
Abstract
The goal of this study was to improve an already established reference method, such as the one devoted to organotin compounds determination (Reference Method for Marine Pollution Studies, No. 59, UNEP). The proposed upgrade consists of replacing the mechanical shaking by ultrasound energy and applying low temperature throughout the whole procedure. The optimization of the new operational conditions was performed by using a factorial design. Quality control was performed using a certified sediment reference material (PACS-2) for sediments (82.5–97% of recovery) and recoveries on spiked samples for suspended particulate matter (SPM) and mussels (94–100%). The proposed procedure was applied to surface sediment samples, SPM, and native bivalve mollusks (Brachidontes rodriguezii) collected in Bahia Blanca estuary, a very industrialized zone. The relative standard deviation (RSD %) of the environmental samples were less than 7.9%. It is important to note that the proposed procedure reduced the sample pretreatment time about seven times.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Organotin compounds (OTCs) are used in a wide range of industrial applications, including stabilizers in the PVC manufacture, biocides, fungicides, miticides, molluscicides, nematocides, ovicides, rodent repellents, catalysts, and wood preservatives (Hoch 2001). In particular, tributyltin (TBT) has been extensively used as biocide mainly in antifouling paints, until their complete ban on January 1, 2008 (Cavalheiro et al. 2016), whereas monobutyltin (MBT) and dibutyltin (DBT) are still widely employed as PVC stabilizers (Hoch 2001). These numerous applications directly or indirectly contribute to a significant input of OTCs to the marine environment.
TBT degrades into the environment, giving rise to DBT and MBT. The final degradation product is inorganic tin (Sn4+), which is stable (Hoch 2001). Degradation of TBT can take place via chemical, photochemical, and/or biological pathways (Furdek et al. 2016; Graceli et al. 2013).
Bioavailability, mobility, toxicity, and physicochemical properties of OTCs are closely linked to their chemical form (de Carvalho and Santelli 2010). The effects caused by TBT are the most known, and they have been a major source of concern (Bao et al. 2011; Meyer et al. 2012; Pagliarani et al. 2010). Monobutyltin is considered the least harmful form, and there is not enough available toxicological data indicating significant toxicity in the marine environment (Herzke et al. 2007). On the contrary, DBT is considerably more toxic than the monosubstituted derivative. The deleterious effects of this compound are probably due to their ability to penetrate cell membranes, interfere with the cell’s electron transfer mechanism, and interact with mitochondrial respiratory complexes (Nesci et al. 2011).
Hence, the determination of the different OTCs and their distribution is relevant and could facilitate a “potential hazard” definition for these compounds (Rosenberg 2005). In this sense, the development of fast, reliable, and accurate analytical methods able to perform the speciation of OTCs in environmental matrices constitutes an area of increasingly active research (de Carvalho and Santelli 2010). Moreover, these analytical methods should comply with the requirements of legislation regarding the minimum permitted levels.
In this context, many analytical methods have been developed for the OTCs analysis mainly based on gas chromatography (GC) coupled to different detectors (de Carvalho and Santelli 2010; Graceli et al. 2013; Morabito et al. 2000), which require a previous derivatization to make the OTCs—typically polar and nonvolatile—compatible with the chromatographic separation, i.e., volatile and thermally stable species (Antizar-Ladislao 2008). Moreover, the derivatization process can be considered a critical step in the analysis because of the possible low yields and analyte loss that may occur during this stage (de Carvalho and Santelli 2010). Several derivatization techniques were used, involving either Grignard reagents (dos Santos et al. 2013; Tsunoi et al. 2002), sodium borohydride (NaBH4) (Gui-bin et al. 2001; Neng et al. 2014), or sodium tetraethylborate (NaBEt4) (Kannan et al. 2010; Lagerström et al. 2017; Okoro et al. 2016; Smitiene et al. 2014). However, the main disadvantage of Grignard derivatization is the need for a careful removal of the excess of reagents, which increase the number of steps and the operating time (Brunori et al. 2006). On the other hand, the major drawback of using alkylation reagents are the complex manipulation under an inert gas atmosphere, low availability, prohibition of air transportation, and high cost of acquisition and storage. Furthermore, specialized technical skills and particular laboratory safety conditions are required (Yañez et al. 2016). By contrast, the advantages of using NaBH4 include the low cost, straightforward sample handling, higher reaction rate, and enhanced yield (Yañez et al. 2016).
In addition, the extraction of the OTCs from the sample matrix is an important issue. Many extraction techniques have been used, assisted by different procedures for improving the contact between sample and extractant, such as shaking (Cassi et al. 2008; Choi et al. 2014; Kim et al. 2017; Okoro et al. 2016), vortex (Bhosle et al. 2004; Dong et al. 2015), and, in the last decades, ultrasound (Filipkowska and Lubecki 2016; Kucuksezgin et al. 2011; Wang et al. 2008; Zhang et al. 2013). The high temperatures and pressures generated during the sonication process (implosion) accelerate and improve the sample treatment in different kind of samples. In this way, higher yields, reduced operation time, shorter reaction times, and milder conditions are achieved (Antizar-Ladislao 2008).
Despite the great amount of methods for OTCs determination found in the literature, only a few standardized methods are available that could provide a systematic and integrated measure of the toxicological and environmental risk assessment (AFNOR NF T 90-250 2005; ISO/TC 147/WG44 2003; UNEP/IOC/IAEA 1994).
