
Abstract
Biological invasions are often characterized by a phase of post-establishment expansion in which the invading species increases its range through colonization of new geographic areas. These expansions are predicted to result in specific genetic signatures, most notably decreased genetic diversity with distance from the point of introduction. The Asian caridean shrimp Palaemon macrodactylus is an introduced species in many regions of the globe. It has most recently invaded the U.S. Atlantic coast, with the first reported sighting in New York in 2001. This study used mitochondrial cytochrome oxidase I sequence data and data for 1598 single-nucleotide polymorphisms generated through restriction enzyme-associated DNA sequencing to evaluate two potential invasion mechanisms describing the spread of P. macrodactylus north of New York: the first based on a single introduction followed by range expansion facilitated by ocean currents, coastal transport, and physical environment; the second based on multiple introductions from overseas into different ports along the coast. We compare population genetic results to predictions of genetic patterns for each of these potential invasion pathways. Results do not support range expansion in which diversity decreases with distance from the point of first detection. Rather, the data suggest that multiple introductions may have taken place with population genomic diversity increasing with distance from New York, and peaks of mitochondrial diversity in populations collected from New York and the Boston–Plymouth coastline. These results indicate that multiple human-mediated dispersal events may be as important as oceanographic and life history considerations during the colonization phases of a marine invasion.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biological invasions threaten biodiversity in terrestrial, aquatic, and marine ecosystems (Bax et al. 2003; Lowry et al. 2013; Thomaz et al. 2014), often negatively affecting ecosystem services and damaging economies (Funk et al. 2014; Walsh et al. 2016). Invasions are both serious concerns for biodiversity conservation, and excellent models for the study of dynamic ecological and evolutionary processes (Lee 2002; Rius et al. 2015; Barrett 2015). Many invasions present an evolutionary paradox because the invading species is ecologically successful despite a high probability of experiencing reduced genetic diversity due to an initial founder event during introduction, often thought to decrease fitness and adaptive potential (Tsutsui et al. 2000; Estoup et al. 2016). The initial introduction of a species is only one of four broadly defined steps characterizing biological invasions: (1) transport, (2) introduction, (3) establishment, and (4) post-establishment spread (Blackburn et al. 2011).
Post-establishment spread, or range expansion, is expected to lead to further decreases in genetic diversity in the invaded region with increasing distance from the point of invasion due to strong genetic drift caused by repeated founder events through space and time (Excoffier et al. 2009). Genetic drift during range expansions can be strong enough to fix otherwise rare alleles near the leading edge of the expansion in a process known as allele surfing (Edmonds et al. 2004; Travis et al. 2007; Hallatschek and Nelson 2008). However, the genetics of range expansion during invasion can be complicated by a variety of demographic phenomena, including multiple introductions, persistent human-mediated transport within the invaded range, or both. Marine examples of this include the invasions of the European green crab Carcinus maenas (Linnaeus, 1758) along the U.S. Atlantic coast (Darling et al. 2008, 2014), the Asian violet tunicate Botrylloides violaceus Oka, 1927 along the U.S. Pacific coast (Bock et al. 2010), and the north Pacific bryozoan Tricellaria inopinata d’Hondt and Occhipinti Ambrogi, 1985 along the U.S. Atlantic coast (Johnson and Woollacott 2015). These complications imposed on an otherwise seemingly clear narrative of post-establishment spread often limit our ability to discern and predict the population genetic patterns of an introduced species.
A recent invader along the U.S. northern Atlantic coast is the caridean shrimp Palaemon macrodactylus Rathbun, 1902, native to China, Japan, and Korea (Ashelby et al. 2013). The species has invaded regions across the globe since the mid-twentieth century, including San Francisco Bay (year of first collection 1957) and other parts of the U.S. Pacific coast, Western Europe (1992), Argentina (2000), and most recently the U.S. Atlantic coast (Ashelby et al. 2013). P. macrodactylus, known as the Asian prawn or Asian shrimp, was first discovered on the Atlantic coast in 2001 in the Bronx River near New York City, incidental to ichthyofaunal surveys (Warkentine and Rachlin 2010). P. macrodactylus is one of at least five palaemonid shrimp—both native and non-native—now in the northeast region of the United States. It was not until 2010, when the discovery of P. macrodactylus was announced, that this shrimp species was included in non-native species surveys elsewhere along the Atlantic coastline, by which time P. macrodactylus was found throughout southern New England in Long Island Sound and Narragansett Bay. In 2012, it was found north of Cape Cod as far as Boston (JTC, unpublished data). P. macrodactylus was also discovered as far south as the Chesapeake Bay in 2007 (Fofonoff et al. 2018). New England and adjacent coastal surveys that were specifically designed to assess the distribution of palaemonid shrimp in the summer of 2014 documented populations of P. macrodactylus from central New Jersey north to Newington, New Hampshire, the northernmost documented location to date (JTC, unpublished data).
Since the discovery of the Western Atlantic P. macrodactylus invasion, only one population genetic study of the species has been published (Lejeusne et al. 2014). This global survey of introduced P. macrodactylus populations used mitochondrial COI data and concluded that the invader had high genetic diversity in all invaded regions globally, indicating limited or no founder events during each invasion. However, their study includes individuals from only one of the many invaded estuaries on the U.S. Atlantic coast, leaving unanswered questions regarding population structuring in this invaded region. Other Palaemon species have shown unexpected population genetic structure within specific regions (e.g., Palaemon elegans Rathke, 1837 in Europe; Reuschel et al. 2010), while others follow expectations set by oceanographic current patterns (e.g., Palaemon floridanus Chace, 1942, in the Caribbean Sea: Baeza and Fuentes 2013).
In the absence of systematic surveys of palaemonid shrimp along the U.S. Atlantic coast in the decades surrounding the appearance of P. macrodactylus in Europe in 1992 (and thus its ready availability to be potentially transported by ships to North America) and its discovery in 2001 in New York, the precise location and timing of introduction in North America is not known. At least two possible colonization histories are possible. The first possible mechanism of spread that was investigated was an expansion scenario, which posits that P. macrodactylus was introduced to New York City (sometime prior to 2001), spread south to Chesapeake Bay (2007) and north to eastern Long Island Sound (2010), and then to Boston (2012) and New Hampshire (2014). This potential pathway of spread is based only on the first collection date of P. macrodactylus at a site, a date not anchored by documented knowledge of the previous absence at each site. This expansion pathway generates explicit genetic expectations, as noted above, for the invaded region based on the assumption that this spread represents repeated founder events through space and time leading to decreased diversity along the expansion axis. A second possible mechanisms of spread is one in which multiple introductions were responsible, at least in part, for the appearance of P. macrodactylus in ports and bays such as Boston, New York, and Chesapeake Bay. Multiple separate introductions would generate a different set of genetic expectations, including potentially distinctive, localized genetic structuring and peaks of genetic diversity.
