
Abstract
Approximately 80% of kidney stone diseases contain calcium. Inherited genetic factors are among the variables that influence the development of calcium-containing kidney stone diseases (CKSD). Previous genome-wide association studies (GWAS) on stone diseases have been reported worldwide; however, these are not focused on calcium-containing stones. We conducted a GWAS to identify germline genetic polymorphisms associated with CKSD in a Medical Center in Taiwan; hence, this study was based primarily on a hospital-based database. CKSD was diagnosed using the chart records. Patients infected with urea-splitting-microorganisms and those with at least two urinary pH value below 5.5 were excluded. None of the patients had cystic stones based on stone analysis. Those over 40 years of age with no history of CKSD and no microscopic hematuria on urinalysis were considered as controls. The DNA isolated from the blood of 14,934 patients (63.7% male and 36.3% female) with CKSD and 29,868 controls (10,830 men and 19,038 women) at a medical center was genotyped for approximately 714,457 single nucleotide polymorphisms (SNPs) with minor allele frequency of ≥ 0.05. We used PLINK 1.9 to calculate the polygenic risk score (PRS) to investigate the association between CKSD and controls. The accuracy of the PRS was verified by dividing it into the training and testing groups. The statistical analyses were calculated with the area under the curve (AUC) using IBM SPSS version 22. We identified 432 susceptibility loci that reached a genome-wide threshold of P < 1.0 × 10− 5. A total of 132 SNPs reached a threshold of P < 5 × 10− 8 using a stricter definition of significance on chromosomes 4, 13, 16, 17, and 18. At the top locus of our study, SNPs in DGKH, PDILT, BCAS3, and ABCG2 have been previously reported. RN7SKP27, HDAC4, PCDH15, AP003068.2, and NFATC1 were novel findings in this study. PRS was adjusted for sex and age, resulting in an AUC of 0.65. The number of patients in the top quartile of PRS was 1.39 folds in the risk of CKSD than patients in the bottom quartile. Our data identified the significance of GWAS for patients with CKSD in a hospital-based study. The PRS also had a high AUC for discriminating patients with CKSD from controls. A total of 132 SNP loci of SNPs significantly associated with the development of CKSD. This first survey, which focused on patients with CKSD, will provide novel insights specific to CKSD and its potential clinical biomarkers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Kidney stone disease (KSD) is a common urological disease with a multifactorial etiology, including a polygenic milieu [1, 2]. Approximately 80% of the major components include calcium containing-stones, such as calcium oxalate and calcium phosphate [3]. Other stone components included uric acid, cysteine, and magnesium ammonia-phosphate [4]. Calcium-containing kidney stone disease (CKSD) has a high recurrence rate up to an estimated 50% within five years [5]. Aside from genetics, there are many systemic diseases associated with CKSD, including gout, diabetes, obesity, metabolic syndrome, hyperparathyroidism, and hyperthyroidism [5, 6]. Environmental and lifestyle factors, such as geographical location, warm weather, and dietary habits are also reported to be associated with CKSD [5]. Therefore, CKSD formation may be a polygenetic factor linked to systemic diseases that may interact with the environment and other factors.
Some previous studies on single-gene polymorphisms (SNPs) have attempted to study the contribution of a single gene to the pathogenesis of KSD, such as IL-18, TAP-2, urokinase, E-cadherin, and vitamin D receptor genes [7,8,9,10,11]. However, there are several limitations, including a less effective single allele and a small population. In 1996, Risch et al. reported that the identification of the entire human genome and its genetic polymorphisms after a human genome project led to new insights into the analysis of risk loci for common diseases [12]. Recently, genome-wide association studies (GWAS) have been conducted to elucidate the influence of multiple factors associated with KSD [13]. Several novel risk loci have been identified in the Japanese and British populations (12,123 cases and 417,378 controls ), including DKGH, CYP24A1, BCR, WDR72, and GPlC1 [14]. The CLDN14 gene polymorphism was identified and studied in a GWAS of 3,773 cases and 42,510 controls from Iceland and the Netherlands [15]. ALPL, CaSR, SLC34A1, TRPV5, and other genes were identified [16]. A GWAS study of a large-scale Japanese population of 11,130 stone patients and 187,639 controls identified 14 significant loci, including nine novel loci [17]. These genetic factors related to the regulation of metabolism and crystallization pathways contributed to the development of KSD. However, GWAS studies specifically focusing on CKSD in other Asian populations, including the Taiwanese population, are lacking. There is also an issue regarding the accuracy of the stone patients and the controls from a self-reported database. In addition, the recruited patients were not accurately classified according to their compositions. Therefore, we aimed to conduct a GWAS focusing on CKSD from a database of a tertiary medical center with the exclusion of other stone compositions, such as infection, uric acid, and cystine stones from the Taiwanese population.