In this study, we proposed some modifications to the Reference Method for Marine Pollution Studies (UNEP/IOC/IAEA 1994) for organotin compounds determination in different environmental samples. The principal changes consisted of implementing low temperature throughout the whole procedure and applying ultrasound energy (US) on the extraction and derivatization steps. The speciation of butyltin compounds (TBT, DBT, and MBT) in MPS, sediments, and mussels samples from the Bahía Blanca Estuary (Argentina) was satisfactorily achieved by using the proposed modifications.
Materials and Methods
Sampling
The Bahía Blanca estuary (38°45′–39°40′S, 61°45′–62°30′W) is located on the southwestern Atlantic Ocean coast, Argentina (Fig. 1). Six sampling sites, affected by different intensities of human and industrial activities, were considered: S1: Rosales Harbor (ship-repair yard); S2: Luis Piedra Buena thermoelectric plant; S3 and S4: Galván Harbor (industrial harbor, gas, and oil loading buoy); S5: Cuatreros Harbor (fishing and recreational port); and S6: Villarino Viejo (rural zone).

Sampling sites at the Bahía Blanca Estuary, Argentina
Sediment samples were collected using a stainless-steel grab sampler and an acrylic corer. The samples were taken in the superficial layer (1–5 cm). Several portions were collected in the same sampling point to achieve representative samples (composite samples). After collection, the samples were refrigerated, stored on solvent-cleaned amber glass containers avoiding exposure to light, and then rapidly transported to the laboratory. For the organotin compound extraction, sediments were frozen at − 20 °C and then lyophilized, powdered, sieved (250 mesh), and stored at 4 °C until analysis.
Mussels (Brachidontes rodriguezii) were collected from natural banks, dock columns, platforms, surface sediments, or in surface water (0–1 m), according to their availability. An average of 60 mussels from each site was used to make a composite sample. Mussels were immediately transferred to the laboratory after collection in a portable cooler. Then, they were washed with distillated water and finally stored at − 20 °C until extraction. Before analysis, mussels were homogenized, lyophilized, smashed in a mortar, and stored at 4 °C.
Suspended particulate matter (SPM) was obtained by filtration of seawater. Typically 4 L of seawater, collected in amber glass bottles, were carried to the laboratory in a portable cooler in the dark. The water was immediately filtered through a 0.4-μm pore size polycarbonate membrane (Millipore® HTTP 04700) with a vacuum pump. Before using, membranes were treated with 0.7% nitric acid, rinse with deionized water, and dried at 50 °C until constant weight. After, they were frozen (− 20 °C), lyophilized, weighed, and stored at 4 °C until used.
Reagents and Standards
Butyltin Trichloride (MBT, 97%), Dibutyltin dichloride (DBT, 97%), Tributyltin chloride (TBT, 96%), and Tetrabutyltin (TeBT, 93%), used as internal standard, were obtained from SIGMA-ALDRICH. Tripropyltin chloride, used as recovery control standard (TPrT, > 99%), was purchased from LGC Promochem. Standard stock solutions of MBT, DBT, TBT, TPrT, and TeBT were prepared in chromatographic grade methanol (U.V.E.) (MERCK) to obtain solutions containing 1 mg mL−1 of tin for each compound.
Before use, all containers were soaked in 10% HNO3 for 24 h and rinsed with doubly distilled water. All standard solutions were stored in amber flask and vials at 0 °C in the darkness (UNEP/IOC/IAEA 1994).
Sodium borohydride (NaBH4, > 96%) was purchased from Fluka Chemika. Sodium hydroxide methanolic solution was prepared by dissolving 1.0 g of sodium hydroxide (98%) in 1 L of chromatographic-grade methanol (U.V.E.) to get a 0.1% solution (UNEP/IOC/IAEA 1994). Chromatographic-grade hexane (U.V.E) was provided by MERCK, and ultrapure deionized water (18.3 Ω cm−1, Barnstead, Dubuque, USA) was used. Silica gel 60 (0.063–0.200 mm) was used for column chromatography (MERCK).
PACS-2 marine sediment reference material (National Research Council of Canada, Ottawa, Canada) was used for assessing the method accuracy.
Apparatus and Instrumentation
An ultrasonic bath TESTLAB (model TB 04 CTPD, with an ultrasound power of 160 W (real ultrasonic power: 100 W) and a frequency of 40 kHz was used. Derivatized compounds were measured by gas chromatography (Agilent 7890 B) coupled to a mass spectrometer (Agilent 5977A). The chromatographic conditions and selected ions are shown in Table 1.
Procedure
The UNEP method is described elsewhere (UNEP/IOC/IAEA 1994). The modified procedure involved approximately 2 g of dry sample (sediment or mussels) or 0.25 g of dry SPM, to which 2 mL of ultrapure deionized water and 100 μL of tripropyltin (surrogate recovery standard) were added in a 50-mL centrifuge tube. The extraction of the OTCs was performed by adding 10 mL of 0.1% sodium hydroxide methanolic solution (4:1 methanol/water) in an ice bath, with application of 8 min of ultrasound (US). Then, 5 mL of hexane and approximately 100 mg of sodium borohydride were added for derivatization of the OTCs. Then, 1 min of US was applied, and the tube was left to stand for 1 h at 0 °C. The samples were dried with anhydrous sodium sulphate and finally centrifuged at 4000 rpm for 20 min at 4 °C to prevent losses by evaporation.
