
Abstract
The cardiac conduction system is a network of distinct cell types necessary for the coordinated contraction of the cardiac chambers. The distal portion, known as the ventricular conduction system, allows for the rapid transmission of impulses from the atrio-ventricular node to the ventricular myocardium and plays a central role in cardiac function as well as disease when perturbed. Notably, its patterning during embryogenesis is intimately linked to that of ventricular wall formation, including trabeculation and compaction. Here, we review our current understanding of the underlying mechanisms responsible for the development and maturation of these interdependent processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ventricular conduction system (VCS) entails components of the cardiac conduction system distal to the atrio-ventricular node and includes the Bundle of His, right, and left bundle branches as well as the Purkinje fiber network (Fig. 1). It is a distinct and essential component of the conduction system that allows for the rapid transmission of electrical impulses originating from the upper chambers. While this intricate lattice of conduction cells represents only ~ 1–2% of the ventricular mass [1], the VCS is crucial for the rapid and near-simultaneous depolarization of both ventricles, from apex to base, thereby allowing for the coordinated contraction of the ventricles. Notably, abrogation of the development and/or normal function of the VCS results in decreased cardiac function and, importantly, can result in lethal ventricular arrhythmias [2].

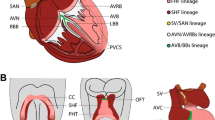
EKG and EGM parameters and VCS Anatomy. a Left: representative tracing from a surface electrocardiogram (ECG). Depolarization of the atrial cardiomyocytes is visualized as the P wave, where the PR interval reflects onset of atrial depolarization to conduction through the AV node. The QRS complex represents ventricular depolarization and the T wave ventricular repolarization. The QT interval entails time of both ventricular depolarization and repolarization. Right: representative intracardiac electrocardiogram (EGM) tracing. His bundle electrogram (HBE) showing an atrial potential followed by a His and finally a ventricular potential. AH interval represents the time for conduction through the AV node. HV interval displays the conduction time through the ventricular conduction system from the His bundle located just below the AV node, to the first identifiable onset of ventricular activation. b Diagram of an adult heart with the cardiac conduction system highlighted. Sino-atrial node (SAN) is the primary pacemaker in a normal heart and is located at the junction of the superior vena cava (SVC) and right atrium (RA). Depolarization subsequently sweeps through the atria inducing contraction. Sole passage of conduction to the ventricles occurs via the atrio-ventricular node (AVN) due to the otherwise electrically inert fibrous tissue of the surrounding atrio-ventricular junction. Conduction is slowed through the AVN and subsequently accelerated through the ventricular conduction system comprising the His bundle (His), right, and left bundle branches (RBB, LBB) and Purkinje fibers (PF). Finally, once the impulse has traversed the Purkinje–myocyte junction (PMJ), the ventricular cardiomyocytes are depolarized in stereotypical apex to base pattern. Conduction intervals covered in a are labeled with respect to conduction through their respective anatomical landmarks
While Purkinje fiber cells are located within the ventricular subendocardium in mammals, they exhibit exquisitely unique anatomic, electrophysiologic, and transcriptional features as compared to the surrounding working myocardium [3]. Since the discovery of Purkinje fiber cells by the Czech Jan Evangelista Purkyně in 1837 [4], it has long been recognized that the establishment of the ventricular conduction system parallels that of ventricular wall formation, both temporally and spatially [5, 6]. These early anatomic observations led to the detailed timeline of their development and have been the foundation for an impressive volume of research to come. More recent work has resulted in the discovery of common progenitor cells and pathways critical to both myocardial trabeculation and compaction of the ventricular wall as well as for VCS development and maturation. In this review, we will provide a brief overview of the intertwined relationship between these two systems throughout development, maturation, and during disease processes alike.
Temporal Regulation of VCS Development
Conduction System Development
At embryonic day 7.5 (e7.5) in mice, correlating roughly to day 22 of gestation (~ d22) in humans, a primitive pacemaker is first observed at the inflow tract region of the linear heart tube, a three-layer structure consisting of endocardium, cardiac jelly, and myocardium [3, 7] (Fig. 2a). The primitive pacemaker initiates an electrical impulse that propagates slowly through the linear heart tube leading to a peristaltic contraction that generates unidirectional blood flow. With looping of the heart tube, there is first evidence of a discrepancy in conduction velocities with a delay occurring within the region of the AV canal at ~ e8.0 in mice (~ d25 in humans). This provides a functional electrophysiologic basis for the atrio-ventricular delay in the mature heart, allowing for sequential contraction of the atria and ventricles and the time necessary for ventricular filling prior to their own contraction.