Materials and methods
Subjects and database information
This study utilized electronic medical records from China Medical University Hospital (CMUH). The individuals with urine pH < 5.5 or those whose urine culture yields Proteus mirabilis, Pseudomonas, Gardnerella, Lactobacillus, and Enterobactericeae, Klebsiella pneumoniae are excluded. Those stone analysis revealed cystine stone was also excluded. We used International Classification of Diseases (ICD)-9th Edition (‘592.0’, ‘592.1’, ‘592.9’, ‘592.90’), and ICD-10th Edition (‘N13.2’, ‘N20.0’, ‘N20.1’, ‘N20.2’, ‘N20.9’, ‘V13.01’, ‘Z87.442’) associated with urolithiasis to identify the case group and aged above 30 years. The control group consists of individuals with negative results for red blood cells (RBCs) in urine and aged above 40 years. After matching for gender, we included 14,934 patients in the case group and 29,868 patients in the control group. This study was approved by the Ethics Review Board of China Medical University Hospital (CMUH111-REC1-026) and was part of the CMUH Precision Medicine Project that began in 2018, which was also approved by the ethics committees of CMUH (CMUH110-REC3-005 and CMUH111-REC1-176).
Genotyping
Genomic DNA was extracted from 200 µl peripheral blood samples of all study participants using the MagCore Genomic DNA Whole Blood Kit from RBC Bioscience, Taiwan, following the manufacturer’s prescribed protocol. To obtain genetic information from the Taiwanese population samples, we employed the Affymetrix Axiom genotyping platform, specifically utilizing the Axiom Taiwan Precision Medicine (TPM) customized SNP array (Thermo Fisher Scientific, Inc., Santa Clara, CA, USA), which covered 714,457 SNPs across the entire human genome.
For data analysis, we utilized PLINK1.9 and excluded samples and SNPs with missing rates greater than 0.1 (--geno 0.1 for SNPs and -mind 0.1 for samples). Additionally, variants with a Hardy-Weinberg equilibrium P-value less than 1e-6 (--hwe 1e-6) and a minor allele frequency (MAF) less than 1e-4 (--maf 0.0001) were also excluded. To improve phasing accuracy, SHAPEIT4 was applied to phase TPM arrays. Subsequently, we employed Beagle5.2 for imputation, which has demonstrated enhanced effectiveness and accuracy compared to other imputation tools. During the imputation process, data were filtered based on an R-square alternate allele dosage of less than 0.3 and a genotype posterior probability of less than 0.9 as the criteria [18].
Genome-wide association study (GWAS)
To identify associated variants, we employed PLINK 1.9 to generate summary statistics for individuals with urolithiasis and the control group. To determine the familial history of second-degree relatives, we collected data for one individual from each familial group within the targeted group, ensuring representation from both cases with different phenotype groups. Family membership was established using PLINK 2.0 KINSHIP analysis. For the case-control-based Genome-Wide Association Study (GWAS), we adopted an additive genetic model. Logistic regression was used to analyze trait associations, adjusting for multiple covariates such as gender, age, and principal components (PCs). To avoid issues of collinearity in linkage disequilibrium (LD) that could lead to overestimation, we selected the most significant variant. Significant associations with urolithiasis were determined based on a p-value threshold of < 1e-05 or a more stringent threshold of 5e-08. To visualize the results, we created Manhattan plots and quantile-quantile plots using the “qqman” R package. Additionally, we presented region plots of the variants of interest using LocusZoom tools.