-
For sediment and SPM samples, the hexane layer containing the derivatized compounds was put into an amber glass vial, and the volume was reduced to 1 mL by a gentle stream of pure nitrogen.
-
For mussel samples, the hexane layer was seeded in silica gel columns to perform the sample clean-up. OTCs were eluted with 4 mL of hexane, and the volume was reduced to 1 mL by a gentle stream of pure nitrogen.
Before injection in the gas chromatograph, 7 μL of TeBT standard solution (internal standard) was added. A comparison between the original UNEP method and the modified one is depicted in Fig. 2.

The UNEP reference method and the introduced modifications
Results and Discussion
Modifications to the UNEP Method
The application of UNEP method (UNEP/IOC/IAEA 1994) to the certified reference material PACS-2 and mussels samples was not satisfactory, mainly due to a considerable difficulty in detecting MBT, probably due to its volatility after derivatization step (Colombini et al. 2004). Also, low recoveries and high relative standard deviation (RSD) values were obtained for TBT and DBT. Accordingly, some modifications were introduced in the UNEP method. The first one was the application of low temperatures (0 °C) throughout the whole procedure, but especially for the derivatization step. Working at low temperatures allowed us to control this stage, avoiding losses of volatile compounds and improving the precision of the results.
Even though MBT was detected after the application of low temperatures, the recoveries were still unsatisfactory. In particular for sediments and SPM, MBT seemed to be more strongly bounded to the sample matrix than DBT and TBT (Quintas et al. 2016). This fact could be attributed to an insufficient desorption of MBT from the sediment. Some studies have indicated that the strength of MBT adsorption is favored by its polarity (Bravo et al. 2015).
Thus, harder extraction conditions were needed to extract quantitatively the MBT. Therefore, we applied US instead of mechanical stirring in order to achieve higher recoveries for the three analytes and better precision (Fig. 3).

OTC levels in PACS-2 and spiked mussels samples, with the corresponding standard deviations
In the case of biological samples, it was essential to perform a clean-up step before the injection in CG–MS. Although UNEP method does not apply a clean-up step, it recommends its use to preserve the working life of the chromatographic column. Despite the fact that methanol induces rapid bursting of the cells and facilitates the homogenization of biological materials due to its osmotic activity (Brunori et al. 2006), sample extracts with high levels of fats can contain substances susceptible to be co-extracted and deposited on the chromatographic column, reducing the efficiency of the separation (de Oliveira et al. 2010; dos Santos et al. 2013). The clean-up step was introduced to minimize these issues.
Optimization of the US Application
The application of US was optimized to maximize the % recovery (response variable). Optimization of the time for extraction (factor A) and derivatization (factor B) in the OTCs extraction/derivatization process was carried out using a full factorial design at two levels, with four central points to estimate the experimental error (Maran et al. 2013). Each point of the experimental design was randomly performed in duplicate. The factor A was studied in the interval between 4 and 8 min, whereas the factor B was studied between 1 and 3 min. The statistical design and analysis of the response variables were supported by statistical graphics software (STATGRAPHICS Plus, version 5.1 STSC, Rockville, MD).
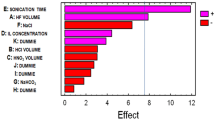
The main effects (A and B) and their interaction (AB) were estimated. For example, Fig. 4 shows the results obtained for DBT, because the results were the same for the three study analytes. The Pareto chart (Fig. 4a) shows the significance of the responses. Only the interaction (AB) can be considered significant, showing a negative effect.

a Pareto chart of the standardized effects in the 22 factorial design for DBT. The red line indicates the confidence level of 95%. b Interaction plot of (A) extraction time versus (B) derivatization time for DBT
The optimum working conditions found after analysis were 8 min for extraction time and 1 min for derivatization time. This behavior could be explained in terms of the interaction AB (Fig. 4b). When A increases, a better response is obtained for the lower value of B. The same results were obtained for the other analyzed compounds (TBT and MBT) and for all the studied sample matrices. Therefore, the optimal conditions were applied for the different analyzed environmental samples.
Analytical Figures of Merit
Quality parameters of the US-UNEP method were evaluated for sediments and mussels. SPM samples were analyzed with the same straight line regression than sediments (Bhosle et al. 2004). The analytical figures of merit are shown in Table 2. Linearity evaluation was performed by applying the ANOVA test, and no lack of fit for the linear models was observed (p > 0.30).
The limit of detection (LOD) for all target analytes were calculated as three times the baseline noise of chromatograms (signal to noise ratio, S/N) (Cassi et al. 2008; Choi et al. 2014) and the limit of quantification (LOQ) as ten times the above mentioned ratio. The LOD and LOQ values were, in all cases, lower for US-UNEP method than for the unmodified UNEP one, for which the LOD values were 5, 10, and 20 ng Sn per gram of sample (either sediment or mussel) for TBT, DBT, and MBT respectively.
Most of the methods found in the literature for OTCs determination using US-assisted pretreatment informed similar limits of detection respect to the method proposed in the current study. Some authors (Filipkowska and Lubecki 2016; Kucuksezgin et al. 2011; Tang and Wang 2007; Tang et al. 2010) reported better detection limits. However, the sonication times were higher, and the overall pretreatment time was much greater than the one used in the proposed method. Also, Wang et al. (2008) report low LOD for OTCs determination in sediments, but the sonication time in the extraction step was 1 h.