Timeline of early ventricular wall and conduction system development. a By embryonic day 7.5 (e7.5) in mice or approximately human gestational day 22 (d22), there is evidence of a primitive pacemaker (purple) located at the inflow portion of the primitive heart tube. The heart tube is composed of endocardial (pink) and myocardial (gray) layers separated by cardiac jelly (blue). b By e9.5 or ~ d26, with looping of the heart, myocardial protrusions extending into the ventricular lumen first begin to appear. At this time, Connexin 40 expression (Cx40, yellow) is detectable throughout the ventricular myocardium, albeit at uniformly low levels. By e10.5 there is a histologically identifiable SA node and there is first evidence of a delay in conduction velocity (purple arrows) occurring within the region of the AV canal (AVC). By e10.5, prior to ventricular septation, the VCS also becomes functionally evident by the reversal of the base-to-apex activation pattern to a “apex-first” electrical activation. c By e12.5, prominent ventricular trabeculae have formed. At this time, there is a clear gradient in Cx40 expression (yellow), with highest found in the subendocardial trabeculae and least in the subepicardial myocardium. Functionally, murine hearts also demonstrate two distinct activation patterns in the LV and RV (straight purple arrows), correlating with the appearance of discrete bundle branches. d By e19 (> d60), trabecular maturation is typically completed in mice with the development of well formed, compact ventricular wall. At this time, the foundation of the entire conduction system has been established, with Cx40 expression (yellow) now restricted to the components of the VCS (His, RBB/LBB, Purkinje fibers). Outflow tract (OFT), right ventricle (RV), left ventricle (LV), right atrium (RA), left atrium (LA)
The VCS is the last component of the cardiac conduction system (CCS) to develop. Functionally, VCS development is first noted in mammals at ~ e10.5 (~ d35 in humans), prior to ventricular septation, by the reversal of the base-to-apex activation pattern to an “apex-first” electrical activation [8] (Fig. 2b). This more mature depolarization sequence allows for the rapid and coordinated contraction of the ventricles from the apex towards the base, and thereby promoting forward flow of blood within the ventricles towards the outflow tracts. By e12.5, murine hearts demonstrate two distinct activation patterns in the LV and RV, correlating with the appearance of discrete bundle branches [8] (Fig. 2c). Elegant cell fate mapping experiments [1] subsequently demonstrated that cells expressing Connexin 40 (Cx40), encoding a high-velocity gap junction protein, give rise to both working myocardium and all cell types of the ventricular conduction system (including His bundle, right, and left bundle branches and Purkinje fiber cells) providing a foundation for the concept of pre-specified common progenitor cells. By e16.5, Cx40+ cells have become restricted to a Purkinje cell fate with limited subsequent proliferative capacity, paralleling the lower proliferative rate in the surrounding trabecular myocardium [9] (Fig. 2d). The VCS, while established prenatally, will continue to undergo extensive functional maturation postnatally [10, 11].
Ventricular Wall Formation
Following cardiac looping, at around e9.0 in mice, cardiac chambers begin to form along with the development of the trabeculae to allow for increased cardiac output and oxygen uptake by the developing trabecular myocardium. During trabeculation, myocardial protrusions extending into the cardiac lumen first begin to appear ~ e9.5 in mice (~ d26 in humans) [12, 13] (Fig. 2b). These protrusions develop radially into a fenestrated network and later undergo remodeling in which the trabeculae cease to grow in the luminal direction and thicken radially, consolidating with the compact myocardium. Trabecular maturation is largely completed by ~ e16.5 in mice (~ d60 in humans) [14] (Fig. 2d). Concomitant with trabecular remodeling or maturation, the compact myocardial layer undergoes extensive proliferation resulting in a dramatic increase in ventricular wall thickness, providing a major contractile force to the functional heart. Similar to the VCS, the compact myocardium will continue to thicken and mature throughout early postnatal development [7].
Cellular Origins of the VCS
While the cardiac conduction system was initially proposed to be derived from neural crest cells given co-expression of classic neuronal markers such as NF-M and Hnk-1 [15], subsequent lineage trace analysis studies have shown that the VCS unquestionably originates from cardiomyocyte progenitors [16,17,18]. Early chick studies performed by Cheng et al. [17] employed viral lineage tracing methods to demonstrate that all components of the CCS, including the VCS, arise from cardiomyogenic progenitors and, later, lineage tracing experiments in mice firmly ruled out a contribution of neural crest derivatives in mammals [18].
