
Abstract
Numerous marine species have been introduced via anthropogenic vectors; however, many species, including highly abundant ones, have uncertain invasion histories. Here, we include the first large-scale population genetics study of the Asian shorecrab (Hemigrapsus sanguineus) in native (east Asia) and non-native (east USA) regions to help resolve questions of source, vector, and multiple introductions. Using a barcoding gene and six microsatellite loci, we analyzed 731 cytochrome oxidase I sequences and 500 microsatellites across 51 populations over two time periods, 2001–2002 and 2013–2014. Explorations of both genetic and historical data indicated that multiple introductions to USA are probable with the greatest genetic diversity concentrating around Long Island (near New York City). Analyses of ballast water flux and genetic data suggest that introductions from Japan via shipping are the likely source and pathway of introduction to USA. In particular, we found unexchanged ballast arriving to USA (notably New York region) from sources throughout Japan, including southern and northern areas where closer genetic connections to USA were detected. Western Europe was also a prominent source of unexchanged ballast, suggesting possible trans-Atlantic transfers. Altogether, extensive biogeographic studies like this help provide integral evidence for resolving key invasion history questions, as well as a baseline understanding of a species’ probable success and prospect for further spread. In turn, while mandatory ballast water exchange in USA has greatly lessened ballast-borne invasions, our study underscores that hotspots of unexchanged ballast can remain potential sources of novel or supplemental introductions worldwide.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Over the past two centuries, myriad anthropogenic vectors have contributed to the global spread of species to novel regions around the world. In marine systems, commercial shipping has been most prominent given its broad scope and global operation (Seebens et al. 2013), accounting for 52–82% of non-indigenous species (NIS) introductions to North America within the past 30 years (Ruiz et al. 2015). Due to the accidental nature of many introduction vectors like shipping, NIS may be associated with extensive data gaps, and some may go undetected for years, decades, or centuries—imposing a critical lag time on their investigation (Carlton 2009; Bortolus et al. 2015; Blakeslee et al. 2016). Further complicating matters is the possibility for multiple introduction events from disparate regions, especially for those vectors that are widespread, continually active, and associated with high propagule pressure (Lejeusne et al. 2014). Yet, despite these inherent challenges, resolving key NIS questions (e.g., what, where, when, how) is vital for understanding an exotic species’ impact, managing/mitigating established NIS, and preventing future introductions.
In recent years, genetic data have become a powerful and increasingly informative tool in NIS investigations, providing integral information on a species’ source, vector, spread, and introduction timing (Geller et al. 2010; Rius et al. 2015). In turn, the mechanism behind, and the recurrence of, species introductions play important roles in shaping the genetic composition of non-native populations (Dlugosch et al. 2015). Although species introductions typically contain a subset of a source genetic pool, strong genetic bottlenecks are not always the norm. In fact, genetic diversity may sometimes be greater in select non-native versus native populations, given high propagule pressure and multiple introduction events inherent in some vectors (Roman and Darling 2007). Multiple introductions can alleviate Allee effects that curtail or slow range expansions in some areas of a species’ non-native range, while introduced populations that benefit from and maintain relatively high levels of standing genetic variation may better adapt to novel selective pressures in the non-native range, thus influencing propagation, spread, and competitiveness (Ellstrand and Schierenbeck 2000; Lee 2002; Kolbe et al. 2008). For example, ongoing population expansions of the European green crab (Carcinus maenas) throughout eastern North America have been abetted by a recent cryptic introduction to Maritime Canada from a different European source, resulting in a reduction in the crab’s initially strong genetic bottleneck by ca. 15% (Roman 2006; Blakeslee et al. 2010; Darling et al. 2014; Tepolt and Palumbi 2015). Thus, characterizing the genetic composition of introduced populations can provide key insights into an NIS’ invasion history and its potential to rapidly adapt to and spread within novel regions. Further, by shedding light on recurring introduction events, genetic reconstructions of a species’ invasion history provide a critical test of the effectiveness of management policies and practices intended to curtail species invasions. In the USA, ballast water exchange (BWE) regulations over the past decade have reduced concentrations of living biota (zooplankton) to ~10% of original concentrations (Minton et al. 2005); however, hotspots of unexchanged ballast water (or residuals in exchanged ballast) remain potential sources of novel or supplemental introductions (NBIC 2016). This is important because even relatively small propagule inoculations (e.g., from unmanaged ballast) within established populations may boost NIS genetic diversity and adaptive advantage (Drake and Lodge 2004).