This study seeks to examine which of the above two potential mechanisms of spread may best explain the nature of the establishment and spread of P. macrodactylus from New York northwards on the Atlantic coast of the United States. In testing these two possible histories, we describe the distribution of genetic diversity in the invaded area between New York and New Hampshire in the context of range expansion expectations. The two potential mechanisms of spread investigated here are not the only possible invasion histories, and this paper does not intend to systematically test the possibilities with rigorous statistical analyses developed to test specific models (e.g., the approximate Bayesian computation method presented in Estoup et al. 2012; Fraimout et al. 2017). While specific statistical tests are possible for the evaluation of some invasion histories, an alternative approach is to use expected genetic outcomes to infer histories (reviewed in Cristescu 2015). Rather, we use genetic expectations of range expansions to determine if P. macrodactylus populations north of New York City likely underwent that process. We implement population genomic techniques to examine potential patterns of local isolation and connectivity in the invaded range. We use both mitochondrial cytochrome oxidase I sequence data and data from 1598 single-nucleotide polymorphisms (SNPs) generated from restriction-enzyme-associated DNA sequencing (RAD-seq). This is the first use of genome-wide SNP markers in an introduced Palaemon species.
Methods
Sample collection
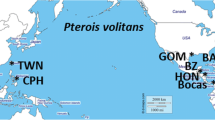
Palaemon macrodactylus samples were collected in 2014, during an intensive summer sampling expedition known as ShrimpEx14, from marina dock fouling communities in New York and New England (Fig. 1) with a hand-held fishing net (45.7 cm diameter ring, 1.0 cm mesh). Sites to the south of Cape Cod included Evers Marina on the Bronx River, New York, New York; Mystic Seaport in Mystic, Connecticut; and Moby Dick Marina in Fairhaven, Massachusetts. Sites to the north of Cape Cod included Brewer Marina in Plymouth, Massachusetts; University of Massachusetts Boston, in Boston, Massachusetts; and Great Bay Marine in Newington, New Hampshire (Table 1). Sites with more established fouling community habitats were often the locations with the greatest abundance of shrimp and were, therefore, targeted for collection. Shrimp distributions in marinas were found to be patchy, leading collection teams involved in the ShrimpEx14 surveys to sample large areas of dock surfaces to acquire the required sample numbers. Shrimp were sorted by gross morphology and color at the sampling locations and those likely to be P. macrodactylus were preserved in 100% ethanol at the sampling site and stored on ice until they were drained of ethanol and stored in a − 20 °C freezer.

Figure generated by the Woods Hole Oceanographic Institution Graphics Department, credit: Jack Cook
Map of the study area with study sites indicated by white circles. Mitochondrial haplotype data are also included for each site. The proportion of sampled individuals with each mitochondrial COI haplotype (Pm18, Pm3, Pm67, Pm1, Pm27, and PmU86) is shown for each population in the color-coded pie charts. NH New Hampshire, MA Massachusetts, CT Connecticut, NY New York. Up is North in this figure.
Palaemon macrodactylus were identified under a dissecting microscope using the defining morphological characteristics of a double row of setae on the ventral side of the rostrum and typically three rostral teeth behind the posterior margin of the orbital socket (González-Ortegón and Cuesta 2006). Length and reproductive status (i.e., gravid, not-gravid) were recorded for each specimen. Size distributions of shrimp included in population genetic analysis varied across sampling location ranging from 1.75 to 5.25 cm total length.
DNA extraction and polymerase chain reaction
Genomic DNA (gDNA) was extracted from a section of abdominal muscle tissue from each individual shrimp using the Omega Insect Extraction Kit (Omega Biotek, Norcross, GA, USA), with a standard protocol including the suggested liquid nitrogen homogenization step. Genomic DNA samples were stored in the kit’s elution buffer at 4°C or -20°C until polymerase chain reactions (PCRs) and RAD sequencing. PCRs to amplify cytochrome oxidase I (COI) were carried out using primers CrustCOIF (5′-TCAACAAATCAYAAAGAYATTGG-3′) and DecapCOIR (5′-AATTAAAATRTAWACTTCTGG-3′) (Lejeusne et al. 2014). The thermocycler temperature profile consisted of 95 °C denaturing step for 3 min, then 30 cycles of 95 °C for 45 s, 48 °C for 60 s, 72 °C for 60 s, followed by a final extension step at 72 °C for 5 min. PCR reactions were purified using a QIAGEN PCR Purification Kit (Qiagen GmbH, Germany) and were sequenced at Eurofins Operon Genomics (Eurofins MWG Operon LLC, Louisville, Kentucky, USA).
During initial optimization of gDNA extraction and amplification protocols, sequencing efforts sometimes produced double peaks (two equally strong sequencing results at one nucleotide position) in the chromatograms for mitochondrial COI (Figure ESM.A1.1, ESM Appendix 1). These double peaks were replicated across individual samples in both directions of sequencing reads and occurred reliably at specific nucleotide locations. In nuclear genes, such results would indicate heterozygosities, but for mitochondrial genes, these results are unexpected because only one copy of each mitochondrial gene is expected to be present, except in rare cases of bi-parental inheritance. We concluded that the consistent double peak results were evidence of a second copy of the gene, or a pseudogene (Williams and Knowlton 2001). In response to this result, we developed the protocol described above, which includes a different type of extraction technique (i.e., different from both the original phenol–chloroform extraction attempts and the Chelex extraction used in previous studies, e.g., Lejeusne et al. (2014)) and a reduced number of amplification cycles during PCR. This protocol eliminated the observed double peaks.
For future work undertaking P. macrodactylus extractions and PCR, materials can be found in the electronic appendices that detail the original methods (ESM Appendix 1).
Mitochondrial sequencing analysis
Mitochondrial DNA sequences were edited and assembled using Geneious 8.1.5 (Kearse et al. 2012). Consensus sequences were then aligned using the Geneious MAFFT alignment plug-in with default settings. The 85 haplotype sequences for P. macrodactylus published in Lejeusne et al. (2014) were downloaded from the NCBI nucleotide database (GenBank Accession Numbers HG792276.1 through HG792360.1, and G792313.1) and aligned to the sequences generated in this study. All sequences were trimmed to 501 base pairs to allow for the inclusion of more individuals (final sequence length was shorter than in Lejeusne et al. (2014), in which 598 base pairs were used). Sequence trimming did not lead to the exclusion of any haplotypes present in individuals sequenced for this study (a conclusion based on the location of the polymorphisms). However, because the sequences were trimmed, the haplotypes previously named Pm55, Pm56, Pm57, Pm58, Pm59, Pm83, and Pm84 (Lejeusne et al. 2014) had variable base pairs trimmed from the dataset. Every defining polymorphism of the sequences in this study fell within the trimmed region of the sequencing reads. Additionally, to test for potential effects of possible pseudogene sequencing (mentioned above) and accurately compare to previously published COI data (from Lejeusne et al. 2014), tests were run excluding nucleotides from analyses that were potentially problematic (e.g., indicating pseudogenes) based on this study’s initial methods analysis (ESM Appendix 1). While removal of possibly problematic bases where double peaks occurred resulted in a reduction in the number of haplotypes, from 85 haplotypes to 22, any alterations in haplotype calling did not substantially change the conclusions of the present study and all nucleotides are included in the following analyses.