Polygenic risk score (PRS) analysis
To compute the Polygenic Risk Score (PRS), we partitioned the cohort randomly into three datasets: the base, training, and testing groups (80%, 10% and 10% of total case and control patients, respectively). Initially, we investigated the association between the studied variables and urolithiasis in the base group using PLINK1.9. Subsequently, we constructed a PRS utilizing the training group and PRSice2 tools while filtering out variants with a Minor Allele Frequency (MAF) greater than 0.01 [19]. As a reference panel, we utilized the 1000 Genomes Phase v.3 data of the East Asian population. The PRS was calculated through z-score normalization. Subsequently, we validated the PRS models in the testing group. To assess the classification accuracy of the PRS, we employed receiver operating characteristic (ROC) curves and calculated the area under the curve (AUC). These statistical analyses were conducted using IBM SPSS Statistics (version 22).
Statistical analysis
Continuous and categorical variables at baseline in the different genotype groups were assessed using appropriate statistical tests, including Student’s t-test, Chi-square test, or Fisher’s exact test. To compare multiple groups, a one-way analysis of variance (ANOVA) with Tukey’s post hoc test was conducted. The significance level for all statistical tests was set at a two-sided level of 0.05. All statistical analyses were carried out using IBM SPSS Statistics (version 22) and R software (version 4.1.0).
Results
GWAS in patients with CKSD
There was 9519 (63.7%) male and 5415 (36.3%) female patients in the stone group (total 14,934 patients). The matched control group had twice the number of patients (29,868), which included 19,038 (63.7%) male and 10,830 (36.3%) female patients. The clinical characteristics of patients with CKSD and control individuals are presented in Table 1. A flowchart of the study sample is presented in Fig. 1.

Flow chart of the genome-wide association studies (GWAS) pipeline. A total of 14,934 patients with CKSD and 29,868 control patients were enrolled. The study population was randomly divided into three groups, namely base, training, and testing, for polygenic risk score (PRS) analysis. A GWAS was performed in the base group, and the results were used to train the PRS model in the training group. The PRS model was then validated in the testing group
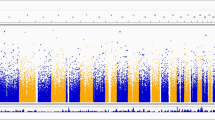
We randomly divided the patients with CKSD and controls into the base group (80% of study cohort) and the replication group (20% of study cohort). A GWAS was conducted in the base group, and 432 SNPs were determined to reach genome-wide significance at a threshold of P < 10− 5. A total of 132 SNPs reached stricter significance at a threshold of P < 5 × 10− 8 on chromosomes 4, 13, 16, 17, and 18. The GWAS results are shown as Manhattan and quantile-quantile (QQ) plots (Fig. 2A). Quantile–quantile plot demonstrating that the observed P values were consistent with the expected values (Fig. 2B). The red line represents the expected values. The region plots of chromosomes 4, 13, 16, and 17 are presented in Supplementary Fig. 1. The results were uploaded and analyzed using LocusZoom, an open tool for analyzing and visualizing GWAS results. The most significant loci according to LocusZoom are listed in Table 2.

Manhattan plot and quantile–quantile plot of CKSD. (A) The Manhattan plot depicts the peaks of SNPs surpassing genome-wide significance levels between the patients with CKSD and control groups. The blue line represents the threshold of P < 1 × 10− 5. The red line represents the threshold of P < 5 × 10− 8. The most significant SNPs in the gene regions of chromosome 2, 4, 13, 16, 17, and 18 were labeled with gene symbols. (B) Quantile–quantile plot demonstrating that the observed P values were consistent with the expected values. The red line represents the expected values
PRS analysis in CKSD
Next, we established a PRS model to assess the cumulative effects of multiple genetic variants in CKSD according to the GWAS results. The patients with CKSD and controls in the replication group were randomly divided into the training group and the testing group (10% of study cohort, respectively). We determined a set of 25 SNPs according to a p-value threshold of 0.0001 and an R2 threshold of 0.00397 to establish the optimal PRS model for the training group (Fig. 3A). The SNPs are listed in Table 3. The PRSs were compared in the CKSD and controls, and a significant difference was observed in the training group (normalized mean PRS ± SD: -0.039 ± 1.006 and 0.076 ± 0.984 in CKSD and control group, respectively; P < 0.001; Fig. 3B). The PRS model was repeated in the testing group and reached significant difference (normalized mean PRS ± SD: -0.029 ± 1.005 in CKSD versus 0.057 ± 0.988 in controls; P = 0.007; Fig. 3C).

