
Abstract
Identifying patterns of genetic structure, diversity, and connectivity in marine species is helpful for conservation purposes. We conducted a range-wide, comparative study of genetic structure and diversity in anadromous and landlocked populations of two Eurasian shad species (Alosa alosa and Alosa fallax). Samples of A. alosa from 13 (N = 660) locations, and A. fallax from 30 (N = 747), were studied with 21 and 18 microsatellite loci, respectively. The average pairwise FST among all anadromous A. alosa sampled in different drainages was 0.083 compared to 0.306 in A. fallax. Genetic clustering analysis identified five to six clusters for A. alosa and up to 16 for A. fallax. In most cases, the clusters identified were comprised of populations that spawn in geographically neighboring drainages. A positive correlation between genetic and geographic distance was found in anadromous A. fallax, but not in A. alosa. Within each species, landlocked populations were more genetically differentiated than anadromous ones. Our results show migration and gene flow are higher in A. alosa than A. fallax, which should be taken into account when determining the geographic scale at which to enact conservation measures to protect each species. Most A. alosa and A. fallax populations were genetically differentiated, indicating that the loss of any could result in a reduction of the adaptive potential for that species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Understanding patterns of genetic structure and gene flow in threatened species is useful for planning and assessing conservation strategies to protect them (Frankham 1995). This is especially the case for anadromous species due to the central role that migration plays in their life histories and population genetics (Schtickzelle and Quinn 2007). For example, some anadromous species actively migrate to the locations where they were born to reproduce (i.e. “natal homing”) (McDowall 2001). Others only migrate short distances at sea and, consequently, return to their natal drainage to spawn regardless of their capacities to home. Such philopatric migratory behaviors restrict gene flow among populations and can promote genetic structure. However, even in species that accurately home and disperse extremely short distances at sea, either by mistake, chance, or design (e.g. to avoid competition), individuals sometimes reproduce in non-natal locations, or “stray”, which tends to have the opposite effect (Salmenkova 2017). In species that usually stray into local drainages, spatial patterns in genetic differentiation such as isolation-by-distance (IBD) are often observed. Data on genetic diversity and structure in an anadromous species can therefore yield important insights into its migratory behaviors and how they impact the exchange of individuals among its populations, or “connectivity”. Such information is crucial for understanding why threatened species are in decline and devising effective strategies to protect them.
For example, knowledge about connectivity can help identify patterns of migration and gene flow that are important for population stability and persistence. Using this information to help maintain natural levels of connectivity has many advantages such as enabling species to re-colonize drainages where local extinctions have occurred (Rieman and Allendorf 2001; Schtickzelle and Quinn 2007; Martin et al. 2015), increasing population growth via recruitment from immigration (Gomulkiewicz et al. 1999), and helping to maintain genetic and phenotypic diversity (Antunes et al. 2006; Keefer and Caudill 2014). Knowledge about connectivity can also be used to predict the geographic scale at which the positive effects of conservation efforts such as habitat restorations or fishing restrictions will impact a species.
Here we present the first range-wide, comparative, study of Alosa alosa and Alosa fallax to help further our understanding of genetic structure and diversity within each species. The allis, A. alosa, and twaite shad, A. fallax, are anadromous species that spawn in some of the same Atlantic drainages along the coast of continental Europe and the United Kingdom (Baglinière et al. 2003; Aprahamian et al. 2003). Alosa fallax is also found in the Baltic, Mediterranean, Adriatic, and Aegean seas. Adult A. alosa reach total lengths of 70 cm, are strictly semelparous, and spawn hundreds of kilometers upstream while the smaller A. fallax (20–60 cm) is primarily iteroparous, and spawns in lower parts of rivers and upper estuaries. Both species can disperse at least 1500 km at sea (Sabatié and Blagliniere 2001), but may often only migrate to waters between 50 and 100 m depth offshore from the watersheds where they spawn (Taverny and Elie 2001). Although A. alosa and A. fallax are primarily anadromous, freshwater populations of each species exist. Freshwater populations of A. alosa were established during the past century when anadromous individuals were trapped behind dams built in several drainages in Portugal (Pereira et al. 1999). Freshwater populations of A. fallax naturally occur in several locations including northern Italy and Ireland (Aprahamian et al. 2003).
Declines and extirpations of A. fallax and especially A. alosa have been observed throughout their ranges and they are now protected in some areas (Baglinière et al. 2003; Mota and Antunes 2011; Davies et al. 2020). Losses in each species appear to be linked to environmental changes such as the building of dams and weirs, and pollution (Costa et al. 2001; Maes et al. 2008; King and Roche 2008). Each species also supports important fisheries that have been overexploited (Baglinière et al. 2003). Improvements in water quality, habitat restorations, stricter harvesting regulations, and other measures taken over the past few decades appear to have had a positive impact on A. alosa and A. fallax (Buysse et al. 2008; Belliard et al. 2009; Sotelo et al. 2014). However, more intensive conservation efforts will almost certainly be necessary to ensure that each species can thrive in many of the locations they inhabited just a few decades ago.
Previous research has shown that genetic structure is generally lower in A. alosa than A. fallax. In one study of A. alosa from throughout the Atlantic that used eight allozyme loci the average (overall) FST was found to be 0.064 (Alexandrino et al. 2006). And, global FST based on 13 microsatellite markers was found to be 0.026 among 15 populations of A. alosa from northern France to northern Portugal (Martin et al. 2015). In contrast, the average FST for A. fallax from the Atlantic and the western Mediterranean was estimated to be 0.288 (Alexandrino et al. 2006). Similarly, pairwise FST estimates between several A. fallax populations from the United Kingdom (UK) ranged from 0 to 0.162 based on data for five microsatellite markers (Jolly et al. 2012). Population structure was also found to be lower in A. alosa than A. fallax in a range-wide study of each species using mitochondrial data (Faria et al. 2006, 2012). Positive correlations between geographic and genetic distance (i.e., “Isolation-By-Distance” or IBD) among populations within A. alosa (Alexandrino et al. 2006; Martin et al. 2015) and A. fallax (Alexandrino et al. 2006; Jolly et al. 2012) have been found and were attributed to natal homing. However, all of these studies included a limited number of highly polymorphic genetic markers and populations, precluding general conclusions across the entire species range.
Here we generated data on 13 A. alosa and 30 A. fallax populations from throughout their ranges using microsatellite loci (21 for A. alosa and 18 for A. fallax). This included three A. alosa and four A. fallax freshwater populations that are now thought to be completely cut off from immigration due to physical barriers (i.e., landlocked). We used this data to test for differences in genetic structure and diversity between A. alosa and A. fallax, and anadromous versus landlocked within each species. We then use our findings to make conservation recommendations for protecting A. alosa and A. fallax.