Summary statistics including nucleotide diversity and pairwise differences for the mitochondrial data, as well as pairwise FST values were calculated using Arlequin v3.5.2.2. A haplotype network was constructed using these results combined with previously published data.
Restriction enzyme-associated DNA (RAD) sequencing
Genomic DNA samples were normalized to a concentration of 20 ng/μl as measured on a QUBIT 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Restriction enzyme-associated DNA sequencing library preparation using the SbfI restriction enzyme (restriction site: 5′-CCTGCAGG-3′) was carried out on concentration-normalized gDNA by Floragenex Inc. (Eugene, OR, USA) in identical fashion to several other recent RAD-seq studies (e.g., Reitzel et al. 2013; Herrera et al. 2015). For library preparation, gDNA was digested with the SbfI restriction enzyme, yielding fragments of various lengths. Barcode tags, ten base pairs in length and specific to individual, as well as an Illumina adaptor, were ligated onto the sticky end of the cut site. Samples were then pooled, sheared, and size selected for optimal Illumina sequencing. Libraries were then enriched through PCR and sequenced by 96-multiplex in a single lane of an Illumina Hi-Seq 2000 sequencer.
RAD data filtering, SNP calling, and population genomic analyses
Using the process_radtags program in Stacks v1.35 (Catchen et al. 2013), raw Illumina reads were filtered for quality with a minimum phred score of 10 in a sliding window of 15% read length (default settings) and sorted by individual barcode. Reads were truncated to 90 base pairs (bp) to remove barcodes and adaptors but leaving the 6 base pair restriction site intact. Putative loci were generated using the denovo_map.pl pipeline in stacks. We used a stack-depth parameter (-m) of 3, meaning that three reads were required to generate a stack (i.e., a locus); a within-individual distance parameter (-M) of 3, allowing for three SNP differences in a read; and a between-individual distance parameter (-n) of 3. The final size of the locus catalog varied as expected with different values for the denovo_map.pl parameters (Figure ESM.A2.2 in ESM Appendix 2).
Population summary statistics including allele frequencies, observed and expected heterozygosities (Hobs and Hexp), nucleotide diversity (π), and FIS, were calculated by the populations program in Stacks, using loci found in five of the six populations and in at least 60% of individuals per population using flags -p 5, -r 0.6. Due to the nature of the sequencing quality and coverage, the populations program was also run for all populations excluding New York, NY, with loci found in all five of the remaining populations, and 60% of individuals (argument flags -p 5, -r 0.6). The decision to exclude New York was based on the fact that the fewest loci were retained for that site; removing it resulted in less of a reduction in loci used when the new constraint that loci must be in all populations was introduced (the -p flag in Stacks populations). These data were used in the principal component analysis. Different -p and -r commands yielded different numbers of loci for analysis (Figure S6). For each RAD-tag, only one SNP was used from the 90 bp sequence using the flag –write_single_snp (specifying that if there were two or more SNPs in the sequence, Stacks would only use the first). Observed and expected heterozygosity (Hobs and Hexp) values were also calculated in the R Package PopGenKit (https://cran.r-project.org/web/packages/PopGenKit/index.html) to provide secondary validations of reported values. Allelic richness (Arich) was calculated in PopGenKit. Genetic diversity summary statistics (Hobs, Hexp, and Arich) were regressed against distance from the New York collection site using the stats package from Scipy (https://scipy.org). The least cost distance dispersal trajectories used in these regressions were calculated using the ‘gdistance’ package in R with a bathymetric constraint from ETOPO1 (van Etten 2015; R Core Team 2016).
Three methods were used to describe the genetic structure of P. macrodactylus populations. First, the smartpca program within EIGENSOFT (Price et al. 2006) was used to perform a principal component analysis (PCA) of genetic diversity. Custom scripts archived as iPython notebooks were used to convert Stacks PLINK output files into EIGENSOFT input files, and to visualize the PCA results (https://github.com/ekbors/p_macrodactylus). The smartpca program within EIGENSOFT was run with one iteration of outlier removal (‘numoutlieriter’ = 1) with otherwise default parameters. To evaluate the impact of missing data on population clustering in EIGENSOFT, the ‘missingmode’ argument was used in certain parameter runs. Missing data appeared to have a significant effect on the population clustering patterns (Figure SI.A4.1 in SI Appendix 4). Therefore, the results from the missing test runs required all further analyses to be run with populations output excluding the New York site and, therefore, analyze only loci in the remaining five populations. Second, fastSTRUCTURE (Pritchard et al. 2000; Hubisz et al. 2009; Raj et al. 2013) was run with the number of genetic lineages (the value of k) set to values between one and ten to assess genetic structure through a hierarchical analysis, and the program chooseK.py was run to select the value of k most consistent with the program’s Bayesian structure model. Third, FST values were calculated by the populations program in Stacks using a p value cutoff of 0.05 with a Bonferroni correction implemented by the program (using the –fst_correction ‘bonferroni_gen’ argument).
In addition to the described approaches of regressing genetic diversity measurements with distance from New York to capture potential range expansion signals in diversity summary statistics, a range expansion-specific analysis was also implemented based on asymmetries in allele frequency data which may indicate the directionality of expansion (Peter and Slatkin 2013). Using an R package developed by Peter and Slatkin (2013), we calculated psi, or the “directionality index” to quantify the relationships between allele frequencies and potential direction and strength of expansion.
Results
Geographic distribution of mitochondrial haplotypes
A 501 base pair region of cytochrome oxidase I (COI) was sequenced for a total of 106 individual Palaemon macrodactylus (19 from New York, New York; 22 from Mystic, Connecticut; 10 from Fairhaven, Massachusetts; 15 from Plymouth, Massachusetts; 19 from Boston, Massachusetts; 21 from Newington, New Hampshire) (Table 2). In the six populations of P. macrodactylus sampled in this study, six haplotypes were identified. For ease of comparison and consistency, the haplotype names used in previously published results (see Lejeusne et al. 2014) are used in this study.