Distribution of PRS in CKSD. (A) Bar plot of p value threshold. The x-axis is p value threshold and the y-axis are R square. (B, C) Distribution of PRSs in CKSD in the training group (B) and the testing group (C). Significance was calculated by Mann-Whitney U test and P value was adjusted with FDR-BH. *** represents P values < 0.005. * represents P values < 0.05
To assess the classification accuracy of the PRS model, we used the receiver operating characteristic (ROC) curve and found an area under the curve (AUC) of 0.535 (Fig. 4A), indicating the ability of the PRS model to discriminate between the patients with CKSD and the controls. The AUC of the PRS combined with sex and age increased to 0.655 (Fig. 4A). We then compared the risk of CKSD in each quantile and found that the patients in the top quantile for the PRSs had 1.39 folds the risk of being classified into the CKSD group compared to the patients in the bottom quantile (p < 0.001, odds ratio [OR] 1.39, 95% confidence interval [95% CI] 1.229–1.578; Fig. 4B). The OR of CKSD increased with the PRS quantiles, indicating that an elevated risk was associated with higher PRSs.

Statistical results and Quantile plots of PRS in CKSD. (A) ROC and AUC analyses for assessment of accuracy of the PRS model and the PRS model combined with age and sex for predicting CKSD. (B) Patients in the replication group were separated into quintiles on the basis of the PRS of CKSD and odds ratios for the likelihood of an association between each quintile of PRSs and CKSD were generated using logistic regression analysis. Values are the odds ratios with 95% confidence intervals
PheWAS analysis in CKSD
To assess the association between genetic variants and various diseases, we used Phenome-wide association studies (PheWAS) to investigate the diseases associated with PRS. PheWAS is a genetic epidemiological method designed to comprehensively assess multiple phenotypic traits, such as diseases, contributing to our understanding of the role of genes in health and disease. We divided all individuals in the replication group into quartiles based on PRSs and conducted a PheWAS analysis. We found an association between the SNPs used to establish the PRS model and the calculi of the kidney and urinary tract (Fig. 5).

PheWAS analysis of CKSD. The Manhattan plot of PheWAS of patients with CKSD by quartile PRS. The X-axis is classifications of physiological function. Red line indicates P value of 0.001, and blue line indicated P value of 0.05
Discussion
Since the causes of stone disease may vary with stone composition, this was the first survey that focused on calcium-containing stone diseases. We identified significant 182 genetic loci of SNPs from a GWAS with stricter significance. In the PRS analysis, after adjusting for age and sex, the area under the curve was 0.652. Our analysis differentiated the controls from the patients with CKSD. There was a 1.39-fold significant risk of CKSD in the top quartile compared with the bottom quartile in the PRS analysis. The highly significant genetic loci were DKGH, PDILT, and BCAS3, which have been previously reported in Japan and Europe [14, 16, 17]. This revealed significant SNPs associated with stone diseases. We also identified novel genetic loci in our enrolled patients focused on calcium-containing diseases instead of all stone diseases, including uric acid, infection, or cystine stones, which might have some confounding effects when performing GWAS and PRS analysis.
The nine lead loci were NFACT1, PCDH15, DGKH, ABCG2, PDILT, BCAS3, HDAC4, RN7SKP27, and AP003068.2. Of these, DGKH, PDILT, and BCAS3 have been reported in the European and Japanese populations. Calcium regulation-related genes include DGKH, NFACT1, and PCDH15. Both BCAS3 and HDAC4 are involved in histone acetyltransferase activity.
One of the top loci was rs682573, which encodes the DGKH gene with an odds ratio of 1.15 that increased the risk of KSD. DGKH, which is associated with stone disease, has been reported in several populations with different reference sequences. Howles et al. reported that the risk allele rs1170174 of the DGKH gene was not associated with stone disease [14]. This gene encodes the diacylglycerol kinase (DGK) enzyme family, which regulates the intracellular concentrations of diacylglycerol and phosphatidic acid. However, an association with urinary calcium excretion in male stone formers among AA homozygotes was reported by Howels et al. in a study with a small sample size [14]. DGKH may require further translational studies to confirm this association.