Methods
Sampling
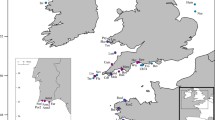
A total of 660 A. alosa from 13 locations, 747 A. fallax from 30, were caught by gill netting or angling from 1991 to 2015 (Fig. 1, Table 1).

Map of sampling locations. Blue (A. alosa) and yellow (A. fallax) circles represent sampling locations. The locations corresponding to each letter (A. alosa) and number code (A. fallax) can be found in Table 1. Green circles on the map represent lakes or reservoirs that were sampled

Graphs from the Bayesian clustering analysis (BAPS) for A. alosa (a) and A. fallax (b), and genetic breaks identified in the BARRIER analysis. Letters (a) and numbers (b) refer to the populations in Table 1 (under “M”) and Fig. 1. Green circles on the map represent landlocked populations. Genetic breaks identified in the Barrier analyses are indicated by white rectangles and for A. alosa (a) are numbered by order of strength where “1” is the strongest
All of the anadromous individuals sampled were caught while migrating upstream except those from six locations indicated in Table 1, which were caught in estuaries or at sea near the mouths of rivers where they are known to spawn. Individuals of both an anadromous and resident population are known to spawn in Lake Skadar, and it was unknown from which of them our samples were obtained. We, therefore, did not include Lake Skadar in any analysis where life history was a factor. In several of the locations sampled, individuals were caught during different years to obtain sufficient sample sizes for population genetic analyses and enable us to examine interannual stability in allele frequencies within spawning sites (Online Resource Table 1). All tissue samples were preserved in 95% ethanol or immediately frozen, and then stored at −80 °C. Different subspecies of A. fallax have been recognized in Lough Leane, Maggiore, Como, Garda, and Skadar, and the Rhone, Herault, and Aude Rivers (Aprahamian et al. 2003 and references there within), but for brevity, we refer to these here as A. fallax.
Molecular Methods and data filtering
DNA was extracted from tissue samples using EasySpin ™ 96-well extraction plates according to the manufacturer’s recommendations. All individuals were genotyped at 24 microsatellite loci that were previously designed for A. alosa, A. fallax (Faria et al. 2004), or A. sapidissima (Julian and Bartron 2007). Subsets of the 24 loci were combined into four “multiplexes” for polymerase chain reaction (PCR). The loci in each PCR multiplex, primers for each locus, and the annealing temperatures used during PCR can be found in Online Resource Table 2. The Multiplex PCR Kit (Qiagen) was used for all PCR reactions according to the manufacturer’s instructions. Multiplexed PCR products were run in an ABI Prism Analyzer with an internal size ladder (Genescan 350 ROX) and allele sizes were calculated using Genescan (Applied Biosystems).
The microsatellite data were screened for potential problems that could impact our analysis. The inbreeding coefficient (FIS) (Weir and Cockerham 1984) for each locus-population combination was calculated using Genepop 4.0.10 (Rousset 2008). Exact tests were conducted using Genepop 4.0.10 to determine if the allele frequencies of any loci significantly departed from Hardy–Weinberg equilibrium expectations. And, linkage disequilibrium between loci was estimated using the same software. The False-Discovery Rate (FDR) (Benjamini and Hochberg 1995) was used to control for multiple testing in the FIS and the linkage disequilibrium analyses described here, and for our examinations of genetic differentiation among temporal samples (AMOVA), pairwise FST, Hardy–Weinberg equilibrium (HWE) and bottlenecks described below.
Hybrids between A. alosa and A. fallax exist in nature and are sometimes difficult to distinguish from the parental species based on morphological characters (Alexandrino et al. 2006; Jolly et al. 2011; Mota and Antunes 2011; Faria et al. 2011; Taillebois et al. 2019). To identify hybrids in our samples we analyzed our microsatellite data using NewHybrids (Anderson and Thompson 2002). Only the data from the 15 loci found to be appropriate for population genetic analysis in both species (see below) were used for this analysis. NewHybrids were run using default genotype frequency classes, uniform priors, a burn-in of 10,000 followed by 30,000 iterations of the data, and set to identify individuals as pure A. alosa, pure A. fallax, F1 hybrids, F2 hybrids, A. alosa backcrosses, or A. fallax backcrosses. All individuals in landlocked A. fallax and A. alosa, where hybridization is unlikely, were found to have a 99% chance or higher of being pure in our NewHybrids analysis. We, therefore, removed all individuals with a less than 99% chance of being pure from all subsequent population analyses. Since the inclusion of hybrids in our analyses of linkage disequilibrium, FIS, and HWE described above could be problematic, we then re-ran each of them using the filtered dataset. We then did a final check of the remaining loci using MICRO-CHECKER 2.2.1 (Van Oosterhout et al. 2004) to test for false and null alleles due to PCR errors.
To check if it was appropriate to combine microsatellite data on samples obtained from the same location at different times, we tested for genetic differences between them using analysis of molecular variance (AMOVA) implemented in Arlequin 3.1 (Excoffier et al. 2005) with 10,000 permutations to assess statistical significance. All samples found to be from the same breeding populations per location were then pooled.
Population structure and migration
Genetic structure among sampling locations was explored by calculating pairwise FST (Weir and Cockerham 1984) using FSTAT 2.9.3.2 (Goudet 1995) with 10,000 permutations to assess statistical significance. In each case where FST was found to be not significant, the statistical power for rejecting the null hypothesis of genetic homogeneity was assessed using simulations done with POWSIM (Ryman and Palm 2006). Briefly, in each simulation, we tested the statistical power of our data to detect allele frequency differences between the pair of populations being examined given their sample sizes and the FST that was estimated for them.
Genetic substructure and gene flow was further explored using several different methods. First, we used BAPS (Corander et al. 2008) to test the hypotheses that the number of genetic clusters (K) in each species was between one and the total number of sampling locations (13 for A. alosa and 30 for A. fallax) without using sampling location as a prior. BAPS was run three times per species, and in each run, each K was tested three times. To complement the BAPS analysis and focus more on gene flow (Bohling et al. 2012), we also analyzed our data using the admixture model in STRUCTURE 2.2 (Pritchard et al. 2010) without a location prior. Ten iterations of STRUCTURE were run per K using a burn-in of 500,000 replicates followed by 1,000,000 additional replicates. The results of the replicate runs were combined with CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007) and visualized using Distruct v1.1 (Rosenberg 2003). Each K was evaluated using the likelihood (Pritchard et al. 2000) and ΔK (Evanno et al. 2005) method. Strong genetic structure and IBD like found here for A. fallax can sometimes bias genetic clustering and admixture analysis (Pritchard et al. 2000; Frantz et al. 2009). We, therefore, used a hierarchical approach to run STRUCTURE for A. fallax. This involved initially analyzing all of the A. fallax populations together and then, based on the results, doing separate runs for subsets of populations based on a K of four (see “Results”) from the full analysis. Lastly, factorial correspondence analysis (FCA) was used to visualize the relationships among individuals of each species based on their multilocus allele frequencies using GENETIX 4.05 (Belkhir et al. 1998). For increased clarity, FCA was also used to analyze the 18 A. fallax populations of the Atlantic and Baltic, and the 12 from the Mediterranean, in two separate analyses.