Connexin 40 (Gja5), the earliest known marker of Purkinje cell differentiation, encodes for a high-velocity gap junction subunit and, in the mature heart, is restricted to the atria and VCS. Its expression is essential to the function of the ventricular conduction system allowing for the rapid transmission of electrical impulses from the AV node to the working myocardium. Embryonically, however, Cx40 is expressed much more broadly [19, 20]. Cx40 transcript and protein can first be visualized at ~ e9.5 in mice throughout the atria and ventricles [20], albeit faintly in the latter (Fig. 2b). By ~ e11.5, Cx40 is expressed transmurally within the ventricles and notably demonstrates enhanced expression in the primitive trabecular network [10, 19]. Over time there is gradual restriction in Cx40 expression with a gradient from the subendocardial trabeculae decreasing towards only faint expression in the subepicardial region (Fig. 2c). By e16.5, Cx40 is present in the trabeculae but absent from the ventricular wall. At e19.0, when the ventricular wall has largely completed compaction, Cx40 expression becomes restricted to the VCS including the His bundle, bundle branches, and Purkinje fiber network [19, 20] (Fig. 2d).
While much is known regarding the cellular origin of the VCS, many questions remain. For example, which cardiomyocytes represent the true VCS progenitors and how might one identify them? What is their transcriptional signature and true proliferative capacity? Do they rely on asymmetric division? While these questions remain to be addressed, prospective Cre fate mapping have provided key insights into the timing of VCS fate commitment by indelibly marking cells expressing Cx40 at various embryonic timepoints [1]. Pulse labeling of Cx40+ cells between e10.5 and e14.5 consistently labeled cells from both the VCS as well as the working myocardium. Notably, however, this fate mapping showed progressive lineage restriction of Cx40+ common progenitors to the VCS over time. Indeed, by e16.5, cells that express Cx40 give rise exclusively to cells of the VCS. This seminal work not only demonstrated that the VCS originates from common Cx40+ cardiomyocyte progenitors within the trabecular myocardium but additionally demonstrated that the ventricular conduction system cells have limited outgrowth following specification. This finding was consistent with prior reports on VCS cell cycle exit following specification [9, 21, 22] and strengthened the widely held belief that the VCS is primarily established through recruitment from a progenitor population rather than by extensive proliferation [8, 17].
Finally, clonal analysis of cell numbers within mixed and unmixed clusters of labeled conductive and working myocardial cells argued for a distinct origin of VCS progenitor cells for the left versus right ventricles, consistent with their well-established origins from first and second heart field, respectively [1]. Later, evidence from additional lineage tracing experiments marking the first and second heart fields emerged supporting this theory [23,24,25]. While the left ventricle, left bundle, and left PF network are derived from Hcn4+ first heart field progenitors, the right ventricle along with the right bundle, and right PF network appear to originate from Isl1+ second heart field [25].
Clearly, additional work will be necessary to identify and characterize the VCS progenitor population from both the right and left ventricular myocardium. Further analysis will not only provide insight into our basic understanding of the development of the conduction system but may also greatly benefit current efforts in the in vitro differentiation of Purkinje fiber cells from human induced-pluripotent stem cells [26, 27]. Specifically, novel techniques such as single-cell RNA sequencing and single-cell time-lapse microscopy will likely provide the resolution needed to clarify some of these outstanding questions previously unattainable through lineage tracing alone [28].
Molecular Regulation of Ventricular Wall and VCS Development
While the developing compact myocardium receives many of its instructions from its interactions with the epicardium, the trabeculae are induced largely by signals derived from the endocardium. The developing Purkinje fiber network, located within the subendocardium, also relies on key interactions with the endocardium. Given the temporal and spatial overlap of ventricular wall and VCS development, it is not surprising that several shared signaling pathways are crucial to the patterning of both the ventricular myocardium and ventricular conduction system (Table 1). Here we focus on a few key pathways relevant to the development the VCS. For a detailed review of the transcriptional regulation of ventricular wall, we refer readers to numerous prior articles on this topic [7, 14, 29] and VCS formation [3, 30,31,32,33].
Notch Signaling
Extensive research from multiple groups have clearly shown Notch to regulate not only trabecular formation but also Purkinje cell development via downstream regulators including Neuregulin 1 and its receptor ErbB, Ephrin B2, and BMP10 [34,35,36,37]. Loss of function mutant mice for either RBPJk, a downstream transcriptional repressor in canonical Notch signaling, and to a lesser extent Notch1, results in poorly formed trabeculae and less-structured myocardium [34]. On the other hand, endothelial-specific gain of function via the conditional deletion of the Notch1 intracellular domain inhibitor, Fkbp1a, results in a hypertrabeculation and non-compaction phenotype [38]. Loss of function murine experiments using Myosin Light Chain 2v (Mlc2v)Cre/+; Notch1f/f or Mlc2vCre; DNMAML (dominant-negative mastermind like, a Notch inhibitor) have clearly shown Notch to be required for AV nodal formation. However these studies also show that the VCS remained functionally intact, with an unchanged HV interval (His bundle to ventricle), a measure of VCS-dependent ventricular activation time [39] (Fig. 1). Conversely, Notch overexpression in the working myocardium during embryogenesis has been shown to be sufficient to induce reprogramming towards a Purkinje-like phenotype [39] and underscores a possible role for Notch signaling in regulating the plasticity of common cardiomyocyte progenitor cells of the VCS. More recent work has further implicated Notch signaling in the modification of the epigenetic landscape of key ion channel genes including potassium channels ITO,f and IK,slow [40].