Quality Control
The traceability of the US-UNEP method was evaluated using the PACS-2 reference material for sediments and spiked samples for the other matrices. The recoveries obtained ranged between 82.5 and 97% for the reference material and between 94 and 100% for the other samples (Fig. 3).
Precision was evaluated under repeatability conditions and was estimated as RSD (%) of three determinations and was found to vary between 1.1 and 1.9% for the determinations in PACS-2 reference material and between 0.9 and 7.1% for the spiked samples. The results were in accordance with analytical validation recommendations (EURACHEM 1998; Thompson et al. 2002).
The recoveries and precision obtained were satisfactory and comparable with other studies found in the bibliography, which also made OTCs determinations on PACS-2 reference material. For instance, Konieczka et al. (2007) and Nichols et al. (2014) applied pressurized liquid extraction and report recoveries only for TBT (104% (RSD: 1.8%) and 72.4% (RSD: 6.8%), respectively), although the method involved several steps and higher times for the extraction/derivatization process. Other authors report very good recoveries and RSD % but applied conventional stirring within an interval of 12–24 h (Bravo et al. 2005, 2012; Pinochet et al. 2009). A different kind of determination of OTCs during a photodegradation study obtained recoveries among 82.7 and 105% with good RSD % values. The extraction also was performed using mechanical stirring during about 12 h (Brosillon et al. 2014). Moreover, to accelerate the extraction process, some authors have applied ultrasound energy. For instance, Yu et al. (2011) studied TBT and DBT with recoveries lower than 87%. Better recoveries were obtained by de Oliveira et al. but RSD % ranged between 5.8 and 25.7 (de Oliveira et al. 2010). Similar results were obtained by Carvalho et al. (2007) but using higher sonication times (approximately 2 h). Finally, Xiao et al. combined US (30 min) and headspace single drop microextraction (HS-SDME). The recoveries ranged from 67.3 to 94.4% with RSD % between 5.2 and 13.6 (Xiao et al. 2008).
Concerning the suspended particulate matter (SPM), other studies in the literature had similar results. dos Santos et al. (2013, 2016) obtained recoveries of 98–110 (RSD < 20%) after applying 5 min of US, three times. Bhosle et al. (2004) informed recoveries of 96% for TBT and 82% for DBT for spiked SPM samples with RSD % between 3 and 7%, using vortex for the extraction/derivatization process. Wang et al. (2008) applied different methods for sediments and SPM. The recoveries obtained ranged between 93 (DBT) and 122 (TBT) %. The extraction was performed using 10 min of mechanical stirring. On the other hand, Tang et al. (2010) used ultrasound energy in the extraction step and the recoveries of the spiked samples ranged between 115 and 141%.
Regarding the samples of mussels, both mechanical shaking and ultrasound are used either in extraction and derivatization steps. Normally, the mechanical shaking was used for long periods of time. Tang et al. obtained recoveries between 83 and 151% for spiked samples after digestion of 1 and 1 h of mechanical shaking (Tang et al. 2010). Kim et al. (2017) reported recoveries for CRM 477 mussels’ tissue reference material among 85 and 116% for TBT, 97 and 115% for DBT, and 74–92% for MBT, using mechanical stirring for 3 h. Other authors employed shaking overnight and obtained recoveries between 70 and 120% for the same reference material (Devier et al. 2005; Turja et al. 2014). As mentioned, several studies used ultrasound with different application times. For instance, Artifon et al. (2016) applied 15 min of US; the recoveries were 84% for TBT and 92% for both DBT and MBT for the CRM 477 reference material, whereas the recoveries for spiked samples were between 88.5 and 109%. Filipkowska and Lubecki (2016) used US for 15 min as well, but the recoveries on spiked samples were lower, i.e., 53% for TBT, 84% for DBT, and 58% for MBT. Other authors used larger times of US application, such as Kucuksezgin et al. (2011), who applied US to the sample for 1.5 h with recoveries of 87% for TBT, 99% for DBT, and 105% for MBT (CRM 477 reference material). Noventa et al. (2015) used even more time of sonication (2 h), and the recoveries obtained for the same reference material were similar (87% for TBT, 92% for DBT, and 132% for MBT).
Application to Environmental Samples
Figure 5 shows the levels of TBT, DBT, and MBT found in the sediment, SPM, and mussel samples in the Bahía Blanca Estuary expressed in ng g Sn g−1 on dry sample basis.
The RSD % values are in accordance with the analysis recommendations for analytical validation, i.e., < 20% (Eurachem 1998; Thompson et al. 2002). In general, for all cases, the mean values of RSD % are lower than those found in the literature with the same derivatization reagent (NaBH4), using either orbital shaking or US assistance (Bhosle et al. 2004; Delucchi et al. 2011; Xiao et al. 2008). The RSD % values obtained for the biological samples were higher than those achieved for sediment and SPM samples, probably due to the addition of a clean-up step, which increased the number of manipulations in the mussel samples.