Five of the six haplotypes were previously reported in Lejeusne et al. (2014) and one haplotype, here named PmU86, was new and unique to New York, NY (n = 1). This is in contrast to previous reports of 11 haplotypes in Mystic, Connecticut, alone, 7 of which were reported to be unique (Lejeusne et al. 2014). Results of the analysis test runs to investigate what the effect of pseudogene sequencing would have been on these results as well as on previously published work, is presented in SI Appendix 1. In short, tests to remove base pairs at locations where double peaks were observed reduced the number of haplotypes previously published from 85 to 22 and the number of loci in this study from 6 to 5.
In the present study, specific haplotypes were present only in one or two populations while others were present in all populations (Fig. 1). The most common haplotypes were Pm18 (n = 70) and Pm3 (n = 22). Pm18 and Pm3 are also the most common haplotypes globally (Lejeusne et al. 2014). Pm27, a haplotype previously only reported in Yamaguchi, Japan, in the native range, was located in Boston and Plymouth, Massachusetts. Pm1 was also only previously reported in the native range, but in this study, it was found in Fairhaven and Boston, Massachusetts. Pm67, a haplotype previously described as unique to the invaded population in the U.S. Northeast, was observed in New York, New York, but not in Mystic, Connecticut, as previously reported (a possible result of sampling effort or an unobserved fluctuation in population size and genetic composition). Individuals sampled from Fairhaven, Massachusetts, and Newington, New Hampshire, were all one of two main haplotypes, Pm18 and Pm3. The two major haplotypes had only one nucleotide difference between them, and all other haplotypes only differed by one nucleotide from either of the major haplotypes (Fig. 2). The one newly reported haplotype sequence in this study was uploaded to GenBank (accession number KY490702.1) and is referred to as PmU86 throughout this study. As noted above, all other sequenced haplotypes from this dataset were present in data previously reported by Lejeusne et al. (2014). Haplotype diversity peaked in New York and Boston while nucleotide diversity and pairwise differences were highest in Plymouth (Table 2).

Haplotype network for mtCOI sequences of Palaemon macrodactylus. The area of each circle reflects the relative proportion of individuals with each haplotype, with PmU86 and Pm67 representing one shrimp each. NH New Hampshire; MA Massachusetts; CT Connecticut; NY New York
There were two samples from Mystic, Connecticut, for which ambiguous bases were present across different sequencing attempts of amplified PCR products. The genomic location of this disagreement in the COI sequences was one for which there was no variability reported in previous studies or in other samples in this study. Therefore, for these two samples, we considered the most parsimonious result to be that the sequences that were consistent with all other samples were correct and that the sequences that would indicate a new polymorphism were, in fact, errors. However, the ambiguity is noteworthy, especially in light of the sequencing variation described above.
RAD-seq efficacy and identification of loci
Illumina sequencing of RAD libraries yielded 122,722,404 raw reads. After processing raw reads with the Stacks program process_radtags, 13.43% of reads were removed due to a missing or ambiguous barcode, 7.16% of reads were removed due to an ambiguous restriction site and 15.32% of reads were removed due to low sequencing quality, leaving 64.10% of reads retained for further processing (Figure ESM.A2.1 in ESM Appendix 2). The percent removed due to low sequencing quality can be considered high and was driven mostly by poor quality in a number of sequencing tiles in the middle of the sequencing reads.
The final catalog used in this study contained 1598 loci. The average depth of sequencing coverage per locus across all individuals was 21.43 with an average standard deviation of sequencing depth of 224.21. For the populations run including five of the six populations and 60% of individuals per population, the number of loci included per population varied greatly (from 969 for New York, New York to 1598 in Newington, New Hampshire), with New York having the fewest loci (Table 3, Table ESM.A2.1 in ESM Appendix 2). For the populations run in which New York was excluded and loci were required to be in all five of the remaining populations, 1092 loci were retained.
Population genomic diversity patterns throughout the invaded range
Genetic diversity statistics either remained consistent or increased with distance from New York (Fig. 3). Observed heterozygosity (Hobs) increased with distance from New York (Fig. 3a). Expected heterozygosity (Hexp) and allelic richness (Arich) had consistent values throughout the sampled region with the exception of small increases in Arich in Boston and Plymouth, MA (Fig. 3b, c). When analyses were run with the requirement that all loci be present in all populations (to eliminate the possibility that missing data were driving this trend for NYC), the patterns remained the same.

Observed heterozygosity, b expected heterozygosity, and c allelic richness plotted against distance between population locations. For a observed heterozygosity, R2 = 0.822 and the p value 0.013. The regressions for other summary statistics were not significant (NEW Newington, New Hampshire; BOS Boston, Massachusetts; PLY Plymouth, Massachusetts; FAI Fairhaven, Massachusetts; MYS Mystic, Connecticut; NYC New York, New York)
In the principal component analysis (PCA) for which New York was omitted and loci were required to be in all populations (to reduce the effects caused by missing data, of which the New York samples were significant contributors), sampling locations demonstrated some clustering (Fig. 4) with 11.6% of the variation described by the first eigenvector, 11% described by the second, and 10.4% the third (ten eigenvectors were calculated by EIGENSOFT indicating that the diversity was almost spread evenly across all ten). There was variation in the structure analysis across different populations runs: some of the data generated by populations led to structure analysis showing geographic structuring and some did not. The variation in these results may be driven by the issue of missing loci in the New York population, but in any case, such variation prevents any definitive conclusion from the fastSTRUCTURE results (Figure ESM.A5.1 and Figure ESM.A5.2 in ESM Appendix 5).

Omitting New York, principal component analysis of select sites made with EIGENSOFT for loci included in the remaining five populations and 60% of the individuals in each (NEW Newington, New Hampshire; BOS Boston, Massachusetts; PLY Plymouth, Massachusetts; FAI Fairhaven, Massachusetts; MYS Mystic, Connecticut). The first eigenvector accounted for 11.6% of the variation, the second eigenvector accounted for 11% of the variation, and the third eigenvector accounted for 10.4% of the variation
Despite the apparent population structuring detected by EIGENSOFT and fastSTRUCTURE, FST values calculated with a Bonferroni correction were either zero, not statistically different than zero, or extremely small. This could indicate that structuring is based more on regional variation in the distribution of diversity, rather than driven by isolation and inbreeding, as FST often measures. The pairwise FST values that were different from zero were Fairhaven-to-Mystic, 7.7 × 10−4; Newington-to-Mystic, 4.6 ×10−4; Newington-to-Plymouth, 1.4 ×10−3; Newington-to-Boston, 4.8 ×10−4.
The values of psi, or the directionality index ranged from − 0.0518 to 0.0375 on one axis. The directionality index indicates that two locations were almost equally likely to be near the “origin” of expansion: New York and Boston (Fig. 5). This suggests that both could be origins of expansion and that other locations may be receiving individuals from those areas.