Id2
While Notch is not ‘required’ for VCS development per se, other factors including the transcriptional repressor Id2 have been shown to be necessary not only for ventricular wall formation but also for the normal patterning of the VCS. Systemic loss of Id2 in mice results in both structural and functional abrogation of the VCS including disrupted formation of the AV bundle and left bundle branch consistent with a widened QRS and RsR′ pattern in lead V6 (or left bundle branch block pattern) by surface electrocardiography [41]. Interestingly, somewhat similar to Notch signaling, Id2 may play a significant role in the specification of the VCS from ventricular cardiomyocyte progenitors. Specifically, the authors demonstrated that Id2 may promote cell cycle exit within committed VCS cells, a known characteristic of VCS occurring between e12 and e14 [9, 17]. With respect to ventricular wall formation, the loss of function of Id2 results in, among other phenotypes, abnormal compact myocardium formation [42].
Irx3
Irx3, a member of the Iroquois family of transcription factors, demonstrates overlapping expression with Id2 within the conduction system and Irx3 loss of function in mice resulted in VCS hypoplasia [11] and abnormal embryonic differentiation as evident by modulation of Cx40 and Cx43 expression [43]. In contrast to Id2 knockouts, Irx3 null mice showed no difference in proliferation or apoptosis rates within the VCS. Instead, their hypoplasia came in the setting of increased proliferation within the working myocardium, which the authors argued may disturb the cell cycle exit required for the recruitment and differentiation of the ventricular conduction cells. The exact mechanism, however, remains unclear. Irx3 has also been implicated in the postnatal maturation of the VCS [11], where mutants demonstrate a progressive reduction in the myocardial wall thickness and the number of His–Purkinje fibers. In addition, Irx3 knock-down mice demonstrate increased frequency of ventricular tachyarrhythmias [44]. While our understanding of postnatal VCS maturation remains incomplete, numerous other transcription factors and signaling mediators including the homeodomain transcription factors Nkx2.5 and Hopx have been implicated in this process.
Nkx2.5/BMP10/Hopx
Seminal work has demonstrated a role for Nkx2.5 in the regulation of normal trabecular formation as well as the conduction system. Expression of Nkx2.5 is enhanced within the VCS during embryogenesis [45] and has been shown to work in concert with the T-box transcription factor Tbx5 to mediate expression of Id2 and other critical factors in VCS differentiation [41]. Conditional knock down of Nkx2.5 within the working myocardium and ventricular conduction system via the MLC2v promoter resulted in hypertrabeculation and myocardial overgrowth [46]. This phenotype could be attributed to the inappropriate perdurance of BMP10 past e15.5, the timepoint in mice at which BMP10 is normally extinguished from the ventricular myocardium. Specifically, the authors show that in vivo overexpression of BMP10 or in vitro exposure of embryonic mouse cardiomyocytes to BMP10 both result in increased proliferation of working cardiomyocytes and a cell overgrowth and hypertrabeculation phenotype. With respect to the conduction system, Nkx2.5 loss of function resulted in the atrophy of both the AV node and His bundle, consistent functionally with a progressive AV block and a prolonged QRS interval [46,47,48]. Notably, haploinsufficiency of Nkx2.5 has been shown by others to result in a reduction in the number of Cx40+ Purkinje fibers [10, 48]. While these studies have elucidated some aspects of the requirement of Nkx2.5 in VCS development, more questions remain. For example, how does the increased proliferation of the working myocardium in Nkx2.5 knock-down mice relate to perturbed VCS specification and cell cycle exit seen in Irx3 null mice? Additionally, does Nkx2.5 play a direct role in the recruitment, proliferation, or retention of ventricular conduction cells? Further investigations will be necessary to clarify the precise roles of these transcription factors.
