, and vascular eye abnormalities. It is hypothesized to arise from failure of the primitive cephalic venous plexus to properly mature affecting the development of nearby skin, eye and brain structures. The result of this failed venous maturation is the lack of superficial venous drainage, which leads to large transhemispheric venous collaterals, prominent ventricular veins and dilated superficial leptomeningeal collateral venous channels. The leptomeningeal angiomatosis often described in this condition is not a proliferation of vessels, but rather leptomeningeal venous channel dilatation and venous pooling due to lack of superficial venous drainage [18]. Inadequate adaptive remodeling leads to chronic venous insufficiency and progressive brain ischemia, volume loss and cortical/subcortical calcification. These characteristic findings of Sturge-Weber are illustrated in Fig. 11.

An 8-year-old boy with Sturge-Weber syndrome. a Axial T1-W contrast-enhanced image demonstrates typical transparenchymal medullary veins and leptomeningeal enhancement. b Axial CT shows typical subcortical mineralization. c A 3-D MR venography shows lack of superficial venous drainage in the affected left cerebral hemisphere, typical of Sturge-Weber syndrome
Severe parenchymal volume loss related to venous injury can be seen in the setting of high flow vein of Galen aneurysm malformations. When vein of Galen malformations are complicated by stenosis of the posterior venous outlets of the brain, the combination of a high flow arteriovenous shunt and cerebral venous outflow restriction often leads to severe cerebral venous hypertension, which is sometimes further aggravated by perfusion steal phenomenon, resulting in rapidly progressive cerebral parenchymal damage. This severe central nervous system injury is sometimes referred to as “melting brain” [19]. On ultrasound imaging, this is characterized by heterogeneous echogenicity of the brain parenchyma with profound volume loss. On MR imaging, characteristic features are severe progressive volume loss, and heterogeneous T1 and T2 signal intensity relating to combined effects of edema, gliosis, hemorrhage and calcification (Fig. 12).

A newborn girl with a vein of Galen arteriovenous malformation. a A 3-D surface rendered MR angiography shows markedly enlarged cortical veins (arrows) secondary to retrograde flow from the high flow fistula. b Axial T1-W image shows the severe volume loss (arrows), often termed “melting brain,” associated with high venous pressures causing severe cerebral ischemia
Calcification/mineralization
Calcification is a nonspecific finding associated with a wide array of brain pathologies. An important cause of mineralization is venous hypertension/insufficiency. Calcifications may be cortically based in a gyral configuration or radially oriented calcifications in the subcortical and periventricular white matter (perimedullary distribution) [20]. These two calcification patterns may coexist. Figure 13 demonstrates a case of a large arteriovenous malformation with shunting and venous outflow obstruction leading to venous congestive encephalopathy and parenchymal calcification.