A heatmap of psi values for all six populations of Palaemon macrodactylus (NEW Newington, NH; BOS Boston, MA; PLY Plymouth, Massachusetts; FAI Fairhaven, Massachusetts; MYS Mystic, Connecticut; NYC New York, New York). Psi values indicate the “directionality index,” a statistic intended to capture asymmetries in allele frequencies indicating directional expansion. As is seen in the heatmap, Boston, Newington, and New York have values at or close to near zero, with Boston’s value slightly positive when others are negative. Note that the values are symmetrical above and below the diagonal axis, except for sign
Discussion
Potential support for multiple introductions rather than a spatial expansion
Results in this study do not support an invasion history in which populations of Palaemon macrodactylus underwent a sequential northward range expansion along the Atlantic coast from New York City, the first-reported location for the species in the United States in 2001. Instead, results suggest evidence for a potential alternative invasion pathway in which multiple introductions account for the spatial spread of P. macrodactylus in the estuarine waters to the north of New York City, with a second introduction possible in the Boston area. This result has potentially broad implications for other cases of post-establishment spread in introduced species and observed range expansions in native species because it is possible that other observed range expansions are the result of human-mediated dispersal.
Both mitochondrial data and RAD-seq data from populations in the invaded range were used to reject the hypothesis that P. macrodactylus underwent a standard range expansion. Our conclusions are, in part, based on the existence of two mitochondrial haplotypes previously only described in the native range in Asia (Lejeusne et al. 2014) in Boston, Plymouth, and Fairhaven, Massachusetts. The peaks in nucleotide diversity in the COI data in Boston–Plymouth and New York also add to the evidence suggesting multiple introductions in those two areas. Further support for a potential pathway of spread in which there is at least one additional introduction in the north comes from the documented increase in observed heterozygosity across the 1598 loci with increasing distance from New York. This trend is contrary to the expectation during a range expansion, in which diversity would be expected to decrease with increasing distance from the point of introduction. These results may demonstrate how the expectations of range expansion can be overwhelmed by the specific context of the invasion—in this case, what appears to be spatial range expansion may have actually been driven by multiple introductions, changing the distribution of genetic diversity in introduced populations. Additionally, the calculations of the directionality index, or psi, identified Boston/Newington and New York as potential locations of the origin of expansion, pointing to the possible existence of two locations of distinct introductions before spread.
Analysis of the subset of 1092 loci generated with RAD-seq and used for PCA (excluding the samples from New York), revealed slight population genetic structuring by location in the invaded range. FST values for the RAD-seq dataset, however, were extremely low and/or statistically indistinguishable from zero. F statistics are used to quantify the level of inbreeding that results from isolation of subpopulations in a structured metapopulation (Hartl and Clark 2007). From these measurements, population structuring is inferred. In cases of range expansion—a clear violation of Hardy–Weinberg equilibrium—one must consider that this framework for evaluating structure may not be appropriate. While the population structure detected using PCA may be driven by isolation of the individual estuaries in which shrimp were collected, it could also be driven by differential introduction patterns across the invaded range. Specifically, shrimp of different genetic identity might exist in Boston and Plymouth, than those in Newington and Mystic because invaders of that genotype may have been introduced more recently in some places and not yet introduced into others. This may not necessarily mean that Newington and Mystic are isolated populations. Rather, it may indicate that the population genetic structure is influenced by the mechanism of the invasions in a subtler manner. Similarly, Mystic and Newington may not be genetically connected oceanographically, but instead, may be experiencing similar effects of post-introduction expansion patterns. Perhaps the two sites were both colonized by the first introduction and underwent bottlenecks or perhaps Newington was colonized from Boston’s invasion and Mystic was colonized from New York’s, and thus they both have decreased diversity and, therefore, cluster together in measures of structure. With time, population homogenization may actually weaken the structural signal, or isolation may strengthen it. At this point in the invasion, however, patterns of structure may be more useful in intuiting the invasion pathway than in inferring mixing or isolation.
Population genetic structure could also be driven by intracoastal human-mediated dispersal after introduction. Recreational boats, commercial fishing vessels, ferries, cargo ships, and touring yachts could all participate in the reshuffling of New England shrimp populations. Many such vessels could transport shrimp in ballast water along the coast, or, for example, in water of tires or other structures used as hull fenders (Lejeusne et al. 2014). This could explain, for example, why some of the mitochondrial haplotypes in Boston were found in Fairhaven and not Plymouth while others were found in Plymouth and not Fairhaven (the coastal order from south to north of these sites is Fairhaven–Plymouth–Boston). In many invasions, multiple vectors are responsible for dispersal (Richardson et al. 2016). Intracoastal dispersal by humans has been shown to be important in other marine invasions, including that of the tunicate Styela clava Herdman, 1881 in the northeastern Pacific Ocean (Darling et al. 2012). Patterns of haplotype distribution could also be due to insufficient sampling of diversity driven by sampling numbers, or sampling of segments of the population that are distributed in patches. For example, if shrimp from one sampling site was only collected from one patch within a marina, but those from another sampling site was collected from five different patches within a marina, then the second set of shrimp might be expected to be more diverse depending on the nature of the patchiness.
While data presented in this study help to understand a possible invasion history of the northward invasion of P. macrodactylus, questions still remain about the rest of the invaded range on the U.S. Atlantic coast. Evaluating the population structure to the south of New York would lend further insight into the invasion dynamics of P. macrodactylus and would present another invasion axis on which to test range expansion expectations.
Oceanographic and life history contexts of the Palaemon macrodactylus invasion
Oceanic currents are major drivers of larval dispersal in the oceans, with current patterns sometimes dictating population genetic structure patterns (White et al. 2010). For example, the coastal polychaete worm Clymenella torquata (Leidy, 1855), has a discontinuity in population genetic structure to the south of Cape Cod—a location hypothesized to be a phylogeographic boundary—that was attributed to converging water masses and not by the physical barrier of the Cape itself (Jennings et al. 2008). However, physical oceanographic studies have revealed that the coastal morphology and current flow patterns in the region are such that the estuaries of Long Island Sound, Boston Harbor, and Cape Cod Bay are tidally forced. Wind, river inputs, and tides drive patterns in Long Island Sound (Whitney et al. 2016), and some more persistent but still variable conditions prevail in Massachusetts waters north of Cape Cod (Jiang et al. 2007). In Cape Cod Bay, seasonal variation has been shown to change the retention patterns of planktonic crustaceans, such as the copepod Calanus finmarchicus (Gunnerus, 1770) (Jiang et al. 2007). Considering these daily and seasonal fluctuations in current patterns, we expect that instead of persistent currents, tidal forcing may play a more important role in the dispersal of larval shrimp in and out of estuaries, making predicting the patterns difficult because of the stochasticity of such processes.