Here, we explore the invasion history of the Asian shore crab, Hemigrapsus sanguineus (De Haan 1835), which to date has lacked a large-scale population genetics investigation in its native or non-native regions, despite being a common and often abundant shorecrab in these areas. Hemigrapsus sanguineus is native to the western Pacific and is generally believed to have been introduced to the western Atlantic via ship’s ballast water ca. 1980s, where it now ranges from Maine to North Carolina. The crab is also non-native in the eastern Atlantic, where it is expanding its European range, recently reaching the UK (Williams and McDermott 1990; Epifanio 2013; Seeley et al. 2015). Hemigrapsus sanguineus lives in coastal and estuarine intertidal habitats consisting of artificial and natural substrates like rock, rubble, jetties, floats and pilings, and has become one of the most abundant shore crabs in northeastern USA, exacting numerous impacts on native and non-native prey and competitors (Fofonoff et al. 2016). To resolve ongoing questions of introduction source, vector, and multiple introductions to the western Atlantic, as well as provide a general biogeographic understanding of the species’ population genetics in native and non-native regions, we used two common genetic markers, a barcoding gene (cytochrome oxidase I) and six microsatellite loci analyzed over two time periods, 2001–2002 and 2013–2014. Moreover, to enhance our understanding of the species’ global movements and likelihood for continued spread, we investigated global sources of ballast water to the eastern USA, focusing on the New York City/Long Island Sound (NYC/LIS) region (based on revealed genetic links), to identify hotspots of managed and unmanaged ballast discharge to the non-native region.
Methods
Sampling, sequencing and analysis
During summers 2013 and 2014, adult H. sanguineus crabs were hand-collected in the intertidal zone in both the native and non-native regions (see Table 1 for locations). Crabs were dissected for their gill tissue, which was preserved (frozen or in 95% ethanol) until DNA extraction using a standard CTAB protocol (France et al. 1996). Additional crabs previously hand-collected from the intertidal zone in 2001–2002 by S. Park and preserved in 95% ethanol, along with two preserved samples collected in 1996 from Shark River Inlet, New Jersey, USA (provided by J. McDermott), were also dissected and DNA-extracted. A 456-bp region of the COI gene was amplified using primers, HSmt-392 (CGAGCAGAATTAAGACAACC) and HSmt-393 (TGAGTAGCATAGTGATAGCTCC) and the PCR profile: 95 °C for 2-min; 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s; and 72 °C for 5-min (Steinberg 2008). Purified amplicons were sequenced at the Research Institute of Molecular Genetics (Kochi University, Japan) or Macrogen (Rockville, Maryland, USA). COI sequences were inspected for ambiguities using Geneious8.1.7 (Biomatters Ltd., Auckland, New Zealand) and aligned without gaps using ClustalW (Larkin et al. 2007). We also included additional overlapping COI sequences (n = 111) previously published for the native range (HQ702865–HQ702892; Yoon et al. 2011). All COI sequences were collapsed into haplotypes using TCS1.21 (Clement et al. 2000), and the output was used to create a map of haplotype frequencies per population and a bubble diagram, showing the relative proportion of each haplotype and their predicted connections (Fig. 1).

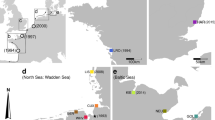
Haplotype frequency maps in a native (Asia) and b introduced (USA) ranges; and c haplotype bubble diagram for both regions. Numbers on each pie chart represent sample sites summarized in Table 1. The pie chart with a white asterisk represents our two 1996 samples from Shark River Inlet, NJ. In (a) and (b), pie charts are relatively sized according to a populations’ sample size. Colored pie pieces (a, b) and haplotype bubbles (c) represent haplotypes observed in both the native and introduced ranges; white pie pieces (a) and white bubbles (c) represent haplotypes only detected in Asia; gray pie pieces (b) and gray bubbles (c) represent haplotypes only detected in USA. Subregional pie charts represent analyses comprised of multiple populations per subregion/region (Table 1); pie charts on top include all haplotypes observed per subregion, while pie charts below portray shared haplotypes only [i.e., just the colored pie pieces in (c)]. The size of haplotype bubbles in (c) demonstrates the number of individuals with that haplotype for both regions based on a size range (see key)
We tested 21 microsatellite primer pairs developed for H. takanoi and H. penicillatus (Makino et al., unpub), finding 6 loci (hp13931, hp01636, hp21388, hp05535, hp06147, ht16728) to successfully amplify H. sanguineus. We performed two separate multiplex PCRs using two primer sets (Table S1) with the Type-It Microsatellite PCR kit (Qiagen, USA). For genotyping, 5′ ends were labeled with fluorescent dyes on forward primers and PIG-tailed on reverse primers (Brownstein et al. 1996). PCR parameters included: activation of Hotstart DNA-Taq polymerase at 95 °C for 5-min; 35 cycles of 95 °C for 30 s, 56 °C for 90 s, and 72 °C for 30 s; and 60 °C for 30-min. Two PCR products for each sample were combined with HiDi-formamide and Gene Scan 500 LIZ size-standard (Applied Biosystems, USA) and analyzed at the Research Institute of Molecular Genetics (Kochi University). Samples were genotyped using Genemapper3.7 (Applied Biosystems). Genetic indices were estimated for each population using Genodive (Meirmans and Van Tienderen 2004). Deviations from Hardy–Weinberg equilibrium were tested using Genepop4.1.4. Using STRUCTURE2.3.3 (Pritchard et al. 2000), a Bayesian clustering algorithm assigned clusters using a model allowing for admixture and correlated allele frequencies, and sampling locations as priors. Models using location as a prior have been found in several cases to reveal informative genetic structure in datasets where the signal of structure is too weak to be detected by standard structure models (e.g., Hubisz et al. 2009). Ten iterations for each population number (K = 1–10) were analyzed with MCMC (burnin = 100,000; total chain length = 1,000,000). A likelihood value and stability for each K (represented as delta K; Evanno et al. 2005) were estimated using STRUCTURE HARVESTER (Earl 2012). K = 3 was selected as the optimum population number. We combined values from different run replicates with CLUMPP (Jakobsson and Rosenberg 2007) and visualized results using DISTRUCT (Rosenberg 2004). Last, since the distribution of shared private alleles (i.e., alleles found in just one population of the source region and shared with individuals in founding populations) can be a powerful predictor of source populations, shared private alleles were estimated between introduced USA and four native subregions (northern, central, southern Japan, and Korea-China), averaged across loci, and standardized using a rarefaction procedure with ADZE1.0 (Szpiech 2008) (Fig. 2).