The nuclear factor of activated T cell cytoplasmic 1 (NFATc1) is a novel and significant genetic locus found in this GWAS that highlights the association between bone and vascular disease; its reported function is involved in osteoclastic activity and atherosclerotic calcification, both of which are associated with calcium regulation [20]. This may also be associated with calcium stone disease. PCDH15 is a member of the cadherin superfamily of membrane proteins. PCDH15 formation mediates calcium-dependent cell-cell adhesion. ABCG2 is a membrane protein belonging to the superfamily of ATP-binding cassette transporters. ABCG2 encoded protein function may play a major role in multidrug resistance in breast cancer [21]. Although there have been no previous reports of stone disease, the ABCG2 rs2231142 variant regulates renal urate excretion [22]. However, our finding for the ABCG2 risk allele was rs2199936, which is a different locus. Further studies on ABCG2 SNPs are required. Both BCAS3 and HDAC4 regulate histone acetylase activity [23]. Their functional role is related to carcinogenesis; however, no association with stone diseases has been reported.
Compared to self-reporting, clinical data from a single medical center have the advantage of verifying clinical information, which can be diagnosed accurately using clinical laboratory data and the final diagnosis from the chart reviews. For an accurate assessment of the disease, the database requires both width (small amounts of data with large numbers of participants) and breatth (large amounts of data with small numbers of participants) [24]. Questionnaires, self-reports, and databases from different sources may have some disadvantages. In addition, the database should include a follow-up period for diseases that may occur within a time interval that can record disease traits more accurately. Although our database has limited breadth, it has sufficient width because of medical records from a hospital base. Therefore, it is important to study a series of GWAS results for various diseases, such as alopecia, hyperthyroidism, early menarche, and body height have been published [25,26,27].
Stone disease can be caused by many pathogens with different genetic backgrounds. Most data sources from the GWAS were obtained from mixed large-scale biobanks, including the UK, Japan, and Taiwan [28]. The confirmation of stone patients was not unique and included medical doctors, images, self-reports, and questionnaires from computer touchscreens. Such a database has discrepancies in selection bias due to patient recruitment, recall bias, and uncertain stone composition [29, 30]. Different stone compositions may be associated with different pathogenic mechanisms, including genetic differences [31]. Infection stones are a major problem that may be caused by a urea-splitting microorganism rather than a genetic background. Uric acid stones are the major form in extremely acidic urine conditions (pH < 5.5). Furthermore, cystic stones are caused by inborn genetic errors [32]. Therefore, patient recruitment is an important issue when focusing on calcium stone disease.
To clarify the data, we recruited patients from a medical center. The diagnosis of stone disease was confirmed through evidence, including doctor’s diagnosis, stone analysis, image reports, and laboratory tests. Laboratory data were collected to rule out infections and uric acid stones. Moreover, patients with cystine stones were excluded. Although the number of patients was limited, our database focused on calcium-containing stones. The control group was also important for recruitment. In our study, we excluded control patients with hematuria from routine urine reports because of possible occult stone disease. Therefore, the database was more reliable. This may have some different SNP loci of SNPs apart from those in the UK and Japan biobanks.
This study included a limited number of patients’ ethnic source from a single medical center. We confirmed the diagnosis of calcium-containing stone disease in well-clarified control patients. Patients with other stone compositions that could alter the results of the genetic analysis were excluded. This decreased the chances of recall and selection bias. Some of the results were similar to the published genetic loci and demonstrated novel loci that were previously neglected.
Conclusion
We reported the first survey of GWAS on CKSD using a hospital-based database, in which some novel genetic loci could be further studied in terms of calcium stone disease formation. PRSs have emerged as accurate and efficient tools for predicting an individual’s health status and susceptibility to disease. By assessing an individual’s genetic composition, PRSs offer valuable insights into potential treatment effectiveness and the likelihood of a positive or negative prognosis, even before symptoms manifest. PRSs enable early identification of a condition before abnormal test results become apparent. The utilization of the PRSs empowers healthcare practitioners to embrace precision medicine, enabling the prescription of personalized preventive strategies tailored to each patient’s unique genetic profile. In this study, we developed multiple PRSs to enhance their discriminatory power in predicting CKSD, with the aim of contributing to the early diagnosis and prediction of this condition.