To help visualize patterns of divergence and genetic breaks within species, a neighbor-joining tree was generated for each species based on Nei’s DST (corrected for sample size bias) (Nei 1973) using PopTree2 (Takezaki et al. 2010) with 1000 bootstrap replicates to evaluate node support. Geographic restrictions to gene flow among anadromous populations within each species were also studied using BARRIER (version 2.2) (Manni et al. 2004). In the BARRIER analysis, the number of loci that supported a particular barrier was calculated assuming three (the default setting) barriers for each species, and those supported by more than half of the loci were reported.
Finally, we used BayesAss 3.03 (Wilson and Rannala 2003) to estimate recent migration rates among populations within each species. Each run of BayesAss was done with a burn-in of one million iterations and 100 million additional ones for collecting data using the following acceptance rates (-a0.7 -f1.0 -m0.3; see the manual for details). Adequate mixing and convergence in the BayesAss simulations were assessed by examining the trace files for each run using Tracer v1.3 (Drummond and Rambaut 2007), and by running the analysis three times per species and comparing the results.
Isolation by distance
We tested for the presence of a positive correlation between genetic and geographic distance, i.e. isolation by distance (IBD), for anadromous populations in each species. The genetic distance estimates used were linearized with the transformation FST/(1−FST) (Slatkin 1973). Geographic distances were based on the shortest path between two locations in marine water shallower than 300 m. This depth cutoff was chosen because adult A. alosa and A. fallax are known to inhabit marine waters up to 300 m (Quignard et al. 1981). The geographic distances were calculated using the Least Cost Path algorithm in qGIS (version 3.161) and a bathymetry map generated in R using the getNOAA.bathy package. Statistical significance of IBD was assessed using mantel tests (Mantel 1967) with 10,000 permutations using the software package IBD: Isolation By Distance (Bohonak 2002). We tested for IBD in each species, and then separately on A. fallax from each the Atlantic and Mediterranean. To avoid biases from including multiple samples from the same spawning populations (see “Results”), and those with extremely low sample sizes (N < 10), we excluded the Nissum, Ringkobing fjords, and the Usk, Wye, Hérault, Tirso, and Pineios Rivers A. fallax samples from our analyses of IBD.
Genetic diversity and bottlenecks
Genetic diversity was studied by estimating the number of alleles (A), expected heterozygosity (HE), and allelic richness (Ar) in each sample. Ar was calculated using the software program hp-rare 1.0 (Kalinowski 2004). Sample size can bias estimates of Ar (Leberg 2002), and we had samples ranging in size from four to 117. We therefore estimated Ar using three different N per species for comparative purposes. The number of alleles (A) and expected heterozygosity (HE) were calculated using FSTAT 2.9.3.2. To test for recent population bottlenecks, we used the software BOTTLENECK 1.2.02 (Piry et al. 1999) with default parameters and assuming loci evolved according to the Two-Phased-Model.
Average HE and Ar (per population) between anadromous A. alosa and A. fallax were compared using a t-test assuming unequal variance. Normality of the HE and Ar data was tested for using QQ-plots, and equality of the variances with the F-test (results not shown). Differences in HE and Ar, by locus, between landlocked and anadromous populations within each species were tested for using a Wilcoxon signed-ranks test. In this analysis, genetic diversity was compared between each landlocked population and the anadromous one to which it was most closely related based on our FST analysis.
Results
Data filtering
Significant allele frequency differences between temporal samples from the same location were not detected using AMOVA (P > 0.05) and were pooled before further analyses (Online Resource Table 1). Linkage disequilibrium was not statistically significant between any microsatellite loci pair. Exact tests for FIS revealed locus Af13, Asa6, and AsaD429 in A. alosa (Online Resource Table 3a) and, Af6, Aa14, Af15, Af20, Asa8, and AsaD429 in A. fallax (Online Resource Table 3b) had significant heterozygote deficiencies in multiple populations and across all populations (per species) and were not used in any subsequent analyses.
To avoid the inclusion of hybrids in our analyses, we removed any individual that was not 0.99% pure based on the NewHybrids analysis from all subsequent population analyses. The number of A. fallax below 0.99 per sampling location, and the original sample sizes, were as follows (in parentheses): Solway Firth (3/16), Scheldt (1/23), Towy (10/48), Severn (5/32), Wye (3/47), Usk (4/32), Minho (2/46), Lima (5/37), Mondego (1/18), Tejo (17/57), Aude (4/24), and Rhone (2/28). Similarly, the number of A. alosa identified as hybrids in each sampling location were Vienne (6/ 92), Charente (6/38), Lima (6/34), Mondego (16/72), Tejo (2/38), and Guadiana (2/12). After filtering these potential hybrids from our dataset, we then re-ran our analyses of linkage disequilibrium among loci and HWE. In all cases, loci and populations were found to be in HWE. The loci found to be suitable for population structure and diversity analysis were also found to be unlinked in all cases (P > 0.05). Our MicroChecker analysis indicated that the 21 remaining loci for A. alosa, and 18 loci for A. fallax, after filtering were appropriate for population genetic analysis.
Population structure and migration
Pairwise FST estimates were significant in 72 of 78 of the cases tested for A. alosa, and 427 of 435 of them for A. fallax Online Resource Table 4a and 4b. The average pairwise FST between anadromous populations of A. alosa was 0.083 (range: − 0.007 to 0.286) while for A. fallax it was 0.306 (range: − 0.0190 to 0.655). The average FST between landlocked and all other populations within each species was 0.149 (range: − 0.007 to 0.321) for A. alosa and 0.538 (range: 0.215 to 0.731) for A. fallax. Our POWSIM analysis indicated that in all cases where FST was found to be not significant, statistical power was lacking due to sample size (Online Resource Table 5). Average FIS per population (Table 1 and Online Resource Table 3) were close to zero (less than ± 0.09) in all cases except for three A. fallax populations that were higher (Solway Firth, Curonian Lagoon, and Izmir) and two that were lower (Lake Maggiore and Pineios River).