STRUCTURE analysis of microsatellite data across the native (Asia) and introduced (USA) ranges (a) and mean number of microsatellite alleles per locus private to the combination of USA populations and regional populations in Asia (b) and between USA populations and local populations in southern Japan (c). Population names and sample sizes associated with abbreviations are in Table 1. Sample sizes in (b) and (c) were rarified to a minimum size (n = 114 and n = 57, respectively)
To estimate haplotype and allelic diversity and quantify the effects of sampling effort, rarefaction curves were constructed for both markers using EstimateS8.20 (Colwell et al. 2012). Rarefaction analyses were also performed using EstimateS8.20 to estimate the number of haplotypes and alleles for each population with a sample size greater than seven individuals at their respective sampling efforts—this was not only to account for differences in sample size across populations but also to determine the number of haplotypes or alleles that may have been missed at a population/region during sampling (Gotelli and Colwell 2001). These analyses were also performed at the broader subregional level, which took into account multiple populations per subregion (see Table 1). In addition, hierarchical analysis of molecular variance (AMOVA) was estimated using ARLEQUIN311 (Excoffier et al. 2005), and the FCT fixation index, which explores differentiation across groups, helped pinpoint close genetic connections and potential source locations at several regional and subregional levels (see Table 2). For COI, we also examined differentiation based on time period. Finally, pairwise ϕSTs were calculated (ARLEQUIN for COI; Genodive for microsatellites) and explored in a non-metric multidimensional scaling (nMDS) analysis (PRIMER6, Plymouth Marine Laboratory, UK) to look for spatial patterns among native and introduced populations, and populations within subregions were also combined to explore pairwise differentiation at the subregional level (Table 1; Fig. 3).

nMDS plots of microsatellite (a, c) and COI (b, d–f) pairwise FST at the population (a, b) and subregional (c–f) levels for the native and introduced ranges. See Table 1 for population and subregional abbreviations and sample sizes. Microsatellite data are displayed in (a, c) at the population level and subregional level, respectively, with symbols and colors representing different regions and subregions in the two ranges. COI data are displayed at the population level (b) with symbols and colors representing different regions and subregions in the two ranges and also at the subregional level (d–f), first with all shared and unshared haplotypes, including China (d), and then with all shared and unshared haplotypes, excluding China (e), and finally with just shared haplotypes, including China (f)
Although we attempted to have equitable numbers of samples and sites for COI and microsatellite data in both the native and introduced ranges, this was not always possible due to lab processing and logistical reasons; e.g., older samples were more degraded than newer samples; the Yoon et al. (2011) dataset only included COI data; and a subset of geographically-spaced sites were chosen for microsatellite analyses. Altogether for COI, our dataset included 16.00 (±1.38) individuals per population in the native region and 12.86 (±1.05) individuals per population in the introduced region; for microsatellites, our dataset included 26.42 (±1.46) individuals per population in the native region and 15.25 (±2.81) individuals per population in the introduced region. Given the fairly weak genetic structure in general in the species (see Results), and the detected genetic bottleneck in the introduced region in particular, we concentrated sampling in the native range. Moreover, because microsatellites generate more alleles per population than COI, we sampled more individuals (on average) per population for this marker, thus providing greater resolution within a population for F-statistics, structure, and private alleles analyses (see Table 1). Overall, paired data between COI and microsatellite markers included a number of geographically spaced sites in both regions: east Asia (n = 12 sites in Japan, S. Korea, and China) and eastern USA (n = 11 sites ranging from Maine to Maryland).