Reproductive strategies, dispersal capabilities, and other life history traits play a crucial role in determining population genetic connectivity and geographic spread in many marine species (Selkoe et al. 2016). In its native range, P. macrodactylus reproduces seasonally from April to October, with two cohorts produced per season throughout their typical 2-year lifespan (Omori and Chida 1988). The shrimps are often found in brackish water in coastal estuaries but there is some evidence that developing larvae and gravid females may migrate to higher salinity waters (Vázquez et al. 2015). In the invaded region of France, for example, evidence suggests that different life stages are spatially segregated within estuaries suggesting that migration occurs (Béguer et al. 2011). Further evidence of offshore mixing of larvae has been reported in the western Mediterranean Sea (Torres et al. 2012). These processes could lead to high levels of mixing in P. macrodactylus.
Coastal environments with high levels of oceanographic mixing and species with longer distance larval dispersal often drive populations towards lower levels of population structure and, notably, towards genetic patterns that do not correlate with Euclidian distance (Cowen et al. 2007; White et al. 2010). However, the P. macrodactylus populations exhibit slight genetic structure, so despite the potential combination of human-mediated dispersal and population mixing, we acknowledge other processes may be responsible for the observed patterns, or that simply not enough time has passed for sufficient mixing to diminish the signals of multiple introductions.
Consistency with other invasions on the U.S. Atlantic coast
Multiple introductions are common in marine invasions (Rius et al. 2015). One of the most well-known non-native crustaceans along the U.S. Atlantic coast, the European green crab Carcinus maenas, has been introduced multiple times (Roman 2006; Darling et al. 2008, 2014). Green crabs were discovered on the Atlantic coast of the United States in 1817, likely through transport in ship fouling assemblages or in solid ballast of cargo or transport vessels (Carlton and Cohen 2003; Roman 2006). C. maenas expanded its range through the Gulf of Maine and its distribution was stable for about a century. In the 1980s, C. maenas was recorded in northern Nova Scotia. The invaders in the northern part of Nova Scotia were not, however, individuals expanding their introduced range from further south (as might be suspected due to climate change) but proved to be new arrivals from Europe (Roman 2006; Darling et al. 2008, 2014). Without genetic data, this second introduction of more diverse European green crabs would not have been distinguishable from a northward expansion—similar to what is reported in this study.
In recent years, the Asian shore crab, Hemigrapsus sanguineus (De Haan, 1835), another introduced crustacean, surpassed C. maenas as the most abundant intertidal crab on the U.S. northern Atlantic coast (Lord and Williams 2016). Introduced in ballast water, H. sanguineus was first collected in 1988 in Cape May County, Delaware, and now occupies a range from South Carolina to Maine (Epifanio 2013). Three H. sanguineus publications discuss invasion population genetics and dynamics: one in the native range (Yoon et al. 2011), one in the invaded region of the U.S. Atlantic coast (Lord and Williams 2016), and another focused on global biogeographic patterns (Blakeslee et al. 2017). These studies revealed population growth as well as expansion. Contrary to the expectations for an introduction followed by a range expansion, COI data for H. sanguineus did not show any specific clines or discontinuity in the population genetics of the invaded range. Although there were other haplotypes present, the populations along the northeast were, like those of P. macrodactylus, dominated by one haplotype.
Another example of an introduced species following an unexpected expansion route in the northern U.S. Atlantic coast is the Asian violet tunicate, Botrylloides violaceus. The violet tunicate is introduced on both coasts of the U.S., having first invaded the U.S. Pacific coast in the mid-twentieth century and the U.S. Atlantic coast after that, likely in the late 1970s (Bock et al. 2010). On the west coast, genetic data point towards punctuated, spatially discontinuous dispersal, likely human-mediated, or potentially to multiple introductions. On the east coast, however, the tunicate followed an isolation-by-distance invasion pattern with a gradient of genetic diversity in microsatellite data from south to north (Bock et al. 2010). This juxtaposition of the same species invading two different coasts with two apparently different expansion patterns—one driven by human-mediated dispersal and one driven by oceanography and life history—highlights how each invasion can be different depending on dispersal vectors and context.
Changing assumptions about temporal genetic structure in introduced species
When a species is newly introduced—like the shrimp P. macrodactylus—the ways in which introduction mechanisms distribute genetic diversity may be most important for genetic structuring. The prevalence of multiple introductions and their potential ability to alter the evolutionary trajectory of an introduced species by dramatically changing the nature and distribution of genetic diversity highlights the need to amend the conception of classical marine population genetics as being driven primarily by physical oceanographic drivers and life-history traits. A new framework should include the dispersal by humans as a driver of population structure, the more so because anthropogenic dispersal of marine species has been in play for centuries if not millennia (Carlton 2009). This is obvious in the case of a secondary invasion but can also occur within a species range. For example, inside what is considered to be the native range of Palaemon elegans—including the Mediterranean, Black, and Baltic Seas—population structure and a recent apparent expansion may be driven by human-mediated dispersal and not oceanographic or biological factors (Reuschel et al. 2010). Additionally, at different times in the history of a metapopulation, different processes are likely to be more important to the shaping of genetic diversity than others.
In the case of C. maenas as well as P. macrodactylus, comparing molecular data from multiple time points is crucial for detecting multiple invasions. To test the hypotheses generated by our understanding of dynamic population processes, repeated genetic assessment of introduced populations (along with knowledge of the quality of baseline survey data) improves our understanding of how an observed pattern that might appear, at a coarse-grained level, to be standard range expansion, may actually be a second introduction (as was observed in P. macrodactylus). The previously published data used in comparison to results of this study are based on samples collected at only one site, making definite inferences of temporal processes difficult. Broader coverage of invaded areas may facilitate more concrete conclusions.
In the case of P. macrodactylus in the northern part of the U.S. Atlantic coast, artifacts of reporting and searching for otherwise largely unstudied crustaceans inadvertently led to an assumption of sequential spread along the coastline. For these reasons, genetic monitoring, or at least periodic genetic studies throughout an invasion will prove highly valuable. While the data presented here suggest that a second introduction is possible, and the previous information regarding haplotype distributions corroborates this explanation of the data, it is still not possible to say exactly when this introduction took place. It is also not possible to determine how long the current population structure will persist as the invasion progresses or if P. macrodactylus will appear to expand northward and southward, or be subjected to yet more new introductions from overseas in the future. Future study of introduced populations to the south of New York City would also help in discerning these dynamics into the future. Comparisons of this system with other regions where P. macrodactylus has invaded will improve our understanding of the spread of this global invader in each invaded region. Additionally, comparisons of RAD-seq data from this invaded region with the native range would improve our understanding of possible sources of secondary introductions. Future work could yield investigations like those Blakeslee et al. (2017), in which the origins of a secondary introduction are traced genetically.