We examined genetic structure in A. alosa and A. fallax using two different clustering algorithms. With BAPS, the number of clusters with the highest posterior probability was five for A. alosa (P = 0.92; Fig. 2a) and 17 for A. fallax (P = 0.97; Fig. 2b). In our STRUCTURE analysis, in all cases, ΔK estimates were highest for a K of two (Online Resource Fig. 1a-e; Online Resource Fig. 2a and 2b). However, the likelihoods for each K generally increased sharply to a K greater than two and then flattened out, except for A. fallax in the north part of the Atlantic and Baltic sea where it was about equal for a K of two and five. We, therefore, present a range of K for each of the STRUCTURE analyses conducted. The clusters identified in our BAPS and STRUCTURE analyses were the same for A. alosa and congruent for A. fallax. However, the BAPS analysis for A. fallax suggested greater fine-scale genetic structure throughout its range than did the STRUCTURE. Based on our STRUCTURE analysis, admixture was highest among neighboring populations and within several geographic regions including areas north and south of northern Iberia (A. alosa and A. fallax) and east and west of Italy in the Mediterranean (A. fallax).
In our FCA analysis of A. alosa, the greatest allele frequency differences were between individuals in the Guadiana drainage (the anadromous Guadiana and landlocked Alqueva) and all other populations (First axis = 4.28% Inertia; Online Resource Fig. 3a). The second axis highlighted the differences between A. alosa north and south of the Bay of Biscay. In the FCA analysis of A. fallax, the first axis highlighted differences between the Atlantic and Mediterranean populations (8.23% Inertia) and the second, between the landlocked Lough Leane and all others (3.31% Inertia) (Online Resource Fig. 3b). When only A. fallax populations in the Atlantic and Baltic Sea were considered, again, Lough Leane was shown to be the most divergent group (5.51% Inertia), and that allele frequency differences between individuals north and south of northern Iberia were substantial (4.20% Inertia; Online Resource Fig. 3c). In the FCA analysis of A. fallax in the Mediterranean, individuals west and east of Italy had the greatest allele frequency differences among them (7.27% Inertia) seconded by those in Corsica and Sardinia (5.17% Inertia) compared to all others (Online Resource Fig. 3d).
The NJ tree of A. alosa populations contained three main clades: (1) the Guadiana and Alqueva, (2) populations south of the Bay of Biscay (Aguieira, Lima, Minho, and Tejo), and (3) those within and north of the Bay of Biscay (Garonne, Charente, Vienne, and Aulne) (Online Resource Fig. 4a). However, only the latter had bootstrap support above 70. The NJ tree of A. fallax contained several well-supported clades and branches, with a major split between the Atlantic and Mediterranean populations (Online Resource Fig. 4b).
We identified geographic barriers to gene flow in A. alosa and A. fallax using the program Barrier. The location of each barrier identified is marked by a white rectangle in Fig. 2a (A. alosa) and Fig. 2b (A. fallax). For A. alosa, where more than one barrier was found, the rectangles are numbered in order of statistical support. In A. alosa, the strongest barrier (“1”) was between the Guadiana and all other populations and was supported by 12 out of 21 loci, followed by the Bay of Biscay (“2”, 10 out of 21 loci) and the Aulne (“3”, 10 out of 21 loci). All other barriers for A. alosa were supported by 5 or fewer loci. In A. fallax, the strongest barrier was found at the Strait of Gibraltar supported by 11 out of 18 loci. All other barriers for A. fallax were supported by 8 or fewer loci and are not shown.
Migration estimates between populations from each of the three BayeAsses runs per species conducted were similar, suggesting the simulation parameters used in the analysis were sufficient to reach convergence. This conclusion was further supported by visual analyses of the trace files for each simulation. We, therefore, present the average of the three migration rate estimates (m) and their 95% credible intervals (Online Resource Table 6a–d). Migration was considered significant if the lower limit of the credible interval for the estimate of m was greater than zero. This occurred in seven cases for A. alosa, and 22 cases for A. fallax. Specifically, migration was detected from two anadromous A. alosa populations (the Garonne and Minho), and five A. fallax ones (Scheldt, Wye, Minho, Guadiana, and Pineios) into neighboring locations. Migration (or translocations) from Lake Garda into Lake Como was also detected.
Isolation by distance
The geographic distances used in our IBD analysis were calculated by measuring the shortest path in water shallower than 300 m between two locations (Online Resource Fig. 5a and 5b ). A pattern of IBD was not found in anadromous A. alosa (r = 0.397, P = 0.0962), but was in A. fallax from throughout their range (r = 0.666, P < 0.001). IBD was also detected in anadromous A. fallax from just the Atlantic and (r = 0.688, P < 0.001), and those only in the Mediterranean (r = 0.528, P = 0.031) (Fig. 3a–d).

Plots of linearized FST with geographic distance (km) (Isolation-By-Distance) in A. alosa and A. fallax. a A. alosa samples from throughout their range, b A. fallax samples from throughout their range, c A. fallax from the Atlantic, d: A. fallax from the Mediterranean and Adriatic Seas. The correlation (r) between genetic distance and geographic distance and its statistical significance (P) are provided for each case
Genetic diversity and bottlenecks
The average within-population HE was 0.452 in anadromous A. alosa versus 0.319 in A. fallax Table 1; Online Resource Table 7a and 7b. The average Ar (based on a sample size of 10) was 3.24 in A. alosa and 2.52 in A. fallax. Using t-tests, HE (P = 0.01) and Ar (P = 0.01) were found to be significantly higher in anadromous A. alosa than A. fallax. Genetic diversity (HE and Ar) was nearly equal in the Guadiana (anadromous) and Alqueva (landlocked) A. alosa populations, but lower in the Aguieira and Castelo de Bode (landlocked) compared to the Mondego and Tejo (anadromous), respectively (Table 2).
However, the differences were only statistically significant in Castelo de Bode/Tejo comparison. In each case, genetic diversity (HE and Ar) was significantly lower in landlocked populations of A. fallax than in the anadromous ones to which they were compared (P < 0.05). Bottlenecks were not detected in any of the A. alosa populations studied, and just one of the A. fallax (Lake Como, P = 0.02).
Discussion
Here we conducted the first range-wide population genetic analysis of anadromous and landlocked populations of A. alosa and A. fallax. Genetic structure was found throughout the ranges of both species. However, patterns of genetic differentiation, gene flow, and diversity markedly differed between the two species. Here we discuss these findings and their implications for conservation.
Population genetic structure in A. alosa
Pairwise FST between populations of A. alosa in France and Scotland, and those in Portugal, were consistently high for the species. This suggests a genetic break exists in the southern Bay of Biscay or northern Spain for A. alosa, as has been seen in other species (Catanese et al. 2017; Robinet et al. 2020). This break was also supported by our cluster, FCA, barrier, migration, and phylogenetic analyses. The absence of any major rivers or known A. alosa populations between the Garonne River in southern France and the Minho River in Portugal may restrict gene flow between these two areas.