Beyond VCS specification by Nkx2.5, the hypoplastic His–Purkinje system observed in the context of Nkx2.5 haploinsufficiency appears to remain functionally intact given the documented normal HV intervals in both mice and humans [48, 49]. Additional studies later demonstrated that Nkx2.5+/− mice have normal Purkinje fiber cell size and electrophysiological properties [10]. Systemic Nkx2.5 null mice die at ~ e10 and therefore cannot be evaluated; however, an intracardiac EP study of conditional postnatal Nkx2.5 knock out mice has not been performed to date to our knowledge [46]. It would be interesting to determine whether complete loss of Nkx2.5 results in significant functional abrogation of the His–Purkinje system or whether the universally observed prolonged QRS interval by surface EKG, in both Nkx2.5+/− and conditional knock out mice simply reflects a decreased number of terminal Purkinje fibers in relation to the volume of working myocardium.
Finally, similar to Nkx2.5 mutants, loss of function of Hopx, a homeodomain-only protein and downstream transcriptional target of Nkx2.5 [50, 51], resulted in delayed withdrawal of the ventricular working myocardium from cell cycle exit and subsequent myocardial overgrowth. Distinctly, however, Hopx null mutants exhibited seemingly normal AV nodal function (unchanged PR intervals and a slightly longer AH interval but not statistically significant), while it was the infra-Hisian conduction system that was primarily affected [52]. This phenotype was functionally evident by prolonged QRS and, importantly, protracted HV intervals consistent with a dysfunctional ventricular conduction system.
Conduction System Development and Disease
The VCS: A Nidus for Arrhythmias
Sudden cardiac death (SCD) remains a significant cause of mortality worldwide and affects roughly 3 million people worldwide yearly, with ~ 353 thousand in the US alone in 2015 [53]. Notably, the majority of cases of SCD (50–85%) are thought to occur as a result of ventricular dysrhythmias, specifically ventricular tachycardia (VT) and fibrillation (VF) [54]. It is well established that the His–Purkinje system is a major source of ventricular arrhythmogenicity in the setting of acquired injury or altered function such as electrolyte disturbances, drug exposure, ischemia, or peri-operative damage [2, 55]. Disruption in the ventricular conduction system can result in both automatic and triggered ventricular arrhythmias as well as serve as the nidus for re-entrant tachycardias, all of which can result in hemodynamic instability and sudden death (Table 1).
Notably, Purkinje cells as well as the Purkinje fiber–myocyte junction (PMJ) are recognized as particularly vulnerable sources of ventricular arrhythmias in the heart [56,57,58,59]. Disruption of the cell–cell communication through gap junction channels via modulation of connexin expression or function, a process known as gap junction remodeling, has been documented in heart disease and is associated with an increased risk of arrhythmias [58, 59].
Genetic Mechanisms Underlying Both VCS Development and Ventricular Dysrhythmias
Several of the aforementioned genes responsible for the normal development of the VCS and ventricular wall formation have also been implicated in conduction pathology. Interestingly, Notch may play a role in preserving the PMJ. Notch overexpression within the ventricular myocardium has been shown to perturb the boundary between VCS and working myocardium, leading to abnormal Purkinje–myocyte junctions [39]. Interestingly, Notch signaling has also known to become transiently activated in the setting of ischemia [60] and could be potentially responsible, in part, for post-infarction ventricular dysrhythmias. Finally, in humans, gene variants in HEY2, a direct Notch target, have been implicated in a propensity of sudden death secondary to ventricular arrhythmias [61].
As mentioned previously, the transcription factor Irx3 is essential for both ventricular wall formation as well as the embryonic development and postnatal maturation of the VCS. Interestingly, analysis of 130 probands with idiopathic VT and no SCN5A mutations revealed two novel, missense IRX3 mutations in which both probands were more prone to NSVT with exercise [44]. Similar to mice, humans with NKX2.5 haploinsufficiency results in structural defects as well as sudden cardiac death and arrhythmias including ventricular tachycardia [47,48,49, 62].
Structure Begets Function
Consistent with an intimate relationship between VCS and ventricular wall formation, there is also a well-established association between LV non-compaction, hypertrabeculation, and the propensity for developing ventricular tachyarrhythmias within humans [2]. Interestingly, histiocytoid cardiomyopathy (HC), while rare, is a prime example of a congenital disorder typified by dysplasia of the ventricular wall and VCS as well as a propensity for lethal ventricular arrhythmias. HC occurs primarily in females less than 2 years of age with an unclear etiology to date, although a mitochondrial cause has been suggested [63]. It commonly presents with both LV non-compaction and Purkinje fiber dysplasia, manifesting clinically with VT and VF [64], and results in sudden death within ~ 20% of cases. Only about 100 cases have been described to date and detail characteristic histologic findings including dysplastic (‘histiocyte-like’) Purkinje fiber cells in addition to LVNC or a dilated CM [63, 64]. The etiology of this fatal disease remains to be elucidated and may provide fresh insight into some of the key early developmental steps required to establish both a normal ventricular wall and VCS.