Levels of TBT, DBT, and MBT—with the respective standard deviations—in samples of sediments (a), Brachidontes rodriguezii mussels (b), and SPM (c), expressed in ng Sn g−1 dry wt (blue square box) TBT, (pink square box) DBT, (violet square box) MBT
The obtained results show that all the samples in all the studied sites have been impacted by the presence of OTCs. The higher values of TBT in sediments could be caused by the presence of numerous boats along the estuary, both commercial and recreational, especially at S4 (Fig. 5a). This site is located in the vicinity of an industrial hub and the discharge of the effluents´ collector, which is a potential source of MBT and DBT. In general, high temperatures and more intense solar radiation in summer provoke an increase in the metabolic activity of microorganisms, which leads to an increase in the TBT degradation and higher values of DBT concentration (Hoch and Schwesig 2004).
In the case of mollusks, in general, lower values of OTCs were obtained. These values are much lower than those found in sediment samples, especially for TBT and DBT. This fact could indicate that B. rodriguezii mussels, as well as other species of bivalve mollusks, have a great capacity to eliminate TBT from their organisms (either by degradation or excretion). The higher the temperature of the mussels’ environment, the greater is the degradation of OTCs (Chandrinou et al. 2007; Hsia and Liu 2003). Finally, as expected, OTCs concentrations obtained in SPM samples are much higher than those found in other matrices. This fact takes place as a consequence of the large surface/volume ratio of the SPM, which makes it a large pollutant sink in the marine environment (Ernst et al. 1993). The OTCs present in the marine ecosystems, due to their hydrophobicity and/or their positive charge, are rapidly adsorbed over the organic and/or mineral phases of the suspended particulate matter (Arnold et al. 1997; Berg et al. 2001). High levels of the three studied compounds were found for SPM at sites S1 and S4. In proximity to S1, sewage discharges could result in high concentrations of DBT and MBT caused by the biodegradation of TBT. In the same way, the high concentrations of DBT found at S4 probably do not only come from the degradation of TBT. There may be additional inputs of DBT, which is a compound used in the PVC industry as a stabilizing agent.
As mentioned, the entire study area seems to be polluted by TBT and its degradation products. As discussed in our previous studies (Quintas et al. 2016), such OTCs distribution could be the result of the tidal currents that characterize the estuary, as well as the dredging processes periodically performed on the Main Channel.
Conclusions
A rapid and reliable method was achieved for OTCs determination with low LOD, high recovery rates, and low RSD %. We have improved the UNEP Reference Method, consuming less time analysis for the determination of the three analytes of interest (TBT, DBT, and MBT) in three environmental compartments (sediment, SPM, and mussels) using the same analytical method.
The use of US energy not only enabled the extraction and derivatization of the analytes but also made it possible to diminish the LOD and the total time of analysis. In the extraction step, the time reduction was from 45 to 8 min and from 15 to 1 min for the derivatization step, i.e., approximately seven times less than the original UNEP Reference Method. This issue is especially important in environmental monitoring programs that are required to face further studies of a wider scope, involving the seasonal behavior of organotin compounds in the area of study.
References
AFNOR NF T 90-250 “Dosage de composés organo-étains dans les sédiments- Méthode par chromatographie en phase gazeuse” (Project 2005)
Antizar-Ladislao B (2008) Environmental levels, toxicity and human exposure to tributyltin (TBT)-contaminated marine environment. A review. Environ Int 34:292–308. https://doi.org/10.1016/j.envint.2007.09.005
Arnold CG, Weidenhaupt A, David MM, Müller SR, Haderlein SB, Schwarzenbach RP (1997) Aqueous speciation and 1-octanol-water partitioning of tributyl-and triphenyltin: effect of pH and ion composition. Environ Sci Technol 31:2596–2602. https://doi.org/10.1021/es970009+
Artifon V, Castro ÍB, Fillmann G (2016) Spatiotemporal appraisal of TBT contamination and imposex along a tropical bay (Todos os Santos Bay, Brazil). Environ Sci Pollut Res 23:16047–16055. https://doi.org/10.1007/s11356-016-6745-7
Bao VWW, Leung KMY, Qiu J-W, Lam MHW (2011) Acute toxicities of five commonly used antifouling booster biocides to selected subtropical and cosmopolitan marine species. Mar Pollut Bull 62:1147–1151. https://doi.org/10.1016/j.marpolbul.2011.02.041
Berg M, Arnold CG, Müller SR, Mühlemann J, Schwarzenbach RP (2001) Sorption and desorption behavior of organotin compounds in sediment-pore water systems. Environ Sci Technol 35:3151–3157. https://doi.org/10.1021/es010010f