Data availability
No datasets were generated or analysed during the current study.
References
Goldfarb DS, Modersitzki F, Asplin JR, Nazzal L (2023) Effect of a high-citrate beverage on urine chemistry in patients with calcium kidney stones. Urolithiasis 51(1):96
Yang S, Tan W, Wei B, Gu C, Li S, Wang S (2023) Association between alcohol and urolithiasis: a mendelian randomization study. Urolithiasis 51(1):103
Finlayson B (1978) Physicochemical aspects of urolithiasis. Kidney Int 13(5):344–360
Evan AP (2010) Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr Nephrol 25(5):831–841
Khan SR, Pearle MS, Robertson WG, Gambaro G, Canales BK, Doizi S, Traxer O, Tiselius HG (2016) Kidney stones. Nat Rev Dis Primers 2:16008
Kramer HJ, Choi HK, Atkinson K, Stampfer M, Curhan GC (2003) The association between gout and nephrolithiasis in men: the Health professionals’ Follow-Up study. Kidney Int 64(3):1022–1026
Lai KC, Lin WY, Man KM, Tsai CH, Chen HY, Tsai FJ, Chen FJ, Chen HY, Liu HP, Ho TJ, Huang PH, Liu PL, Lin FY, Shen JL, Liu JT, Chen YH, Chen WC (2010) Association of interleukin-18 gene polymorphisms with calcium oxalate kidney stone disease. Scand J Urol Nephrol 44(1):20–26
Huang SH, Chen RH, Wan L, Tsai FJ, Chen WC (2005) TAP 2 gene Msp-I polymorphism might be associated with calcium oxalate stone disease. Urol Int 75(3):264–268
Tsai FJ, Lin CC, Lu HF, Chen HY, Chen WC (2002) Urokinase gene 3’-UTR T/C polymorphism is associated with urolithiasis. Urology 59(3):458–461
Tsai FJ, Wu HC, Chen HY, Lu HF, Hsu CD, Chen WC (2003) Association of E-cadherin gene 3’-UTR C/T polymorphism with calcium oxalate stone disease. Urol Int 70(4):278–281
Amar A, Afzal A, Hussain SA, Hameed A, Khan AR, Shakoor M, Abid A, Khaliq S (2020) Association of vitamin D receptor gene polymorphisms and risk of urolithiasis: results of a genetic epidemiology study and comprehensive meta-analysis. Urolithiasis 48(5):385–401
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273(5281):1516–1517
Palsson R, Indridason OS, Edvardsson VO, Oddsson A (2019) Genetics of common complex kidney stone disease: insights from genome-wide association studies. Urolithiasis 47(1):11–21
Howles SA, Wiberg A, Goldsworthy M, Bayliss AL, Gluck AK, Ng M, Grout E, Tanikawa C, Kamatani Y, Terao C, Takahashi A, Kubo M, Matsuda K, Thakker RV, Turney BW, Furniss D (2019) Genetic variants of calcium and vitamin D metabolism in kidney stone disease. Nat Commun 10(1):5175
Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, Sulem P, Halldorsson BV, de Vegt F, d’Ancona FC, den Heijer M, Franzson L, Christiansen C, Alexandersen P, Rafnar T, Kristjansson K, Sigurdsson G, Kiemeney LA, Bodvarsson M, Indridason OS, Palsson R, Kong A, Thorsteinsdottir U, Stefansson K (2009) Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet 41(8):926–930
Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, Haraldsdottir E, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Masson G, Holm H, Gudbjartsson DF, Thorsteinsdottir U, Indridason OS, Palsson R, Stefansson K (2015) Common and rare variants associated with kidney stones and biochemical traits. Nat Commun 6:7975
Tanikawa C, Kamatani Y, Terao C, Usami M, Takahashi A, Momozawa Y, Suzuki K, Ogishima S, Shimizu A, Satoh M, Matsuo K, Mikami H, Naito M, Wakai K, Yamaji T, Sawada N, Iwasaki M, Tsugane S, Kohri K, Yu ASL, Yasui T, Murakami Y, Kubo M, Matsuda K (2019) Novel risk loci identified in a Genome-Wide Association Study of Urolithiasis in a Japanese Population. J Am Soc Nephrol 30(5):855–864