Conclusion
Recent studies have revealed a clear interdependence between the formation of the VCS and ventricular wall. Temporal and spatial associations of their embryonic patterning are evident and indication of their interconnectedness is supported by their common origins and their co-regulation by a core set of transcription factors. Disruptions in these developmental processes have been directly associated with clinically significant ventricular arrhythmias and sudden cardiac death, further arguing for additional research into their development and maturation. An interesting and relevant question that remains to be further clarified is the identity and molecular constitution of the common progenitor of the VCS. Additional insights can now be gleaned using the emerging single-cell expression profiling and analytical technologies. These tools may prove essential to not only further defining our basic understanding of VCS development but also help to advance our abilities in the in vitro differentiation of ventricular conduction system components and working myocardial cells alike from human stem cell sources.
References
Miquerol L, Moreno-Rascon N, Beyer S et al (2010) Biphasic development of the mammalian ventricular conduction system. Circ Res 107:153–161. https://doi.org/10.1161/CIRCRESAHA.110.218156
Haissaguerre M, Vigmond E, Stuyvers B et al (2016) Ventricular arrhythmias and the His-Purkinje system. Nat Rev Cardiol 13:155–166. https://doi.org/10.1038/nrcardio.2015.193
van Weerd JH, Christoffels VM (2016) The formation and function of the cardiac conduction system. Development 143:197–210. https://doi.org/10.1242/dev.124883
Steiner I (1988) History of Purkinje’s fibers. Cesk Fysiol 37:509–512
Davies F (1930) The conducting system of the bird’s heart. J Anat 64:129–146
Sommer JR, Johnson EA (1968) Cardiac muscle. A comparative study of Purkinje fibers and ventricular fibers. J Cell Biol 36:497–526
Samsa LA, Yang B, Liu J (2013) Embryonic cardiac chamber maturation: trabeculation, conduction, and cardiomyocyte proliferation. Am J Med Genet C 163C:157–168. https://doi.org/10.1002/ajmg.c.31366
Rentschler S, Vaidya DM, Tamaddon H et al (2001) Visualization and functional characterization of the developing murine cardiac conduction system. Development 128:1785–1792
Sedmera D, Reckova M, DeAlmeida A et al (2003) Spatiotemporal pattern of commitment to slowed proliferation in the embryonic mouse heart indicates progressive differentiation of the cardiac conduction system. Anat Rec 274:773–777. https://doi.org/10.1002/ar.a.10085
Meysen S, Marger L, Hewett KW et al (2007) Nkx2.5 cell-autonomous gene function is required for the postnatal formation of the peripheral ventricular conduction system. Dev Biol 303:740–753. https://doi.org/10.1016/j.ydbio.2006.12.044
Kim K-H, Rosen A, Hussein SMI et al (2016) Irx3 is required for postnatal maturation of the mouse ventricular conduction system. Sci Rep 6:19197. https://doi.org/10.1038/srep19197
Sedmera D, Pexieder T, Vuillemin M et al (2000) Developmental patterning of the myocardium. Anat Rec 258:319–337
Moorman AFM, Christoffels VM (2003) Cardiac chamber formation: development, genes, and evolution. Physiol Rev 83:1223–1267. https://doi.org/10.1152/physrev.00006.2003
Zhang W, Chen H, Qu X et al (2013) Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C 163C:144–156. https://doi.org/10.1002/ajmg.c.31369
Gorza L, Schiaffino S, Vitadello M (1988) Heart conduction system: a neural crest derivative? Brain Res 457:360–366
Gourdie RG, Mima T, Thompson RP, Mikawa T (1995) Terminal diversification of the myocyte lineage generates Purkinje fibers of the cardiac conduction system. Development 121:1423–1431
Cheng G, Litchenberg WH, Cole GJ et al (1999) Development of the cardiac conduction system involves recruitment within a multipotent cardiomyogenic lineage. Development 126:5041–5049
Jiang X, Rowitch DH, Soriano P et al (2000) Fate of the mammalian cardiac neural crest. Development 127:1607–1616
Delorme B, Dahl E, Jarry-Guichard T et al (1995) Developmental regulation of connexin 40 gene expression in mouse heart correlates with the differentiation of the conduction system. Dev Dyn 204:358–371. https://doi.org/10.1002/aja.1002040403
Delorme B, Dahl E, Jarry-Guichard T et al (1997) Expression pattern of connexin gene products at the early developmental stages of the mouse cardiovascular system. Circ Res 81:423–437