Individuals from most of the anadromous populations of A. alosa that we examined were included in just two genetic clusters identified in our BAPS (and STRUCTURE) analysis. This suggests there is considerable gene flow in this species, especially within the regions represented by these two major genetic clusters. One of the two clusters is comprised of populations from France and the UK, and the other from those in Portugal. Genetic differentiation among populations included in each of these two clusters was low (FST < 0.027), but usually significant, especially for the Mondego in Portugal. This suggests that while straying among populations within these two regions is common, individuals regularly return to their natal river to spawn. The two anadromous populations not included in these two major clusters (the Guadiana and Aulne) spawn in drainages at the edges of the current range of A. alosa, which may have contributed to their geographic isolation and divergence.
The close genetic relationships (FST < 0.014) between the Solway Firth population in the U.K. and most of those sampled in France suggests that straying in this region may occur over thousands of kilometers. This result was somewhat unexpected because Solway Firth in Scotland is more than 1500 km from the mouths of the Vienne, Charente, and Garonne Rivers in France and geographically closest to the Aulne River. Yet, all of these populations were several-fold more genetically distinct from the Aulne (FST > 0.05) than to each other. This may be explained connectivity that occurs in shallower waters off the western coast of the UK, away from the Aulne (Fig. 1). In addition, genetic diversity in the Aulne was the second-lowest among all of the anadromous A. alosa populations studied, suggesting it may be a relatively small population that has rapidly diverged due to increased genetic drift. It is also possible that some or all of these populations have recently been (re)colonized and are not yet at migration-drift equilibrium.
The three landlocked populations of A. alosa we examined were more closely related to the anadromous ones in each of the drainages they inhabit than any other we studied (Tables 3A and 3B, Online Resource Fig. 3a and 3b). These results confirm that these three landlocked populations were independently derived from anadromous fish that were trapped within each reservoir when the dams were built. The three landlocked populations had the highest FST values for the species, which could be due to founder effects, bottlenecks, genetic isolation, or a combination of these factors. Our BOTTLENECK analysis did not show that population instability has affected genetic diversity in any of these three populations. Shad spawn in large schools (Baglinière et al. 2003; Aprahamian et al. 2003) and reproduction is unlikely to occur with only a few individuals. If spawning behavior creates a lower limit to the Ne of colonizing populations, founder effects may not have had a significant impact on genetic diversity in landlocked populations of A. alosa. This hypothesis is supported by our finding of genetic diversity (Ar and HE) in the Alqueva and Aguieira is about equal to that in the anadromous populations that founded them (Table 2). Construction of the dams on the Tejo, Mondego, and Guadiana Rivers began in 1945, 1972, and 1998 and formed the Castelo de Bode, Aguieira, and Alqueva Reservoirs, respectively. The divergence between the landlocked Castelo de Bode population and the Tejo, which founded it, was more than three times higher than between the Aguieira and Mondego or Alqueva and Guadiana (same relationship). These results suggest that the Castelo de Bode population has significantly diverged in just a few decades due to genetic isolation. This rapid evolution may also be due to the Castelo de Bode having a lower carrying capacity (and population size) than the Tejo River, which could accelerate the loss of genetic diversity and divergence due to drift.
Population genetic structure A. fallax
Pairwise FST between A. fallax populations from the Atlantic and Mediterranean were generally greater than 0.1, indicating that a genetic break exists within A. fallax at the Strait of Gibraltar. This break was also supported by our clustering, FCA, Barrier, and phylogenetic analyses, and suggests that gene flow between these two areas is restricted. However, the divergence between A. fallax from these two areas may also be due to past vicariance during glacial periods (Faria et al. 2012), as has been found for a variety of other marine species (Patarnello et al. 2007).
As evidenced by the strong patterns of IBD found in A. fallax, and our clustering, phylogenetic, and FCA analyses, populations of A. fallax were usually most closely related to their nearest geographic neighbors. This was found to be the case throughout its range, and within the Mediterranean and Atlantic, suggesting it is a general feature of the species. One exception to this was the close relationship between where the A. fallax population from Solway Firth (Scotland) and the Scheldt (Belgium), Nissum (Denmark), and Ringkobing (Denmark) in the North Sea. These populations clustered together in our BAPS and STRUCTURE analyses (Fig. 2b, Online Resource Fig. 2b), and pairwise FST among them were below 0.05 (Online Resource Table 4b). Given the observed divergences of other populations from the UK (e.g. the Towy and Severn), the close relationships among the Solway, Scheldt, and Ringkobing populations suggest gene flow has occurred along the north side of Scotland and down the east side of the U.K. into the North Sea. This adds support to the hypothesis that migration in shad occurs offshore in this area, as we suggested earlier based on our analysis of A. alosa from Solway Firth.
Like in A. alosa, genetic differences between landlocked and anadromous populations of A. fallax were among the highest observed for the species. The average pairwise FST between A. fallax in the Italian glacial lakes (the Como, Garda, and Maggiore) and the anadromous one in the Po was 0.348. Divergence of A. fallax in Lough Leane from nearby anadromous A. fallax in the UK and northern Europe was also substantial (average FST > 0.300), as has previously been observed (Coscia et al. 2010). All four freshwater A. fallax populations studied are now thought to be geographically isolated, which has likely contributed to their divergence from anadromous populations. Like in A. alosa, freshwater populations of A. fallax may have a lower carrying capacity and consequently size than anadromous populations, which has contributed to their divergence. FST for the two most closely related anadromous and freshwater populations of A. fallax that we studied (the Lake Garda and Po populations) was severalfold lower than any other pair. As the Lake Garda and Po populations could exchange migrants as recently as 50 years ago via the Mincio River, their close relationship suggests that connectivity is a strong determinant of rates of divergence in landlocked populations of A. fallax.
Genetic diversity in A. alosa and A. fallax
Genetic diversity (HE and Ar) was found to be higher in populations of A. alosa than A. fallax (Table 1), which is consistent with findings from microsatellite data analyzed in Jolly et al. (2012), but conflicts with previous studies based on mtDNA and allozymes (Faria et al. 2012; Alexandrino et al. 2006). The markers used in each of these studies are influenced by mutation, genetic drift, and selection in different ways (Galtier et al. 2009), which may explain these inconsistent results. Our results suggest that, despite this potential species-wide difference, contemporary admixture in A. alosa has resulted in greater within-population genetic diversity in A. alosa than A. fallax. However, this and other hypotheses should be further tested with additional markers across the genome.