Bhosle NB, Garg A, Jadhav S, Harjee R, Sawant SS, Venkat K, Anil AC (2004) Butyltins in water, biofilm, animals and sediments of the west coast of India. Chemosphere 57:897–907. https://doi.org/10.1016/j.chemosphere.2004.06.037
Bravo M, Lespes G, De Gregori I, Pinochet H, Gautier MP (2005) Determination of organotin compounds by headspace solid-phase microextraction–gas chromatography–pulsed flame-photometric detection (HS-SPME–GC–PFPD). Anal Bioanal Chem 383:1082–1089. https://doi.org/10.1007/s00216-005-0131-5
Bravo M, Valenzuela A, Fuentes E, Quiroz W (2012) Critical evaluation of fiber coatings for organotin determination by using solid phase microextraction in headspace mode. J Chromatogr 1223:9–14. https://doi.org/10.1016/j.chroma.2011.12.038
Bravo MA, Flores M, Parra S, Quiroz W, Maxwell P, Mester Z (2015) Chemometric optimization of an extraction procedure using tartaric acid for butyltin compounds from sediment samples by GC-PFPD. J Chil Chem Soc 60:2803–2806. https://doi.org/10.4067/S0717-97072015000100006
Brosillon S, Bancon-Montigny C, Mendret J (2014) Study of photocatalytic degradation of tributyltin, dibutylin and monobutyltin in water and marine sediments. Chemosphere 109:173–179. https://doi.org/10.1016/j.chemosphere.2014.02.008
Brunori C, Ipolyi I, Massanisso P, Morabito R (2006) New trends in sample preparation methods for the determination of organotin compounds in marine matrices. In: Konstantinou IK (ed) Antifouling paint biocides. Springer, Berlin, pp 51–70
Carvalho PN, Pinto LF, Basto MCP, Vasconcelos MTS (2007) Headspace solid-phase micro-extraction and gas chromatography-ion trap tandem mass spectrometry method for butyltin analysis in sediments: optimization and validation. Microchem J 87:147–153. https://doi.org/10.1016/j.microc.2007.07.003
Cassi R, Tolosa I, de Mora S (2008) A survey of antifoulants in sediments from Ports and Marinas along the French Mediterranean coast. Marine Poll Bull 56:1943–1948. https://doi.org/10.1016/j.marpolbul.2008.07.011
Cavalheiro J, Sola C, Baldanza J, Tessier E, Lestremau F, Botta F, Preud’hommea H, Monperrusa M, Amouroux D (2016) Assessment of background concentrations of organometallic compounds (methylmercury, ethyllead and butyl-and phenyltin) in French aquatic environments. Water Res 94:32–41. https://doi.org/10.1016/j.watres.2016.02.010
Chandrinou S, Stasinakis AS, Thomaidis NS, Nikolaou A, Wegener JW (2007) Distribution of organotin compounds in the bivalves of the Aegean Sea, Greece. Environ Int 33:226–232. https://doi.org/10.1016/j.envint.2006.09.010
Choi JY, Hong GH, Ra K, Kim KT, Kim K (2014) Magnetic characteristics of sediment grains concurrently contaminated with TBT and metals near a shipyard in Busan, Korea. Marine Poll Bull 85:679–685. https://doi.org/10.1016/j.marpolbul.2014.03.029
Colombini V, Bancon-Montigny C, Yang L, Maxwell P, Sturgeon RE, Mester Z (2004) Headspace single-drop microextration for the detection of organotin compounds. Talanta 63:555–560. https://doi.org/10.1016/j.talanta.2003.11.035
de Carvalho Oliveira R, Santelli RE (2010) Occurrence and chemical speciation analysis of organotin compounds in the environment: a review. Talanta 82:9–24. https://doi.org/10.1016/j.talanta.2010.04.046
de Oliveira CR, dos Santos D, dos Santos Madureira LA, de Marchi MRR (2010) Speciation of butyltin derivatives in surface sediments of three southern Brazilian harbors. J Hazard Mater 181:851–856. https://doi.org/10.1016/j.jhazmat.2010.05.091
Delucchi F, Narvarte MA, Amin O, Tombesi NB, Freije H, Marcovecchio J (2011) Organotin compounds in sediments of three coastal environments from the Patagonian shore, Argentina. Int J Environ Waste Manag 8:3–17. https://doi.org/10.1007/s10661-006-9547-4
Devier MH, Augagneur S, Budzinski H, Le Menach K, Mora P, Narbonne JF, Garrigues P (2005) One-year monitoring survey of organic compounds (PAHs, PCBs, TBT), heavy metals and biomarkers in blue mussels from the Arcachon Bay, France. J Environ Monit 7:224–240. https://doi.org/10.1039/B409577D
Dong CD, Chen CF, Chen CW (2015) Composition and source of butyltins in sediments of Kaohsiung Harbor, Taiwan. Estuar Coast Shelf Sci 156:134–143. https://doi.org/10.1016/j.ecss.2014.08.002
dos Santos DM, de Marchi MRR, Godoi AFL, Turra A, Montone RC (2013) Matrix effect on butyltin analysis of sediments and fish tissues by GC-PFPD. J Braz Chem Soc 24:998–1005. https://doi.org/10.5935/0103-5053.20130129
dos Santos DM, Turra A, de Marchi MRR, Montone RC (2016) Distribution of butyltin compounds in Brazil’s southern and southeastern estuarine ecosystems: assessment of spatial scale and compartments. Environ Sci Pollut Res 23:16152–16163. https://doi.org/10.1007/s11356-016-6720-3
Ernst W, Boon JP, Weber K (1993) Occurrence and fate of organic micropollutants in the North Sea. In: Salomons W, Bayne BL, Duursma EK, Förstner U (eds) Pollution of the North Sea. Springer, Berlin, pp 284–299