Conclusions
This study represents the first use of genome-wide population genomic markers generated by RAD-sequencing in an introduced Palaemon shrimp. Similar to other introduced species, P. macrodactylus has likely been introduced multiple times in the invaded range along the U.S. northern Atlantic coast, as is reflected in patterns of genetic diversity. Data presented here indicate that invasion context is crucial to making predictions about genetic diversity. In addition to oceanographic dispersal mechanisms and life histories, human-mediated dispersal may play an important role in shaping the diversity of marine species. Furthermore, the age of metapopulations may partially determine which force plays the most important role in driving population structure. At the current time, our understanding of the population dynamics of marine invasions is not generalizable across species. Through more intense study, continued research through time, and comparisons among multiple introduced species, our understanding of marine invasions will improve.
References
Ashelby CW, Johnson ML, De Grave S (2013) The global invader Palaemon macrodactylus (Decapoda, Palaemonidae): an interrogation of records and a synthesis of data. Crustaceana 86:594–624
Baeza JA, Fuentes MS (2013) Phylogeography of the shrimp Palaemon floridanus (Crustacea: Caridea: Palaemonidae): a partial test of meta-population genetic structure in the wider Caribbean. Mar Ecol 34:381–393
Barrett SCH (2015) Foundations of invasion genetics: the Baker and Stebbins legacy. Mol Ecol 24:1927–1941
Bax N, Williamson A, Aguero M, Gonzalez E, Geeves W (2003) Marine invasive alien species: a threat to global biodiversity. Mar Policy 27:313–323
Béguer M, Bergé J, Martin J, Martinet J, Pauliac G, Girardin M, Philippe Boët P (2011) Presence of Palaemon macrodactylus in a European estuary: evidence for a successful invasion of the Gironde (SW France). Aquat Invasions 6:301–318
Blackburn TM, Pyšek P, Bacher S, Carlton JT, Duncan RP, Jarošík V, Wilson JRU, Richardson DM (2011) A proposed unified framework for biological invasions. Trends Ecol Evol 26:333–339
Blakeslee AMH, Kamakukra Y, Onufrey J, Makino W, Urabe J, Park S, Keogh CL, Miller AW, Minton MS, Carlton JT, Miura O (2017) Reconstructing the invasion history of the Asian shorecrab, Hemigrapsus sanguineus (De Haan 1835) in the Western Atlantic. Mar Biol 164:47
Bock DG, Zhan A, Lejeusne C, MacIsaac HJ, Cristescu ME (2010) Looking at both sides of the invasion: patterns of colonization in the violet tunicate Botrylloides violaceus. Mol Ecol 20:503–516
Carlton JT (2009) Deep invasion ecology and the assembly of communities in historical time. In: Rilov G, Crooks JA (eds) Biological invasions in marine ecosystems. Springer, Berlin, pp 13–56
Carlton JT, Cohen AN (2003) Episodic global dispersal in shallow water marine organisms: the case history of the European shore crabs Carcinus maenas and C. aestuarii. J Biogeogr 30:1809–1820
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Cowen RK, Gawarkiewic G, Pineda J, Thorrold SR, Werner FE (2007) Population connectivity in marine systems an overview. Oceanography 20:14–21
Cristescu ME (2015) Genetic reconstructions of invasion history. Mol Ecol 24:2212–2225
Darling JA, Bagley MJ, Roman J, Tepolt CK, Geller JB (2008) Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol Ecol 17:4992–5007
Darling JA, Herborg L-M, Davidson IC (2012) Intracoastal shipping drives patterns of regional population expansion by an invasive marine invertebrate. Ecol Evol 2:2557–2566
Darling JA, Tsai YHE, Blakeslee AMH, Roman J (2014) Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab Carcinus maenas. R Soc Open Sci 1:140202
Edmonds CA, Lillie AS, Cavalli-Sforza LL (2004) Mutations arising in the wave front of an expanding population. Proc Natl Acad Sci USA 101:975–979
Epifanio CE (2013) Invasion biology of the Asian shore crab Hemigrapsus sanguineus: a review. J Exp Mar Biol Ecol 441:33–49
Estoup A, Lombaert E, Marin JM, Guillemaud T, Pudlo P, Robert CP, Cornuet JM (2012) Estimation of demo-genetic model probabilities with Approximate Bayesian Computation using linear discriminant analysis on summary statistics. Mol Ecol Resour 12:846–855
Estoup A, Ravigné V, Hufbauer R, Vitalis R, Gautier M, Facon B (2016) Is there a genetic paradox of biological invasion? Annu Rev Ecol Evol Styst 47:51–72
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Fofonoff PW, Ruiz GM, Steves B, Simkanin C, Carlton JT. (2018) Palaemon macrodactylus, in California. National Exotic Marine and Estuarine Species Information System. http://invasions.si.edu/nemesis/. Accessed 30 Mar 2018
Fraimout A, Debat V, Fellous S, Hufbauer RA, Foucaud J, Pudlo P, Marin JM, Price DK, Cattel J, Chen X, Depra M, Francois Duyck P, Guedot C, Kenis M, Kimura MT, Loeb G, Loiseau A, Martinez-Sanudo I, Pascual M, Polihronakis Richmond M, Shearer P, Singh N, Tamura K, Xuereb A, Zhang J, Estoup A (2017) Deciphering the Routes of invasion of Drosophila suzukii by Means of ABC Random Forest. Mol Biol Evol 34:980–996
Funk JL, Matzek V, Bernhardt M, Johnson D (2014) Broadening the case for invasive species management to include impacts on ecosystem services. Bioscience 64:58–63
González-Ortegón E, Cuesta JA (2006) An illustrated key to species of Palaemon and Palaemonetes (Crustacea: Decapoda: Caridea) from European waters, including the alien species Palaemon macrodactylus. J Mar Biol Assoc UK 86:93–102
Hallatschek O, Nelson DR (2008) Gene surfing in expanding populations. Theor Popul Biol 73:158–170
Hartl DL, Clark AG (2007) Principles of population genetics. Sinauer Associates Incorporated, Sunderland
Herrera S, Watanabe H, Shank TM (2015) Evolutionary and biogeographical patterns of barnacles from deep-sea hydrothermal vents. Mol Ecol 24:673–689
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Jennings RM, Shank TM, Mullineaux LS, Halanych KM (2008) Assessment of the Cape Cod phylogeographic break using the bamboo worm Clymenella torquata reveals the role of regional water masses in dispersal. J Hered 100:86–96
Jiang M, Brown MW, Turner JT, Kenney RD, Mayo CA, Zhang Z, Zhou M (2007) Springtime transport and retention of Calanus finmarchicus in Massachusetts and Cape Cod Bays, USA, and implications for right whale foraging. Mar Ecol Prog Ser 349:183–197
Johnson C, Woollacott R (2015) Analyses with newly developed microsatellite markers elucidate the spread dynamics of Tricellaria inopinata d’Hondt and Occhipinti-Ambrogi, 1985—a recently established bryozoan along the New England seashore. Aquat Invasions 10:135–145
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:386–391
Lejeusne C, Saunier A, Petit N, Béguer M, Otani M, Carlton JT, Rico C, Green AJ (2014) High genetic diversity and absence of founder effects in a worldwide aquatic invader. Sci Rep 4:1–9. https://doi.org/10.1038/srep05808