Ballast water discharge
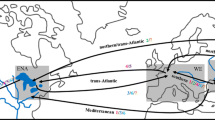
Given ballast water is the putative vector for H. sanguineus’ introduction to North America (Fofonoff et al. 2016), we analyzed ballast water delivery and management data collected by the National Ballast Information Clearinghouse (NBIC) over a 10-year period (2005–2014; NBIC 2016), corresponding to the timing that BWE has occurred in the USA and the timing of our recent genetic samples. Since 1999, all overseas commercial ships have been required to report their ballast water discharge history in USA waters, and since 2004, USA regulations have required overseas ships arriving in all USA ports to perform open ocean BWE to reduce planktonic concentrations prior to discharge of coastal ballast water (i.e., entrained within 200 nmi of shore). Despite these regulations, not all ships either (a) report their ballast water management (i.e., since 2005, ~94% reporting compliance has been achieved in the NYC/LIS region), or (b) perform BWE prior to discharge of overseas ballast water, sometimes due to time and/or space-constrained shipping routes (Miller et al. 2011). Altogether, ~15% unexchanged ballast water discharge volume exists in the region. Thus, though reduced, substantial quantities of biota are still undoubtedly transferred to the USA via ballast water, either as residuals following BWE or through unexchanged ballast water (NBIC 2016). Because we detected elevated genetic diversity of H. sanguineus in the NYC/LIS region (ostensibly arising from multiple introductions; see below), we investigated the global sources and volumes of ballast water (both exchanged and unexchanged) to this area, interrogating 70,398 ship arrivals in the process. To provide a single estimate of ballast water flux integrating the unexchanged and exchanged overseas coastal ballast water, discharge volumes were calculated as the total volume of unexchanged ballast water discharge plus 10% of the total volume of exchanged ballast water (sensu Muirhead et al. 2015). Source locations of ballast water delivered to the region were mapped using ArcMap 10.3.1 (ESRI, USA) and scaled by the total effective discharge volume originating at that location (Fig. 4).

Global sources and volumes of ballast water (BW) delivered to Long Island Sound/New York City region between 2005 and 2014 (a). Detailed inset maps highlight ballast water sources from (b) west European waters and (c) east Asian waters. Effective ballast water volumes (=unexchanged ballast water + 10% of exchanged ballast water) were calculated for all discharges and plotted using data from the National Ballast Information Clearinghouse (NBIC 2016). Maps were produced using the Winkle Tripel projection
Results
Haplotype and allelic richness
Our analyses include 731 COI sequences and 500 microsatellites across 51 populations. See Table 1 for summary data. In total, we detected 84 COI haplotypes: 78 in the native range (72 only in Asia), 12 in the introduced range (6 only in USA), and 6 shared between ranges (GenBank Accession #s: KY062668- KY062752). See Table S2 for occurrences of haplotypes per population. For microsatellites, 109 alleles were detected across 6 loci (Table S3): 105 in the native range (38 only in Asia), 71 in the introduced range (4 only in USA), and 67 shared between them. Total native haplotype and allelic richness were significantly greater than introduced richness (COI: χ 2=48.4; p < 0.001; microsatellites: χ 2=6.57; p = 0.010). Several USA populations had higher expected haplotype richness than the average expected richness across all USA populations, and 4 out of 5 of these were in LIS. In fact, LIS expected richness (6.27 ± 1.04) in 2014 was 1.5 times greater than all other USA populations (4.26 ± 0.58). Similarly, four out of the five populations with the highest average allelic richness in USA in 2014 were in LIS, which was 1.3 times higher (7.75 ± 0.35) than all other 2014 USA populations (6.03 ± 0.67) and 1.6 times higher than populations closest to the species’ initial introduction report (4.92 ± 0.25) (Table 1; Fig. 1). Subregional analyses similarly found the LIS area to have the greatest observed and expected haplotype richness compared to other USA subregions. For example, there were 4.56 (±0.38) haplotypes observed in the LIS subregion and 6.27 (±1.04) haplotypes predicted, compared to 3.50 (±0.38) observed and 4.83 (±0.72) predicted in the north subregion (Table 1; Fig. 1).
In rarefaction analyses, rarefaction curves in Asia (Figure S1a) suggested further sampling would reveal more diversity, but differences between observed and predicted richness were less substantial for microsatellite loci. Particular to the non-native range, estimator curves suggested capture of much of the haplotype and allelic richness in the USA across our whole dataset (Figure S1b). Comparisons of the older versus newer time period found expected versus observed haplotype richness for the 2001 time period to be similar (2001 observed = 3 versus 2001 expected = 3.30 ± 0.15), while for 2014, about 1.4 times more haplotypes were predicted than were actually observed (2014 observed = 12 versus 2014 expected = 16.99 ± 0.73).
Haplotype frequencies
Among six shared haplotypes, H7 and H52 dominated, making up 71% (H7 = 27%, H52 = 44%) of the total diversity, but with differences between the ranges: in Asia, H7 = 19% and H52 = 47% of native diversity, while in USA, H7 = 40% and H52 = 38% of non-native diversity. Two other shared haplotypes (H23, H29) were in higher frequencies in non-native (8, 9%) versus native ranges (0.1, 4%), and were ubiquitous throughout the introduced range, but H23 was found in just two northern Japanese and one Korean population, while H29 was ubiquitous throughout Japan. H21 and H74 were rare in both ranges (native frequencies = 0.2% for both; introduced frequencies = 1 and 0.7%) and in the non-native range were only found in LIS. The six haplotypes only detected in the USA were all singleton occurrences, except H48, which made up 1.4% of USA diversity and only detected in LIS. The other five USA haplotypes were detected in Maine (n = 2), Long Island (n = 1), and New Jersey (n = 1) (Fig. 1). Finally, our two 1996 preserved samples represented two shared haplotypes, H21 and H52; the latter is a common haplotype throughout both regions and the former is a rare haplotype in both regions and only detected in northern Japan in our contemporary sampling of the native range.