Erokhina IL, Rumyantsev PP (1988) Proliferation and biosynthetic activities of myocytes from conductive system and working myocardium of the developing mouse heart. Light microscopic autoradiographic study. Acta Histochem 84:51–66. https://doi.org/10.1016/S0065-1281(88)80010-2
Sedmera D, Thompson RP (2011) Myocyte proliferation in the developing heart. Dev Dyn 240:1322–1334. https://doi.org/10.1002/dvdy.22650
Miquerol L, Bellon A, Moreno N et al (2013) Resolving cell lineage contributions to the ventricular conduction system with a Cx40-GFP allele: a dual contribution of the first and second heart fields. Dev Dyn 242:665–677. https://doi.org/10.1002/dvdy.23964
Später D, Abramczuk MK, Buac K et al (2013) A HCN4 + cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat Cell Biol 15:1098–1106. https://doi.org/10.1038/ncb2824
Liang X, Wang G, Lin L et al (2013) HCN4 dynamically marks the first heart field and conduction system precursors. Circ Res 113:399–407. https://doi.org/10.1161/CIRCRESAHA.113.301588
Tsai S-Y, Maass K, Lu J et al (2015) Efficient generation of cardiac Purkinje cells from ESCs by activating cAMP signaling. Stem Cell Rep 4:1089–1102. https://doi.org/10.1016/j.stemcr.2015.04.015
Maass K, Shekhar A, Lu J et al (2015) Isolation and characterization of embryonic stem cell-derived cardiac Purkinje cells. Stem Cells 33:1102–1112. https://doi.org/10.1002/stem.1921
Li G, Xu A, Sim S et al (2016) Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell 39:491–507. https://doi.org/10.1016/j.devcel.2016.10.014
Paige SL, Plonowska K, Xu A, Wu SM (2015) Molecular regulation of cardiomyocyte differentiation. Circ Res 116:341–353. https://doi.org/10.1161/CIRCRESAHA.116.302752
Munshi NV (2012) Gene regulatory networks in cardiac conduction system development. Circ Res 110:1525–1537. https://doi.org/10.1161/CIRCRESAHA.111.260026
de la Pompa JL, Epstein JA (2012) Coordinating tissue interactions: notch signaling in cardiac development and disease. Dev Cell 22:244–254. https://doi.org/10.1016/j.devcel.2012.01.014
Christoffels VM, Smits GJ, Kispert A, Moorman AFM (2010) Development of the pacemaker tissues of the heart. Circ Res 106:240–254. https://doi.org/10.1161/CIRCRESAHA.109.205419
Hatcher CJ, Basson CT (2009) Specification of the cardiac conduction system by transcription factors. Circ Res 105:620–630. https://doi.org/10.1161/CIRCRESAHA.109.204123
Grego-Bessa J, Luna-Zurita L, del Monte G et al (2007) Notch signaling is essential for ventricular chamber development. Dev Cell 12:415–429. https://doi.org/10.1016/j.devcel.2006.12.011
Rentschler S, Morley GE, Fishman GI (2002) Molecular and functional maturation of the murine cardiac conduction system. In: Cold Spring Harbor symposia on quantitative biology, vol 67. Cold Spring Harbor Laboratory Press, New York, pp 353–361
Hertig CM, Kubalak SW, Wang Y, Chien KR (1999) Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J Biol Chem 274:37362–37369
Gourdie RG, Wei Y, Kim D et al (1998) Endothelin-induced conversion of embryonic heart muscle cells into impulse-conducting Purkinje fibers. Proc Natl Acad Sci USA 95:6815–6818
Chen H, Zhang W, Sun X et al (2013) Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 140:1946–1957. https://doi.org/10.1242/dev.089920
Rentschler S, Yen AH, Lu J et al (2012) Myocardial Notch signaling reprograms cardiomyocytes to a conduction-like phenotype. Circulation 126:1058–1066. https://doi.org/10.1161/CIRCULATIONAHA.112.103390
Khandekar A, Springer S, Wang W et al (2016) Notch-mediated epigenetic regulation of voltage-gated potassium currents. Circ Res 119:1324–1338. https://doi.org/10.1161/CIRCRESAHA.116.309877
Moskowitz IPG, Kim JB, Moore ML et al (2007) A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 129:1365–1376. https://doi.org/10.1016/j.cell.2007.04.036
Jongbloed MRM, Vicente-Steijn R, Douglas YL et al (2011) Expression of Id2 in the second heart field and cardiac defects in Id2 knock-out mice. Dev Dyn 240:2561–2577. https://doi.org/10.1002/dvdy.22762
Zhang S-S, Kim K-H, Rosen A et al (2011) Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc Natl Acad Sci USA 108:13576–13581. https://doi.org/10.1073/pnas.1106911108
Koizumi A, Sasano T, Kimura W et al (2016) Genetic defects in a His-Purkinje system transcription factor, IRX3, cause lethal cardiac arrhythmias. Eur Heart J 37:1469–1475. https://doi.org/10.1093/eurheartj/ehv449