EURACHEM (1998) The fitness for purpose of analytical methods. A laboratory guide to method validation and related topics. LGC (Teddington) Ltd, London 75 pp
Filipkowska A, Lubecki L (2016) Endocrine disruptors in blue mussels and sediments from the Gulf of Gdańsk (Southern Baltic). Environ Sci Pollut Res 23:13864–13876. https://doi.org/10.1007/s11356-016-6524-5
Furdek M, Mikac N, Bueno M, Tessier E, Cavalheiro J, Monperrus M (2016) Organotin persistence in contaminated marine sediments and pore waters: in situ degradation study using species-specific stable isotopic tracers. J Hazard Mater 307:263–273. https://doi.org/10.1016/j.jhazmat.2015.12.07
Graceli JB, Sena GC, Lopes PFI, Zamprogno GC, da Costa MB, Godoi AFL et al (2013) Organotins: a review of their reproductive toxicity, biochemistry, and environmental fate. Toxicol 36:40–52. https://doi.org/10.1016/j.reprotox.2012.11.008
Gui-bin J, Qun-fang Z, Ji-yan L, Di-jing W (2001) Occurrence of butyltin compounds in the waters of selected lakes, rivers and coastal environments from China. Environ Pollut 115:81–87. https://doi.org/10.1016/S0269-7491(01)00088-4
Herzke D, Schlabach M, Mariussen E (2007) Literature survey of polyfluorinated organic compounds, phosphor containing flame retardants, 3-nitrobenzanthrone, organic tin compounds, platinum and silver. Universita NILU
Hoch M (2001) Organotin compounds in the environment—an overview. Appl Geochem 16:719–743. https://doi.org/10.1016/S0883-2927(00)00067-6
Hoch M, Schwesig D (2004) Parameters controlling the partitioning of tributyltin (TBT) in aquatic systems. Appl Geochem 19:323–334. https://doi.org/10.1016/S0883-2927(03)00131-8
Hsia MP, Liu SM (2003) Accumulation of organotin compounds in Pacific oysters, Crassostrea gigas, collected from aquaculture sites in Taiwan. Sci Total Environ 313:41–48. https://doi.org/10.1016/S0048-9697(03)00329-2
ISO/TC 147/WG44 (2003) Water quality-Determination of selected organotin compounds by gas chromatography
Kannan K, Takahashi S, Fujiwara N, Mizukawa H, Tanabe S (2010) Organotin compounds, including butyltins and octyltins, in house dust from Albany, New York, USA. Arch Environ Contam Toxicol 58:901–907. https://doi.org/10.1007/s00244-010-9513-6
Kim T, Jeon S, Hong S, Song SJ, Kwon BO, Ryu J, Khim JS (2017) Spatiotemporal distributions of butyltin compounds in various intertidal organisms along the Samcheok and Tongyeong coasts of Korea. Chemosphere 172:268–277. https://doi.org/10.1016/j.chemosphere.2016.12.152
Konieczka P, Sejerøe-Olsen B, Linsinger TP, Schimmel H (2007) Determination of tributyltin (TBT) in marine sediment using pressurised liquid extraction–gas chromatography–isotope dilution mass spectrometry (PLE–GC–IDMS) with a hexane–tropolone mixture. Anal Bioanal Chem 388:975–997. https://doi.org/10.1007/s00216-007-1285-0
Kucuksezgin F, Aydin-Onen S, Gonul LT, Pazi I, Kocak F (2011) Assessment of organotin (butyltin species) contamination in marine biota from the Eastern Aegean Sea, Turkey. Marine Poll Bull 62:1984–1988. https://doi.org/10.1016/j.marpolbul.2011.06.020
Lagerström M, Strand J, Eklund B, Ytreberg E (2017) Total tin and organotin speciation in historic layers of antifouling paint on leisure boat hulls. Environ Pollut 220:1333–1341. https://doi.org/10.1016/j.envpol.2016.11.001
Maran JP, Mekala V, Manikandan S (2013) Modeling and optimization of ultrasound-assisted extraction of polysaccharide from Cucurbita moschata. Carbohydr Polym 92:2018–2026. https://doi.org/10.1016/j.carbpol.2012.11.086
Meyer A, Strajhar P, Murer C, Da Cunha T, Odermatt A (2012) Species-specific differences in inhibition of human and zebrafish 11b hydroxysteroid dehydrogenase 2 by thiram and organotins. Toxicology 301:72–78. https://doi.org/10.1016/j.tox.2012.07.001
Morabito R, Massanisso P, Quevauviller P (2000) Derivatization methods for the determination of organotin compounds in environmental samples. TrAC Trends Analyt Chem 19:113–119. https://doi.org/10.1016/S0165-9936(99)00196-X
Neng NR, Santalla RP, Nogueira JMF (2014) Determination of tributyltin in environmental water matrices using stir bar sorptive extraction with in situ derivatisation and large volume injection-gas chromatography–mass spectrometry. Talanta 126:8–11. https://doi.org/10.1016/j.talanta.2014.03.021
Nesci S, Ventrella V, Trombetti F, Pirini M, Pagliarani A (2011) Tributyltin (TBT) and mitochondrial respiration in mussel digestive gland. Toxicol In Vitro 25:951–959. https://doi.org/10.1016/j.tiv.2011.03.004
Nichols DS, Jordan TB, Kerr N (2014) Determination of tributyltin in marine sediment and waters by pressurised solvent extraction and liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 406:2993–2998. https://doi.org/10.1007/s00216-014-7683-1
Noventa S, Barbaro J, Formalewicz M, Gion C, Rampazzo F, Brusà RB, Gabellini M, Berto D (2015) A fast and effective routine method based on HS-SPME–GC–MS/MS for the analysis of organotin compounds in biota samples. Anal Chim Acta 858:66–73. https://doi.org/10.1016/j.aca.2014.11.028