Genetic diversity was found to be lower in landlocked versus anadromous populations of each species, except in the cases of the most recently geographically isolated A. alosa populations (the Alqueva and Aguieira). In the Alqueva and Aguieira, genetic diversity was not found to be significantly different than in the anadromous populations that founded them, suggesting that they were not greatly impacted by founder effects. If founder effects were similarly low in the Castelo de Bode population (A. alosa), and the four freshwater populations of A. fallax that were studied, loss of genetic diversity in them is likely due to their geographic isolation, and genetic drift over time. However, this idea should be tested using simulations. A bottleneck was detected in the Lake Como population, suggesting a recent decrease in population size has reduced genetic diversity in this population. Since bottlenecks can be difficult to detect with less than tens or hundreds of loci (Peery et al. 2012), their potential impact on genetic diversity should be further evaluated for these populations using a more powerful dataset.
Philopatric behavior in A. alosa and A. fallax
A pattern of IBD was not found in A. alosa but was in A. fallax. This result conflicts with previous studies where IBD was detected in each species (Alexandrino et al. 2006; Jolly et al. 2012). However, it is consistent with estimates of migration in A. alosa based on otolith microchemistry where returning spawners were often from non-neighboring populations (Martin et al. 2015). It also fits with our BayesAss analysis where migration from the Minho River into the neighboring Lima and Mondego Rivers was observed in both species and was higher in A. alosa than A. fallax (Online Resource Tables 6a and 6b). Migration from the Minho River was also detected further south than the Mondego River (in the Tejo River) in A. alosa, but not A. fallax. These findings suggest that straying occurs more frequently, and over greater distances, in A. alosa than A. fallax. However, we cannot exclude the possibility that differences in genetic structure between A. alosa and A. fallax are due to them having different demographic histories, such as if A. alosa populations were founded, or re-colonized, more recently than A. fallax.
It is possible that genetic structure in A. alosa and A. fallax is not due to natal homing, but rather limited dispersal distances. However, A. alosa and A. fallax are thought to widely disperse at sea (Aprahamian et al. 2003; Martin et al. 2015; Davies et al. 2020), including sea migrations up to 1500 km (Sabatié and Blagliniere 2001). They also stay at sea for several years before returning to spawn (Lassalle et al. 2008). Given the time each species spends at sea, and that they inhabit marine waters with different bathymetries, currents, and patterns of upwelling, it is likely that individuals from most populations migrate, or are transported, various distances at sea to feed. If so, then the strong pattern of IBD found for A. fallax, and its consistency in the Atlantic and Mediterranean, is likely at least partially due to natal homing. Similarly, the genetic structure in A. alosa shown here, and IBD found in previous studies, are consistent with them having some capacity to home. Our results show that if natal homing is occurring in each species, fidelity is lower in A. alosa than A. fallax. This may be due to differences in their abilities to home, dispersal distance at sea, or other factors such as a higher evolutionary cost of emigration in A. fallax.
Management recommendations
We have conducted, to our knowledge, the most complete assessment of genetic structure and diversity in European shads to date. Information on the migratory behaviors of anadromous species including salmon (Banks et al. 2000; Hendry et al. 2004), sturgeon (Stabile et al. 1996), and other shad (Waters et al. 2000; Hasselman et al. 2010) is regularly used to devise and assess management plant to assess them. The results of our work combined with that of numerous other authors highlighted above should similarly serve as valuable resources for conservation managers focused on protecting A. alosa and A. fallax and other species in their communities.
The Allis and Twaite shad could be managed together to some extent due to their similar life histories and overlapping ranges. However, our results show that conservation units (Moritz 1994; Waples and Lindley 2018) should span larger areas or include more populations in A. alosa than A. fallax. Since straying is likely more common and occurs over greater distances, in A. alosa than A. fallax, positive effects of immigration (see Introduction) are expected to have a more geographically localized impact in A. fallax than A. alosa. Our analysis of migration showed that gene flow from the Garonne and Minho (A. alosa), and the Scheldt, Wye, Minho, Guadiana, and Rhone and (A. fallax) into neighboring populations is substantial (Online Resource Table 6a and 6b). This indicates that these populations are significant sources of migrants within each species and may be especially important for maintaining natural levels of connectivity within each of them. Other studies have identified populations with high immigration rates such as those in the Nivelle (Taillebois et al. 2019) and Severn (Jolly et al. 2012; Martin et al. 2015; Davies et al. 2020) Rivers that too should be protected. However, given that most populations of A. alosa and especially A. fallax were genetically distinct, each may play an important role in the population dynamics of each species and should be conserved.
Genetic diversity was found to be higher in populations of anadromous A. alosa compared to A. fallax, which we concluded is due at least in part to different degrees of admixture and migration within each species. Consequently, reduced connectivity via immigration could have a particularly strong negative impact on genetic diversity in populations of A. fallax. Genetic diversity was lower in all three landlocked A. fallax populations that were studied when compared to anadromous ones, as was it for the oldest of the three A. alosa that we examined. Since all of the landlocked populations that we studied are currently geographically isolated, and some had reduced genetic diversity, efforts should be made to preserve what variation remains in each of them. This should include the removal of dams and weirs that impede or block gene flow into freshwater populations, or the installation of effective fish passes for these species to mitigate their effects.
Code and data availability
The microsatellite data and the code used for generating bathymetry maps used in this study are available in Dryad (https://datadryad.org).