STRUCTURE analysis and shared private alleles
All six microsatellite loci were polymorphic with 10–30 alleles per locus. Significant deviations from HWE were detected in locus hp13931 at HY2 and STO, locus hp21388 at IWA, and locus ht16728 at TTT, AK, HI2, SUG, IWA, AMA, and SMP. STRUCTURE analysis exhibited two clusters (Fig. 2a) in the native range and one in the non-native range. Japan, Korea, and China (HUI) formed a cluster (blue) and China (TTT) formed a distinct cluster (green). USA populations formed another cluster (red), while TP1, TP2, and SMP populations had mixed probabilities of clustering in the Japan–Korea–China (HUI) group and the USA group. The number of shared private alleles was greater in the USA/southern Japan combination (0.40 ± 0.17) versus USA/northern Japan (0.16 ± 0.10), USA/central Japan (0.23 ± 0.15) and USA/Korea-China (0.05 ± 0.02). For local populations in southern Japan, there were few shared private alleles between USA and SUG (0.02 ± 0.01), IWA (0.05 ± 0.05), NB2 (0.002 ± 0.002), and other native populations (0.12 ± 0.07), but there were a relatively large number of shared private alleles between USA and AMA (0.41 ± 0.22) (Fig. 2b, c).
Regional and pairwise differentiation
Summary data for regional differentiation are displayed in Table 2. For COI, there were three non-significant (p > 0.05) pairwise comparisons of regional differentiation and eleven for nuclear microsatellites. The two markers were consistent with non-significant differentiation between Asia and LIS, northern Japan and southern USA, and northern Japan and LIS. Microsatellite markers also demonstrated non-significant differentiation between both central and southern Japan with southern USA, and between southern Japan and LIS. Comparisons of time periods (2001–2002 and 2013–2014) in the COI dataset demonstrated significant (P < 0.05) differentiation between the time periods for USA but not for Asia. As such, we combined our temporal data for Asia across our various analyses (including those discussed above), but for USA, we separated some analyses by time period to look for differential temporal patterns (e.g., Figs. 1, 3).
nMDS plots of pairwise ϕST data were suggestive of closer spatial relationships between LIS populations in USA and those from both southern and northern Japan (Fig. 3), and when subregional analyses included shared haplotypes only, JAP-N and USA-LIS overlapped (Fig. 3f). For COI, two of the three Chinese populations were most disparate from USA populations, while in the microsatellite analysis, both Chinese populations were most separated from USA populations. Finally, in both the COI and microsatellite subregional plots, there was clear separation between the older (2001) USA samples from the newer (2014) samples (Fig. 3).
Ballast Water Discharge
When global coastal ballast water flux to NYC/LIS was plotted, the effective ballast water discharge from coastal waters originating from overseas during the 10-year period from 2005 to 2014 exceeded 2.8 million MT (and >12 million MT total discharge), and this water emanated from all corners of the globe (Fig. 3a). Included are tens of thousands of MT of effectively unexchanged ballast water from Japan, Korea, and China (Fig. 3c), suggesting the possibility for multiple H. sanguineus introductions during the time between 2001 and 2014. However, there was also significant ballast water flux (hundreds of thousands of MT) from Europe (France, Netherlands, Belgium, Germany, and UK, Fig. 3b) where H. sanguineus has also invaded and continues to spread (Epifanio 2013; Seeley et al. 2015).
Discussion
Our study represents the first large-scale biogeographic exploration of the population genetics of the abundant Asian shorecrab in native east Asian and non-native east USA ranges, imparting a greater understanding of the crab’s geographic source region, its vector of introduction, and the likely existence of multiple introductions over time. It also provided a biological test case evaluating the effectiveness of, and suggesting some limitations to, BWE management at curtailing successful establishments of entrained propagules (i.e., via multiple introduction events). Below, we further discuss these results and the significance for H. sanguineus’ introduction, and how it may inform our understanding of the anthropogenic transport of marine species worldwide.
Multiple introductions and lessening of a bottleneck
We detected a strong genetic bottleneck in the introduced range of H. sanguineus, where predicted haplotype richness was 18 times lower than the native range across all samples, versus ~1.5 times lower for microsatellites (marker differences are probably because mtDNA has one-quarter the effective population size of nuclear microsatellite markers; Moore 1995). For COI, this bottleneck differed somewhat between time periods, presumably as a result of subsequent introductions—a trend that has also been observed in several other species introductions (Roman and Darling 2007). In our study, just three haplotypes were detected in 2001 in USA (~2 on average per population), and these were also the three most commonly observed in Japan. By 2014, 12 haplotypes had been detected (~4 on average per population), and these 9 additional haplotypes were either rare or undetected in east Asia, the latter likely a result of a diverse native range (Figure S1). Interestingly, however, one of these 9 haplotypes (H21) was detected in a sampled crab from 1996 (Fig. 1), but was not detected in our 2001 data. In contrast to COI, changes in microsatellite allelic richness with time were not as dramatic: across 6 loci, an average of 5 alleles were detected in 2001 versus 7 in 2014. Altogether, while these temporal differences could suggest enhanced diversity as a result of multiple introductions, they may also reflect differences in sampling effort between the time periods, i.e., we were unable to produce the same range or number of sequences in the earlier time period compared to the later one. However, given the continued active operation of shipping between the regions over time, delivering unmanaged ballast water (plus residual biota in exchanged ballast water) to the USA from sources in east Asia, multiple introduction events appear likely in this system (see further discussion below).