A 14-year-old boy with seizures. a, b Axial CT images show symmetrical perimedullary, subcortical and deep basal ganglia mineralization. c Axial T2-W image shows an arteriovenous malformation nidus (arrow) in the left frontal lobe and extensive white matter signal abnormality. d A 3-D MR venography demonstrates enlarged cortical vein collaterals (open arrow), thrombosed left transverse sinus, and marked stenosis at the junction of the right sigmoid sinus and jugular bulb (arrow). Overall, findings are consistent with a large arteriovenous malformation with shunting and venous outflow obstruction leading to venous congestive encephalopathy
The pathophysiology of brain parenchymal calcification in venous hypertension is not well understood. Proposed pathophysiological mechanisms for cortical calcifications include alterations of calcium homeostasis secondary to ischemia and seizure-induced excitotoxic injury. Hypoxic-ischemic injury causes deregulation of neuronal and microglial calcium homeostasis and leads to intracellular calcium precipitation [21]. In Sturge-Weber, the development of calcifications has been temporally associated with seizure onset. Some hypothesize that seizure in Sturge-Weber causes excitotoxic injury to brain cells with little or no vascular reserve, causing synergistic damage to the cortex [22]. The proposed pathophysiological mechanisms for perimedullary subcortical and deep white matter calcifications include increased vessel permeability and microvenous stasis. Increased permeability of cerebral vessels causes abnormal passage of proteins and calcium into brain cells and interstitium with crystallization. Venous hypertension may cause microvenous stasis with subsequent thrombosis and eventual calcification. Calcifications often occur at the medullary vein-venous watershed in the subcortical region, a characteristic location in venous hypertension [23].
Conclusion
Acute and chronic venous injury in children is seen in a diverse group of disorders. Brain parenchymal injury occurs along common pathophysiological pathways. Understanding the characteristic imaging patterns of acute and chronic injury will help the imaging professional accurately identify venous injury.
References
Meder JF, Chiras J, Roland J et al (1994) Venous territories of the brain. J Neuroradiol 21:118–133
Fries G, Wallenfang T, Hennen J et al (1992) Occlusion of the pig superior sagittal sinus, bridging and cortical veins: multistep evolution of sinus-vein thrombosis. J Neurosurg 77:127–133
Leach JL, Fortuna RB, Jones BV, Gaskill-Shipley MF (2006) Imaging of cerebral venous thrombosis: current techniques, spectrum of findings, and diagnostic pitfalls. Radiographics 26(Suppl 1):S19–S41 discussion S42–43
Tsai FY, Wang AM, Matovich VB et al (1995) The staging of acute dural sinus thrombosis - correlation with venous-pressure measurements and implications for treatment and prognosis. AJNR Am J Neuroradiol 16:1021–1029
Gotoh M, Ohmoto T, Kuyama H (1993) Experimental study of venous circulatory disturbance by dural sinus occlusion. Acta Neurochir 124:120–126
Yoshikawa T, Abe O, Tsuchiya K et al (2002) Diffusion-weighted magnetic resonance imaging of dural sinus thrombosis. Neuroradiology 44:481–488
Lovblad KO, Bassetti C, Schneider J et al (2000) Diffusion-weighted MRI suggests the coexistence of cytotoxic and vasogenic oedema in a case of deep cerebral venous thrombosis. Neuroradiology 42:728–731
Forbes KP, Pipe JG, Heiserman JE (2001) Evidence for cytotoxic edema in the pathogenesis of cerebral venous infarction. AJNR Am J Neuroradiol 22:450–455
Ducreux D, Oppenheim C, Vandamme X et al (2001) Diffusion-weighted imaging patterns of brain damage associated with cerebral venous thrombosis. AJNR Am J Neuroradiol 22:261–268
Thomalla G, Schwark C, Sobesky J et al (2006) Outcome and symptomatic bleeding complications of intravenous thrombolysis within 6 hours in MRI-selected stroke patients: comparison of a German multicenter study with the pooled data of ATLANTIS, ECASS, and NINDS tPA trials. Stroke 37:852–858
Kumral E, Polat F, Uzunkopru C et al (2012) The clinical spectrum of intracerebral hematoma, hemorrhagic infarct, non-hemorrhagic infarct, and non-lesional venous stroke in patients with cerebral sinus-venous thrombosis. Eur J Neurol 19:537–543
Volpe JJ (2009) Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 8:110–124
Raybaud C, Ahmad T, Rastegar N et al (2013) The premature brain: developmental and lesional anatomy. Neuroradiology 55(Suppl 2):23–40
Arrigoni F, Parazzini C, Righini A et al (2011) Deep medullary vein involvement in neonates with brain damage: an MR imaging study. AJNR Am J Neuroradiol 32:2030–2036
Marti-Bonmati L, Menor F, Mulas F (1993) The Sturge-Weber syndrome - correlation between the clinical status and radiological CT and MRI findings. Childs Nerv Syst 9:107–109
Tournut P, Turjman F, Guibal AL et al (1992) MRI in Sturge-Weber syndrome. J Neuroradiol 19:285–292
Recinos PF, Rahmathulla G, Pearl M et al (2012) Vein of Galen malformations: epidemiology, clinical presentations, management. Neurosurg Clin N Am 23:165–177
Parsa CF (2013) Focal venous hypertension as a pathophysiologic mechanism for tissue hypertrophy, port-wine stains, the Sturge-Weber syndrome, and related disorders: proof of concept with novel hypothesis for underlying etiological cause (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc 111:180–215
Meila D, Grieb D, Melber K et al (2016) Hydrocephalus in vein of Galen malformation: etiologies and therapeutic management implications. Acta Neurochir 7:1279–1284
Ramonet D, Pugliese M, Rodriguez MJ et al (2002) Calcium precipitation in acute and chronic brain diseases. J Physiol Paris 96:307–312
Rodriguez MJ, Ursu G, Bernal F et al (2001) Perinatal human hypoxia-ischemia vulnerability correlates with brain calcification. Neurobiol Dis 8:59–68
Wu J, Tarabishy B, Hu JN et al (2011) Cortical calcification in Sturge-weber syndrome on MRI-SWI: relation to brain perfusion status and seizure severity. J Magn Reson Imaging 34:791–798
Bansal A, Gailloud P, Jordan L, Ruiz DS (2009) Regression of cerebral calcifications after endovascular treatment in a case of vein of Galen arteriovenous malformation. Case Report J Neurosurg Pediatr 4:17–20
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None.
Additional information
CME activity
This article has been selected as the CME activity for the current month. Please visit the SPR Web site at www.pedrad.org on the Education page and follow the instructions to complete this CME activity.
Rights and permissions
About this article

Cite this article
Linscott, L.L., Leach, J.L., Jones, B.V. et al. Imaging patterns of venous-related brain injury in children. Pediatr Radiol 47, 1828–1838 (2017). https://doi.org/10.1007/s00247-017-3975-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-017-3975-x