Okoro HK, Fatoki OS, Adekola FA, Ximba BJ, Snyman RG (2016) Spatio-temporal variation of organotin compounds in seawater and sediments from Cape Town harbour, South Africa using gas chromatography with flame photometric detector (GC-FPD). Arab J Chem 9:95–104. https://doi.org/10.1016/j.arabjc.2013.05.014
Pagliarani A, Nesci S, Trombetti F, Ventrella V (2010) Organotin effects on membrane-bound ATPase activities. In: Chin HF (ed) Organometallic compounds: preparation structure and properties. Nova Science Publishers Inc., New York, pp 225–253
Pinochet H, Tessini C, Bravo M, Quiroz W, De Gregori I (2009) Butyltin compounds and their relation with organic matter in marine sediments from San Vicente Bay-Chile. Environ Monit Assess 155:341–353. https://doi.org/10.1007/s10661-008-0439-7
Quintas PY, Oliva AL, Arias A, Domini CE, Alvarez MB, Garrido M, Marcovecchio JE (2016) Seasonal changes in organotin compounds in sediments from the Bahía Blanca Estuary. Environ Earth Sci 75:1–13. https://doi.org/10.1007/s12665-016-5471-2
Rosenberg E (2005) Speciation of tin. In: Cornelis R, Caruso J, Crews H, Heumann K (eds) Handbook of elemental speciation II—species in the environment, food, medicine and occupational health, Chichester, Chapter 2.20
Smitiene V, Semasko I, Vickackaite V (2014) Speciation of methyltins by dispersive liquid–liquid microextraction and gas chromatography with mass spectrometry. J Sep Sci 37:1989–1995. https://doi.org/10.1002/jssc.201400074
Tang CH, Wang WH (2007) Optimization of an analytical method for determining organotin compounds in fish tissue by base-hydrolysis pretreatment and simultaneous ethylation–extraction procedures. Anal Chim Acta 581:370–376. https://doi.org/10.1016/j.aca.2006.08.029
Tang CH, Hsu CH, Wang WH (2010) Butyltin accumulation in marine bivalves under field conditions in Taiwan. Mar Environ Res 70:125–132. https://doi.org/10.1016/j.marenvres.2010.03.011
Thompson M, Ellison SL, Wood R (2002) Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl Chem 74:835–855. https://doi.org/10.1351/pac200274050835
Tsunoi S, Matoba T, Shioji H, Harino H, Tanaka M (2002) Analysis of organotin compounds by Grignard derivatization and gas chromatography–ion trap tandem mass spectrometry. J Chromatogr A 962:197–206. https://doi.org/10.1016/s0021-9673(02)00534-4
Turja R, Höher N, Snoeijs P, Baršienė J, Butrimavičienė L, Kuznetsova T, Kholodkevich SV, Budzinskig MH, Lehtonen KK (2014) A multibiomarker approach to the assessment of pollution impacts in two Baltic Sea coastal areas in Sweden using caged mussels (Mytilus trossulus). Sci Total Environ 473:398–409. https://doi.org/10.1016/j.scitotenv.2013.12.038
UNEP/IOC/IAEA (1994) (United Nations Environment Programme/Intergovernmental Oceanographic Commission/International Atomic Energy Agency) Determination of organotins in environmental samples. Reference methods for marine pollution studies, no. 59. UNEP, pp 28–30
Wang X, Hong H, Zhao D, Hong L (2008) Environmental behavior of organotin compounds in the coastal environment of Xiamen, China. Marine Pollut Bull 57:419–424. https://doi.org/10.1016/j.marpolbul.2008.04.034
Xiao Q, Hu B, He M (2008) Speciation of butyltin compounds in environmental and biological samples using headspace single drop microextraction coupled with gas chromatography-inductively coupled plasma mass spectrometry. J Chromatogr A 1211:135–141. https://doi.org/10.1016/j.chroma.2008.09.089
Yañez J, Riffo P, Mansilla HD, Bravo M, Quiroz W, Santander P (2016) Speciation analysis of organotin compounds (OTCs) by a simultaneous hydride generation–liquid/liquid extraction and GC–MS determination. Microchem J 126:460–465. https://doi.org/10.1016/j.microc.2016.01.002
Yu ZH, Zhang J, Wang XR (2011) Speciation analysis of organotin compounds in sediment by hyphenated technique of high performance liquid chromatography-inductively coupled plasma mass spectrometry. Chin J Anal Chem 39:544–547. https://doi.org/10.1016/S1872-2040(10)60434-3
Zhang K, Shi J, He B, Xu W, Li X, Jiang G (2013) Organotin compounds in surface sediments from selected fishing ports along the Chinese coast. Chin Sci Bull 58:231–237. https://doi.org/10.1007/s11434-012-5406-6
Acknowledgements
The authors thank W. Melo for providing the estuary map. This research was supported by a doctoral Grant funded by the National Council of Scientific and Technological Research (CONICET-Argentina) and was part of the Ph.D. Thesis of Pamela Y. Quintas. Funding was provided through research Grants PIP D-738 2011, PICT 2010-1302, and PGI 24/Q054 from CONICET, ANPCyT, and the National South University, respectively.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Quintas, P.Y., Oliva, A.L., Alvarez, M.B. et al. Fast and Feasible Ultrasound-Assisted Pretreatment for the Determination of Organotin Compounds in Environmental Samples. Arch Environ Contam Toxicol 74, 645–655 (2018). https://doi.org/10.1007/s00244-017-0494-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-017-0494-6