Interestingly, the greatest genetic diversities found in USA populations were in the LIS subregion (Fig. 1), about 250-km north of H. sanguineus’ first record in Cape May, NJ (Williams and McDermott 1990). In fact, closer populations to the first record (New Jersey and Maryland) were 1.6–2.0 times less diverse than LIS. FCT analyses also suggested less differentiation (greater genetic connections) between Japan and LIS populations than many other USA populations (Table 2), suggesting that the NYC/LIS region could be a recipient area for contemporary introductions, especially given New York City’s prominence as a major shipping port. In fact, another recent species invasion to the broader NYC/LIS region is also from east Asia: the Oriental grass shrimp Palaemon macrodactylus was first noted in the Bronx River Estuary (NY) in 2001 and has since been discovered in several populations along Long Island Sound and in the Chesapeake Bay (Warkentine and Rachlin 2010; Fofonoff et al. 2016; JTC and M. Roy, unpublished).
Evidence for multiple introductions and lessening of a species’ genetic bottleneck with time has also been well-studied in the globally invasive green crab (C. maenas), which overlaps extensively with H. sanguineus in its introduced USA range. Carcinus maenas has had two major introduction events from the eastern to the western Atlantic: the first in the early 1800s to mid-Atlantic/New England, and the second in the 1990s to eastern Nova Scotia (Carlton and Cohen 2003; Roman 2006). The admixture of genotypes from these two introductions (Pringle et al. 2011) could affect population and community dynamics in the system and potentially alleviate deleterious fitness effects associated with genetic bottlenecks. Discerning multiple introduction events not only provides a greater understanding of the potential dissipation of a bottleneck, but also the types and abundances of genotypes that work together to influence phenotype in a species’ new range (Darling et al. 2014; Tepolt and Palumbi 2015).
Asian source locations for USA introduction
Our genetic analyses suggest Japan as the probable source region for the USA introduction. This is because Japanese subregions appeared most genetically connected to USA subregions in numerous analyses from both markers (e.g., haplotype map, FCT, nMDS, STRUCTURE, and private alleles; Table 2; Fig. 1–3). Two northern and two southern Japanese populations particularly stood out: in the north, Tateyamasaki and Kesennuma were both spatially close in nMDS analyses to high diversity LIS populations, and Tateyamasaki was the only Asian population with the rare haplotype (H21) detected in one of our two 1996 New Jersey samples and in three of our 2014 LIS samples. In subregional nMDS analyses of shared haplotypes only, the northern Japan subregion and the USA LIS subregion overlapped, further suggesting JPN-N as a possible source area (Fig. 3). In the southern Japanese subregion, the populations Iwashima and Amakusa were spatially close in nMDS analyses and also prominent in STRUCTURE and private alleles analyses. It is possible that longer breeding seasons in southern latitudes of the crab’s native range (Fukui 1988) may increase the temporal availability of H. sanguineus larvae for ship’s ballast tanks.
Interestingly, central Japanese populations near prominent seaports (Tokyo, Kawasaki, Yokohama) were not indicated as probable source locations, as we may have expected. Lower densities of H. sanguineus may occur in these areas due to limitations in suitable habitat, where much of the natural shoreline has been converted to concrete (Ogura et al. 2010). However, it is also possible that we missed sampling existing populations that nevertheless became entrained in ship’s ballast water, and the detection of six unshared haplotypes in USA may point to this possibility. Further sampling of Chinese populations would also bolster our confidence that China is a less likely source region than Japan—the few Chinese populations we were able to sample appeared more disparate from USA populations than the majority of those from Japan and South Korea. Even so, enhanced sampling in China is required to fully resolve the role this region may play in the genetic composition of USA H. sanguineus.