Thomas PS, Kasahara H, Edmonson AM et al (2001) Elevated expression of Nkx-2.5 in developing myocardial conduction cells. Anat Rec 263:307–313
Pashmforoush M, Lu JT, Chen H et al (2004) Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117:373–386
Tanaka M, Berul CI, Ishii M et al (2002) A mouse model of congenital heart disease: cardiac arrhythmias and atrial septal defect caused by haploinsufficiency of the cardiac transcription factor Csx/Nkx2.5. In: Cold Spring Harbor symposia on quantitative biology, vol 67, pp 317–325
Jay PY, Harris BS, Maguire CT et al (2004) Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest 113:1130–1137. https://doi.org/10.1172/JCI19846
Schott JJ, Benson DW, Basson CT et al (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281:108–111
Chen H, Shi S, Acosta L et al (2004) BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131:2219–2231. https://doi.org/10.1242/dev.01094
Shin CH, Liu Z-P, Passier R et al (2002) Modulation of cardiac growth and development by HOP, an unusual homeodomain protein. Cell 110:725–735
Ismat FA, Zhang M, Kook H et al (2005) Homeobox protein Hop functions in the adult cardiac conduction system. Circ Res 96:898–903. https://doi.org/10.1161/01.RES.0000163108.47258.f3
Benjamin EJ, Blaha MJ, Chiuve SE et al (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135:e146–e603. https://doi.org/10.1161/CIR.0000000000000485
John RM, Tedrow UB, Koplan BA et al (2012) Ventricular arrhythmias and sudden cardiac death. Lancet 380:1520–1529. https://doi.org/10.1016/S0140-6736(12)61413-5
Scheinman MM (2009) Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm 6:1050–1058. https://doi.org/10.1016/j.hrthm.2009.03.011
Kim EE, Shekhar A, Lu J et al (2014) PCP4 regulates Purkinje cell excitability and cardiac rhythmicity. J Clin Invest 124:5027–5036. https://doi.org/10.1172/JCI77495
Herron TJ, Milstein ML, Anumonwo J et al (2010) Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 7:1122–1128. https://doi.org/10.1016/j.hrthm.2010.06.010
Morley GE, Danik SB, Bernstein S et al (2005) Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc Natl Acad Sci USA 102:4126–4129. https://doi.org/10.1073/pnas.0500881102
Behradfar E, Nygren A, Vigmond EJ (2014) The role of Purkinje-myocardial coupling during ventricular arrhythmia: a modeling study. PLoS ONE 9:e88000. https://doi.org/10.1371/journal.pone.0088000
Gude NA, Emmanuel G, Wu W et al (2008) Activation of Notch-mediated protective signaling in the myocardium. Circ Res 102:1025–1035. https://doi.org/10.1161/CIRCRESAHA.107.164749
Bezzina CR, Barc J, Mizusawa Y et al (2013) Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 45:1044–1049. https://doi.org/10.1038/ng.2712
Benson DW, Silberbach GM, Kavanaugh-McHugh A et al (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573. https://doi.org/10.1172/JCI8154
Xie H, Chen X, Chen N, Zhou Q (2017) Sudden death in a male infant due to histiocytoid cardiomyopathy: an autopsy case and review of the literature. Am J Forensic Med Pathol 38:32–34. https://doi.org/10.1097/PAF.0000000000000289
Finsterer J (2008) Histiocytoid cardiomyopathy: a mitochondrial disorder. Clin Cardiol 31:225–227. https://doi.org/10.1002/clc.20224
Acknowledgements
This work was supported by the NIH Office of Director’s Pioneer Award LM012179-03, the American Heart Association Established Investigator Award 17EIA33410923, the Department of Pediatrics and Division of Pediatric Cardiology at Lucille Packard Children’s Hospital, the Stanford Cardiovascular Institute, the Stanford Division of Cardiovascular Medicine, Department of Medicine, the Institute for Stem Cell Biology and Regenerative Medicine, and an endowed faculty scholar award from the Stanford Child Health Research Institute/Lucile Packard Foundation for Children (S.M.W). This was also supported by the Training Grant (T32) entitled Research Training in Myocardial Biology at Stanford (NIH 2 T32 HL094274) (W.R.G.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
W.R.G. declares that he has no conflict of interest. S.M.W. declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Goodyer, W.R., Wu, S.M. Fates Aligned: Origins and Mechanisms of Ventricular Conduction System and Ventricular Wall Development. Pediatr Cardiol 39, 1090–1098 (2018). https://doi.org/10.1007/s00246-018-1869-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-018-1869-9