References
Alexandrino PB, Faria R, Castro F et al (2006) Interspecific differentiation and intraspecific substructure in two closely related clupeids with extensive hybridization, Alosa alosa and Alosa fallax. J Fish Biol 69:242–259. https://doi.org/10.1111/jfb.2006.69.issue-sb
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1219–1229
Antunes A, Faria R, Johnson WE et al (2006) Life on the edge: the long-term persistence and contrasting spatial genetic structure of distinct brown trout life histories at their ecological limits. J Hered 97:193–205. https://doi.org/10.1093/jhered/esj014
Aprahamian MW, Aprahamian C, Baglinière JL, et al (2003) Alosa alosa and Alosa fallax spp. Literature review and bibliography. University of Porto. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/290354/sw1-014-tr-e-e.pdf
Baglinière JL, Sabatié MR, Rochard E, et al (2003) The allis shad Alosa alosa: Biology, ecology, range, and status of populations. In: American Fisheries Society Symposium, pp 85–102
Banks MA, Rashbrook VK, Calavetta MJ et al (2000) Analysis of microsatellite DNA resolves genetic structure and diversity of chinook salmon (Oncorhynchus tshawytscha) in California’s Central Valley. Can J Fish Aquat Sci 57:915–927. https://doi.org/10.1139/cjfas-57-5-915
Belliard J, Marchal J, Ditche JM et al (2009) Return of adult anadromous allis shad (Alosa alosa L.) in the river Seine, France: a sign of river recovery? River Res Appl 25:788–794. https://doi.org/10.1002/rra.1221
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B-Methodol 57:289–300
Bohonak A (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Hered 93:153–154
Buysse D, Coeck J, Maes J (2008) Potential re-establishment of diadromous fish species in the River Scheldt (Belgium). Hydrobiologia 602:155–159. https://doi.org/10.1007/s10750-008-9292-5
Catanese G, Watteaux R, Montes I et al (2017) Insights on the drivers of genetic divergence in the European anchovy. Sci Rep 7:65–75. https://doi.org/10.1038/s41598-017-03926-z
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539. https://doi.org/10.1186/1471-2105-9-539
Coscia I, Rouzic A, King JJ et al (2010) A highly permeable species boundary between two anadromous fishes. J Fish Biol 77:1137–1149. https://doi.org/10.1111/j.1095-8649.2010.02768.x
Costa A, Almeida C, Domingos I et al (2001) Present status of the main shads’ populations in Portugal. Bull Fr Pêche Piscic 2001:1109–1116
Davies P, Britton RJ, Nunn AD et al (2020) Novel insights into the marine phase and river fidelity of anadromous twaite shad Alosa fallax in the UK and Ireland. Aquat Conserv Mar Freshw Ecosyst 30:1291–1298. https://doi.org/10.1002/aqc.3343
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:117693430500100. https://doi.org/10.1177/117693430500100003
Faria R, Wallner B, Weiss S (2004) Isolation and characterization of eight dinucleotide microsatellite loci from two closely related clupeid species (Alosa alosa and A. fallax). Mol Ecol 2004:586–588. https://doi.org/10.1111/men.2004.4.issue-4
Faria R, Weiss S, Alexandrino PB (2006) A molecular phylogenetic perspective on the evolutionary history of Alosa spp. (Clupeidae). Mol Phylogenet Evol 40:298–304. https://doi.org/10.1016/j.ympev.2006.02.008
Faria R, Pinheiro A, Gabaldón T et al (2011) Molecular tools for species discrimination and detection of hybridization between two closely related Clupeid fishes Alosa alosa and A. fallax. J Appl Ichthyol 27:16–20. https://doi.org/10.1111/j.1439-0426.2011.01846.x
Faria R, Weiss S, Alexandrino PB (2012) Comparative phylogeography and demographic history of European shads (Alosa alosa and A. fallax) inferred from mitochondrial DNA. BMC Evol Biol 12:194. https://doi.org/10.1186/1471-2148-12-194
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305–327. https://doi.org/10.1146/annurev.ge.29.120195.001513
Frantz AC, Cellina S, Krier A et al (2009) Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? J Appl Ecol 46:493–505. https://doi.org/10.1111/j.1365-2664.2008.01606.x
Galtier N, Nabholz B, Glémin S, Hurst GDD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18:4541–4550. https://doi.org/10.1111/j.1365-294X.2009.04380.x
Gomulkiewicz R, Holt RD, Barfield M (1999) The effects of density dependence and immigration on local adaptation and niche evolution in a black-hole sink environment. Theor Popul Biol 55:283–296. https://doi.org/10.1006/tpbi.1998.1405
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate f-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Hasselman DJ, Bradford RG, Bentzen P (2010) Taking stock: defining populations of American shad (Alosa sapidissima) in Canada using neutral genetic markers. Can J Fish Aquat Sci 67:1021–1039
Hendry A, Bohlin T, Jonsson B, Ole KB (2004) To sea or not to sea. Evolution illuminated: salmon and their Relatives. Oxford University Press, Oxford, pp 92–125
Jolly M, Maitland P, Genner M (2011) Genetic monitoring of two decades of hybridization between allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet 12:1–14. https://doi.org/10.1007/s10592-011-0211-3
Jolly MT, Aprahamian MW, Hawkins SJ et al (2012) Population genetic structure of protected allis shad (Alosa alosa) and twaite shad (Alosa fallax). Mar Biol 159:675–687. https://doi.org/10.1007/s00227-011-1845-x
Julian SE, Bartron ML (2007) Microsatellite DNA markers for American shad (Alosa sapidissima) and cross-species amplification within the family Clupeidae. Mol Ecol Notes 7:805–807. https://doi.org/10.1111/j.1471-8286.2007.01710.x
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543. https://doi.org/10.1023/B:COGE.0000041021.91777.1a
Keefer ML, Caudill CC (2014) Homing and straying by anadromous salmonids: a review of mechanisms and rates. Rev Fish Biol Fish 24:333–368. https://doi.org/10.1007/s11160-013-9334-6
King JJ, Roche WK (2008) Aspects of anadromous Allis shad (Alosa alosa Linnaeus) and Twaite shad (Alosa fallax Lacépède) biology in four Irish Special Areas of Conservation (SACs): status, spawning indications and implications for conservation designation. Hydrobiologia 602:145–154. https://doi.org/10.1007/s10750-008-9280-9
Lassalle G, Trancart T, Lambert P (2008) Latitudinal variations in age and size at maturity among allis shad Alosa alosa populations. J Fish Biol 73:1799–1809. https://doi.org/10.1111/j.1095-8649.2008.02036.x
Leberg PL (2002) Estimating allelic richness: effects of sample size and bottlenecks. Mol Ecol 11:2445–2449. https://doi.org/10.1046/j.1365-294x.2002.01612.x
Maes J, Stevens M, Breine J (2008) Poor water quality constrains the distribution and movements of twaite shad Alosa fallax fallax (Lacépède, 1803) in the watershed of river Scheldt. Hydrobiologia 602:129–143. https://doi.org/10.1007/s10750-008-9279-2
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (Genetic, Morphologic, Linguistic) variation: how barriers can be detected by using monmonier’s algorithm. Hum Biol 76:173–190. https://doi.org/10.1353/hub.2004.0034
Mantel N (1967) Assumption-free Estimators using U statistics and a relationship to the Jackknife Method. Biometrics 23:567–571. https://doi.org/10.2307/2528016