Liu TY, Lin CF, Wu HT, Wu YL, Chen YC, Liao CC, Chou YP, Chao D, Chang YS, Lu HF, Chang JG, Hsu KC, Tsai FJ (2021) Comparison of multiple imputation algorithms and verification using whole-genome sequencing in the CMUH genetic biobank. Biomed (Taipei) 11(4):57–65
Choi SW, O’Reilly PF (2019) PRSice-2: polygenic risk score software for biobank-scale data. Gigascience 8(7)
Gu W, Wang Z, Sun Z, Bao Z, Zhang L, Geng Y, Jing L, Mao X, Li L (2020) Role of NFATc1 in the bone-vascular Axis Calcification Paradox. J Cardiovasc Pharmacol 75(3):200–207
Nakanishi T, Ross DD (2012) Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer 31(2):73–99
Woodward OM, Köttgen A, Coresh J, Boerwinkle E, Guggino WB, Köttgen M (2009) Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A 106(25):10338–10342
Wang Z, Qin G, Zhao TC (2014) HDAC4: mechanism of regulation and biological functions. Epigenomics 6(1):139–150
Abhishek A, Roddy E, Doherty M (2017) Gout - a guide for the general and acute physicians. Clin Med (Lond) 17(1):54–59
Yang JS, Liu TY, Chen YC, Tsai SC, Chiu YJ, Liao CC, Tsai FJ (2023) Genome-Wide Association Study of Alopecia Areata in Taiwan: the conflict between individuals and Hair Follicles. Clin Cosmet Investig Dermatol 16:2597–2612
Liu TY, Liao WL, Wang TY, Chan CJ, Chang JG, Chen YC, Lu HF, Yang HH, Chen SY, Tsai FJ (2022) Genome-wide association study of hyperthyroidism based on electronic medical record from Taiwan. Front Med (Lausanne) 9(830621
Chiou J-S, Cheng C-F, Liang W-M, Chou C-H, Wang C-H, Lin W-D, Chiu M-L, Cheng W-C, Lin C-W, Lin T-H, Liao C-C, Huang S-M, Tsai C-H, Lin Y-J, Tsai F-J (2022) Your height affects your health: genetic determinants and health-related outcomes in Taiwan. BMC Med 20(1):250
Palmer LJ (2007) UK Biobank: bank on it. Lancet 369(9578):1980–1982
Swanson JM (2012) The UK Biobank and selection bias. Lancet 380(9837):110
Collins R (2012) What makes UK Biobank special? Lancet 379(9822):1173–1174
Howles SA, Thakker RV (2020) Genetics of kidney stone disease. Nat Rev Urol 17(7):407–421
Evan AP, Worcester EM, Coe FL, Williams J Jr., Lingeman JE (2015) Mechanisms of human kidney stone formation. Urolithiasis 43(10 1):19–32
Funding
This study was supported in part by the China Medical University Hospital (DMR-113-053 and CMU112-S-04).
Author information
Authors and Affiliations
Contributions
Wen-Chi Chen and Yu-Chia Chen took the primary role in writing the manuscript. Wen-Chi Chen designed most of the experiments, with Yu-Chia Chen, Yung-Hsiang Chen, Ting-Yuan Liu, and Chang-Hai Tsai, and supervised the final experiments. Wen-Chi Chen and Fuu-Jen Tsai obtained the funding and conceived, designed, and supervised the study. Fuu-Jen Tsai provided the original ideas for this study.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethics approval
This study was approved by the Ethics Review Board of China Medical University Hospital (CMUH111-REC1-026) and was part of the CMUH Precision Medicine Project that began in 2018, which was also approved by the ethics committees of CMUH (CMUH110-REC3-005 and CMUH111-REC1-176).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, WC., Chen, YC., Chen, YH. et al. Identification of novel genetic susceptibility loci for calcium-containing kidney stone disease by genome-wide association study and polygenic risk score in a Taiwanese population. Urolithiasis 52, 94 (2024). https://doi.org/10.1007/s00240-024-01577-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00240-024-01577-0