Lord JP, Williams LM (2016) Increase in density of genetically diverse invasive Asian shore crab (Hemigrapsus sanguineus) populations in the Gulf of Maine. Biol Invasions 19(4):1153–1168. https://doi.org/10.1007/s10530-016-1304-1
Lowry E, Rollinson EJ, Laybourn AJ, Scott TE, Aiello-Lammens ME, Gray SM, Mickley J, Gurevitch J (2013) Biological invasions: a field synopsis, systematic review, and database of the literature. Ecol Evol 3:182–196
Omori M, Chida Y (1988) Life history of a caridean shrimp Palaemon macrodactylus with special reference to the difference in reproductive features among ages. Nippon Suisan Gakkaishi 54:365–375
Peter BM, Slatkin M (2013) Detecting range expansions from genetic data. Evolution 67:3274–3289
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raj A, Stephens M, Pritchard JK (2013) fastSTRUCTURE: variational inference of population structure in large SNP datasets. Genetics 197:573–589
Reitzel AM, Herrera S, Layden MJ, Martindale MQ, Shank TM (2013) Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol Ecol 22:2953–2970
Reuschel S, Cuesta JA, Schubart CD (2010) Marine biogeographic boundaries and human introduction along the European coast revealed by phylogeography of the prawn Palaemon elegans. Mol Phylogenet Evol 55:765–775
Richardson MF, Sherman CDH, Lee RS, Bott NJ, Hirst AJ (2016) Multiple dispersal vectors drive range expansion in an invasive marine species. Mol Ecol 25:5001–5014
Rius M, Turon X, Bernardi G, Volckaert FAM, Viard F (2015) Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol Invasions 17:869–885
Roman J (2006) Diluting the founder effect: cryptic invasions expand a marine invader’s range. Proc R Soc Lond Biol Sci 273:2453–2459
Selkoe KA, D’Aloia CC, Crandall ED, Iacchei M, Liggins L, Puritz JB, von der Heyden S, Toonen RJ (2016) A decade of seascape genetics: contributions to basic and applied marine connectivity. Mar Ecol Prog Ser 554:1–19
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. Accessed 28 June 2016
Thomaz SM, Kovalenko KE, Havel JE, Kats LB (2014) Aquatic invasive species: general trends in the literature and introduction to the special issue. Hydrobiologia 746:1–12
Torres AP, Dos Santos A, Cuesta JA, Carbonell A, Massuti E, Alemany F, Reglero P (2012) First record of Palaemon macrodactylus Rathbun, 1902 (Decapoda, Palaemonidae) in the western Mediterranean. Mediterr Mar Sci 13:278–282
Travis JM, Munkemuller T, Burton OJ, Best A, Dytham C, Johst K (2007) Deleterious mutations can surf to high densities on the wave front of an expanding population. Mol Biol Evol 24:2334–2343
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proc Natl Acad Sci USA 97:5948–5953
van Etten J (2015) gdistance: Distances and Routes on Geographical Grids. R package version 1.1-9. https://CRAN.R-project.org/package=gdistance. Accessed 28 June 2016
Vázquez MG, Bas CC, Kittlein M, Spivak ED (2015) Effects of temperature and salinity on larval survival and development in the invasive shrimp Palaemon macrodactylus (Caridea: Palaemonidae) along the reproductive season. J Sea Res 99:56–60
Walsh JR, Carpenter SR, Vander Zanden MJ (2016) Invasive species triggers a massive loss of ecosystem services through a trophic cascade. Proc Natl Acad Sci USA 113:4081–4085
Warkentine BE, Rachlin JW (2010) The first record of Palaemon macrodactylus (Oriental Shrimp) from the eastern coast of North America. Northeast Nat 17:91–102
White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ (2010) Ocean currents help explain population genetic structure. Proc R Soc Lond Biol 277:1685–1694
Whitney MM, Ullman DS, Codiga DL (2016) Subtidal exchange in eastern Long Island Sound. J Phys Oceanogr 46:2351–2371
Williams ST, Knowlton N (2001) Mitochondrial pseudogenes are pervasive and often insidious in the snapping shrimp genus Alpheus. Mol Biol Evol 18:1484–1493
Yoon M, Hong S-E, Kwon Nam Y, Kim DS (2011) Genetic diversity of the Asian shore crab, Hemigrapsus sanguineus, in Korea and Japan inferred from mitochondrial cytochrome coxidase subunit I gene. Anim Cells and Syst 15:243–249
Acknowledgements
We thank the exuberant field assistance provided by the many members of the “ShrimpEX2014” research team, based out of the Williams College-Mystic Seaport Maritime Studies Program in Mystic, Connecticut. Support was provided for the ShrimpEX2014 research program by the Massachusetts Office of Coastal Zone Management and the Northeast Sea Grant Consortium. This research is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. Funding for the RAD-sequencing was provided in part by the Woods Hole Sea Grant Program, and the Woods Hole Oceanographic Institution Biology Department. Additional support was provided by the National Science Foundation (OCE-1131620 to TMS) and the James Education Fund for Ocean Exploration within the Ocean Exploration Institute of the Woods Hole Oceanographic Institution. We would like to thank the editors and reviewers for Marine Biology who contributed insight and perspective to this manuscript.
Author information
Authors and Affiliations
Contributions
Eleanor K. Bors (EKB) conceived of the project with substantial input from James T. Carlton (JTC) and Timothy M. Shank (TMS). JTC and EKB planned and undertook sample collection. EKB undertook the laboratory work and data analysis for the genetic and genomic data. EKB, JTC, and TMS discussed interpretation of the data. JTC and TMS both provided support for the project from assistance in the field to laboratory materials.
Corresponding authors
Ethics declarations
Conflict of interest
All funding is listed in the acknowledgements section. The authors declare that they have no conflict of interest.
Ethical guidelines
All guidelines for the sampling of organisms were followed. Collecting permits were obtained for New Hampshire, Massachusetts, Rhode Island, Connecticut, and New York, copies of which are available upon request.
Data availability statement
Data have been made available online on GenBank (BioProject ID PRJNA521400). As noted in the text, some accession numbers are already available online for mitochondrial data. Scripts used in analysis are available on the author’s GitHub page accessible via the following link: https://github.com/ekbors/p_macrodactylus-.
Additional information
Responsible Editor: M. Rius.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewed by undisclosed experts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bors, E.K., Carlton, J.T. & Shank, T.M. Multiple spatially distinct introductions and not range expansion may explain colonization history in a non-native marine shrimp. Mar Biol 166, 67 (2019). https://doi.org/10.1007/s00227-019-3514-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-019-3514-4