Martin J, Rougemont Q, Drouineau H et al (2015) Dispersal capacities of anadromous Allis shad population inferred from a coupled genetic and otolith approach. Can J Fish Aquat Sci. https://doi.org/10.1139/cjfas-2014-0510
McDowall (2001) Anadromy and homing: two life-history traits with adaptive synergies in salmonid fishes? Fish Fish 2:78–85. https://doi.org/10.1046/j.1467-2979.2001.00036.x
Moritz C (1994) Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Mota M, Antunes C (2011) First report on the status of Allis shad (Alosa alosa) in the Minho River (Northwestern Iberian Peninsula). J Appl Ichthyol 27:56–69. https://doi.org/10.1111/j.1439-0426.2011.01853.x/full
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Patarnello T, Volckaert FAMJ, Castilho R (2007) Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographical break? Mol Ecol 16:4426–4444. https://doi.org/10.1111/j.1365-294X.2007.03477.x
Peery MZ, Kirby R, Reid BN et al (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Pereira MJC, Cowx IG, Luís TS et al (1999) Observations on the ecology of a landlocked population of allis shad in Aguieira Reservoir, Portugal. J Fish Biol 55:658–664. https://doi.org/10.1111/j.1095-8649.1999.tb00706.x
Piry S, Luikart G, Cornuet J-M (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Pritchard JK, Stephens M, Donnelly P (2000) Inference of Population Structure Using Multilocus Genotype Data. Genetics 155:945–959
Pritchard J, Wen X, Falush D (2010) Structure. Version 2.3. https://burfordreiskind.com/wp-content/uploads/Structure_Manual_doc.pdf
Quignard JP, Douchement C (1981) Alosa alosa (Linneaus 1758). In: The freshwater fishes of Europe, vol 2. Clupeidae, Anguillidae. H. Hoestlandt (eds), pp 89–126
Rieman BE, Allendorf FW (2001) Effective population size and genetic conservation criteria for bull trout. North Am J Fish Manag 21:756–764. https://doi.org/10.1577/1548-8675(2001)021%3c0756:EPSAGC%3e2.0.CO;2
Robinet T, Roussel V, Cheze K, Gagnaire P (2020) Spatial gradients of introgressed ancestry reveal cryptic connectivity patterns in a high gene flow marine fish. Mol Ecol 29:3857–3871. https://doi.org/10.1111/mec.15611
Rosenberg NA (2003) distruct: a program for the graphical display of population structure: PROGRAM NOTE. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Ryman N, Palm S (2006) POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6:600–602. https://doi.org/10.1111/j.1471-8286.2006.01378.x
Sabatié MR, Blagliniere JL (2001) Some ecobiological traits in Moroccan shads: a cultural and socio-economic value interest which has disappeared. Bull Fr Pêche Piscic 74:1–16
Salmenkova EA (2017) Mechanisms of homing in salmonids. Biol Bull Rev 7:287–298. https://doi.org/10.1134/S2079086417040077
Schtickzelle N, Quinn TP (2007) A metapopulation perspective for salmon and other anadromous fish. Fish Fish 8:297–314
Slatkin M (1973) Gene flow and selection in a cline. Genetics 1973:733–756
Sotelo G, Andree K, López MA et al (2014) The puzzling demographic history and genetic differentiation of the twaite shad (Alosa fallax) in the Ebro River. Conserv Genet 15:1037–1052. https://doi.org/10.1007/s10592-014-0597-9
Stabile J, Waldman JR, Parauka F, Wirgin I (1996) Stock structure and homing fidelity in Gulf of Mexico sturgeon (Acipenser oxyrinchus desotoi) based on restriction fragment length polymorphism and sequence analyses of mitochondrial DNA. Genetics 144:767–775
Taillebois L, Sabatino S, Manicki A et al (2019) Variable outcomes of hybridization between declining Alosa alosa and Alosa fallax. Evol Appl 2019:12889. https://doi.org/10.1111/eva.12889
Takezaki N, Nei M, Tamura K (2010) POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol Biol Evol 27:747–752. https://doi.org/10.1093/molbev/msp312
Taverny CC, Elie PP (2001) Répartition spatio-temporelle de la grande Alose Alosa alosa (Linné, 1766) et de L’Alosa feinte Alosa fallax (Lacépede, 1803) dans le golfe de Gascogne. Bull Francais De La Peche Et De La Piscicult 362(363):803–821
Van Oosterhout C, Hutchinson W, Wills D, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Waples RS, Lindley ST (2018) Genomics and conservation units: the genetic basis of adult migration timing in Pacific salmonids. Evol Appl 11:1518–1526. https://doi.org/10.1111/eva.12687
Waters JM, Epifanio JM, Gunter T (2000) Homing behaviour facilitates subtle genetic differentiation among river populations of Alosa sapidissima: microsatellites and mtDNA. J Fish Biol. https://doi.org/10.1111/j.1095-8649.2000.tb00760.x/abstract
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution. https://doi.org/10.2307/2408641
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Acknowledgements
We dedicate this manuscript to the memory of Paulo Alexandrino, who inspired us to study European shads and was a remarkable researcher, mentor, and friend. We thank the many fishermen and researchers that helped us to collect samples for this study over the years, especially M.W. Aprahamian, C. Antunes, M. Barbosa, D. Bekkevold, P. Beeck, D. Bobori, R. Cannas, D. Clavé (and all of MIGADO), M. Le Corre, J. Y. Menella, D. Mordak, F.N. Marzano, M. Mota, A. Piccinini, J. Raeymaekers, R. Sabatié, S. Weiss, and T. Zolubas. We also thank Walt Eanes, Mike Bell, Stephan Munch, and several anonymous reviewers for helping revise an earlier version of this manuscript, and Paulo Pereira for assistance making the bathymetry maps used in this study.
Funding
This work was funded in part by the EU Project AARC—Atlantic Aquatic Resources Conservation, project nº 2009-1/079, Atlantic Area Programme. Funds were also provided by FEDER unds through the Operational Programme for Competitiveness Factors -COMPETE and by National Funds through FCT—Foundation for Science and Technology under the «Refª do Projecto– Ref. FCT e ref. Compete»; PTDC/BIA-BEC/105447/2008 -FCOMP-01-0124-FEDER-008972—The role of ecological speciation and selection in the divergence of shads Alosa spp. RF was financed by FCT under the Programa Operacional Potencial Humano – Quadro de Referência Estratégico Nacional from the European Social Fund and the Portuguese Ministério da Educação e Ciência through the postdoctoral fellowships SFRH/BPD/26384/2006 and SFRH/BPD/89313/2012.
Author information
Authors and Affiliations
Contributions
All three authors designed, and collected samples for, this study. Laboratory work was conducted by JD, SJS and RF. All analyses were completed by SJS and RF. The paper was written by Stephen J. Sabatino with extensive contributions and editing from PA, and RF.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
All samples were collected in accordance with EU ethical standards.
Consent for publication
All authors have read and approved this manuscript for publication.
Additional information
Responsible Eitor: C. Eizaguirre.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewers: J. G. Prunier and an undisclosed expert.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sabatino, S.J., Faria, R. & Alexandrino, P.B. Genetic structure, diversity, and connectivity in anadromous and freshwater Alosa alosa and A. fallax. Mar Biol 169, 2 (2022). https://doi.org/10.1007/s00227-021-03970-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-021-03970-4