Introduction vector
Our results suggest several source locations and multiple introduction events are likely for the USA introduction of H. sanguineus from Asia, and this is further supported by our analyses of BWE between the regions. The likely introduction vector for these multiple events is commercial shipping in ballast water tanks (Fofonoff et al. 2016). The crab has a relatively long-lasting planktonic larval stage (up to 44 days) (Epifanio et al. 1998), allowing for larval entrainment in ballast tanks for the duration of a Japan–USA voyage. Although ballast water regulations have become more stringent over the past decade in USA, thereby reducing the potency of ballast water as a vector (Minton et al. 2005), BWE has not been applied to all ballast water sourced in Japan and its surroundings that are then discharged into the NYC/LIS region (see Fig. 3). Further, BWE does not remove 100% of live planktonic organisms from ballast water tanks, so residual discharge may still contribute to species introductions, albeit at much reduced levels (NBIC 2016). Our ballast water discharge analysis demonstrated unexchanged ballast coming from sources throughout Japan, including southern and northern regions where we detected closer genetic connections to USA populations. Interestingly, however, western Europe was also one of the most prominent sources of unexchanged ballast water discharge to USA waters. Non-native H. sanguineus populations have been present in western Europe since the late 1990s (Epifanio 2013), indicating opportunities for trans-Atlantic transfer. Since we have not genetically compared these regions, we cannot directly speak to the nature of gene flow between Europe and America, but we also cannot discount the possibility for genetic exchange given the extent of ballast water flux between the regions. A thorough exploration is needed to resolve this question, as well as the source(s) of European introductions, and whether additional Asian and trans-Atlantic ballast water transfer could continue to enhance genetic diversity in the expanding introduced Atlantic ranges. Moreover, it is also possible that gene flow could be moving in the opposite direction—from USA to Asia—through active shipping vectors, and common established alleles in the USA are being moved back to Asia. However, as these common alleles tend to be shared between the two ranges, this may not dramatically affect native gene pool dynamics, unless such alleles are transported to more differentiated Asian populations (e.g., TTT in China; Fig. 3).
Other shipping related vectors, specifically hull fouling and niche spaces on ships (e.g., sea-chests), could serve as additional mechanisms of introduction or gene flow for H. sanguineus in USA. In particular, Micu et al. (2010) speculated that recreational yacht fouling may have introduced H. sanguineus to the Black Sea, while Gollasch (1999) reported that a related species, H. penicillatus, was found in empty barnacle tests fouling the hull of a newly arrived vessel in Germany from Japan. Furthermore, several studies (Coutts et al. 2003; Coutts and Dodgshun 2007; Frey et al. 2014) have reported a wide diversity of crab species comprising fouling communities in sea-chests (recesses in ships’ hulls serving as intake reservoirs). However, the modern-day role of ship fouling in contributing to global gene flow of H. sanguineus and many other species requires further investigation. On the other hand, an unlikely modern introduction vector for the crab’s USA introduction is intercontinental (interoceanic and transoceanic) movement with commercial oysters. Widespread global movements of live adult oysters and their associated biota ceased almost 50 years ago (Carlton 1992; Ruesink et al. 2005; Miura et al. 2006), making oysters a seemingly asynchronous vector with the appearance of H. sanguineus outside of east Asia commencing in the 1980s–1990s.
Ecological importance
Hemigrapsus sanguineus is a highly abundant crab in its established USA range, and population densities often exceed those of its native range. In fact, Klassen (2012) found peak densities across multiple USA studies to be about three times greater than those in native locations. More specifically, in Amakusa, Japan, H. sanguineus densities were reported as ~30 crabs/m2 in a single boulder layer treatment and ~80 crabs/m2 in a double boulder layer (Takada 1999). In contrast, an 8-year study of a non-native LIS population found H. sanguineus densities at or greater than 80 crabs/m2, while concurrently, a native mud crab species Eurypanopeus depressus declined in abundance by 95% (Kraemer et al. 2007). Similarly, a 12-year investigation of New England (USA) sites revealed declines of many coastal and estuarine intertidal species (panopeid mud crabs, Littorina spp. snails, Cancer spp. crabs, and spider crabs) concomitant with H. sanguineus population expansion in the area (O’Connor 2014). These results are consistent with several other analyses showing high densities of H. sanguineus correlating with lower densities of native prey species, including barnacles, polychaetes, blue mussels, ephemeral algae, and commercial bivalves (Tyrrell et al. 2000; DeGraaf and Tyrrell 2004; Freeman and Byers 2006; Griffen and Byers 2006). Indeed, within our own study, we found H. sanguineus to be patchily distributed in the native range and sampling effort was often >1 h to collect sufficient numbers for genetic analysis, while sampling the introduced range sometimes produced the same numbers in <10 min.
As summarized by Epifanio (2013), H. sanguineus’ demographic success in USA could be for many reasons, including its higher fecundity and longer breeding season than native species, its superior competition for shelter, its strong aggregation behavior, its predation of co-occurring crab competitors, and its release from natural enemies, like parasites. Regarding the latter, H. sanguineus is infected with nine parasites in its native range, including two rhizoceophalan castrators (Blakeslee et al. 2009). Native rhizocephalan prevalence occasionally reaches levels as high as 80% (such as in Amakusa, Japan), suggesting a substantial negative effect on H. sanguineus demography in its native range (Yamaguchi et al. 1994). In contrast, only three parasites have been detected in the introduced range, none of which are castrators (Kroft and Blakeslee 2016). Reproductively and energetically “healthier” populations of H. sanguineus in non-native regions could thus provide a competitive edge over native species burdened by typical native parasite loads (Torchin et al. 2005; Blakeslee et al. 2013). However, further study in this area is needed.
Conclusions and implications
Our H. sanguineus biogeographic study in native versus non-native regions demonstrates the utility of genetic data in resolving integral NIS questions, such as source, vector, and timing, while also revealing the existence of multiple introductions from multiple source populations despite increasingly stringent ballast water regulation. This lessening of a bottleneck may also contribute to H. sanguineus’ continued Atlantic spread, especially if genetic admixture from differentially adapted native populations provided greater tolerances to novel habitats. Past and ongoing ecological research has documented significant impacts of H. sanguineus in eastern USA communities that may become enhanced with genetically diverse introduced populations amalgamated from multiple Asian sources. Moreover, recently published microsatellite primers tested on invasive H. sanguineus in Europe (Poux et al. 2015) could further elucidate whether patterns observed in USA populations are similar to those in Europe, or if European invasion history greatly differs from USA. Given evidence for strong ballast water flux between Europe and America, a detailed look at European populations is an important future endeavor. Last, we must acknowledge that while widespread, mandatory, open ocean BWE appears to have lessened global ballast water-borne invasions substantially, the vector remains active, especially in parts of the world where no regulations apply. As such, the apparent continued introduction of H. sanguineus as a result of ballast water is further testament to the need for management solutions that enable ships to operate anywhere in the world without concern for species transport in their ballast tanks.
References
Blakeslee AMH (2016) Parasites and genetics in marine invertebrate introductions: signatures of diversity declines across systems. In: Canning Clode J (eds) Biological invasions in changing ecosystems: vectors, ecological impacts, management and predictions. De Gruyter Open, Warsaw, pp 138–182
Blakeslee AMH, Keogh CL, Byers JE, Kuris AM, Lafferty KD, Torchin ME (2009) Differential escape from parasites by two competing introduced crabs. Mar Ecol Prog Ser 393:83–96
Blakeslee AMH, McKenzie CH, Darling JA, Byers JE, Pringle JM, Roman J (2010) A hitchhiker’s guide to the Maritimes: anthropogenic transport facilitates long-distance dispersal of an invasive marine crab to Newfoundland. Divers Distrib 16:879–891
Blakeslee AMH, Fowler AE, Keogh CL (2013) Marine invasions and parasite escape: updates and new perspectives. Adv Mar Biol 66:87–169
Bortolus A, Carlton JT, Schwindt E (2015) Reimagining South American coasts: unveiling the hidden invasion history of an iconic ecological engineer. Divers Distrib 21:1267–1283
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1010
Carlton JT (1992) Introduced marine and estuarine mollusks of North America: an end-of-the-20th-century perspective. J Shellfish Res 11:489–505
Carlton JT (2009) Deep invasion ecology and the assembly of communities in historical time. In: Rilov G, Crooks JA (eds) Biological invasions in marine ecosystems: ecological, management, and geographic perspectives. Springer-Verlag, Berlin Heidelberg, pp 13–56
Carlton JT, Cohen AN (2003) Episodic global dispersal in shallow water marine organisms: the case history of the European shore crabs Carcinus maenas and. C aestuarii. J Biogeogr 30:1809–1820
Clement M, Posada DC, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Colwell RK, Chao A, Gotelli NJ, Lin S-Y, Mao, C X, Chazdon RL, Longino JT (2012) Models and estimators linking individual-based and sample-based rarefaction, extrapolation, and comparison of assemblages. J Plant Ecol 5:3–21
Coutts ADM, Dodgshun TJ (2007) The nature and extent of organisms in vessel sea-chests: A protected mechanism for marine bioinvasions. Mar Pollut Bull 54:875–886
Coutts ADM, Moore KM, Hewitt CL (2003) Ships’ sea-chests: an overlooked transfer mechanism for non-indigenous marine species? Mar Pollut Bull 46:1504–1515
Darling JA, Tsai YHE, Blakeslee AM, Roman J (2014) Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab. Carcinus maenas R Soc Open Sci 1:140202
DeGraaf JD, Tyrrell MC (2004) Comparison of the feeding rates of two introduced crab species, Carcinus maenas and Hemigrapsus sanguineus, on the blue mussel, Mytilus edulis. Northeast Nat 11:163–167
Dlugosch KM, Anderson SR, Braasch J, Cang FA, Gillette HD (2015) The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Mol Ecol 24:2095–2111
Drake JM, Lodge DM (2004) Global hot spots of biological invasions: evaluating options for ballast–water management. Proc R Soc Lond B Biol Sci 271:575–580
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellstrand NC, Schierenbeck KA (2000) Hybridization as a stimulus for the evolution of invasiveness in plants? Proc Natl Acad Sci 97:7043–7050
Epifanio CE (2013) Invasion biology of the Asian shore crab Hemigrapsus sanguineus: a review. J Exp Mar Biol Ecol 441:33–49
Epifanio CE, Dittel AI, Park S, Schwalm S, Fouts A (1998) Early life history of Hemigrapsus sanguineus, a non-indigenous crab in the Middle Atlantic Bight (USA). Mar Ecol Prog Ser 170:231–238
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver 30: An integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fofonoff, PW, Ruiz, GM, Steves, B and Carlton, JT (2016) National Exotic Marine and Estuarine Species Information System. http://invasionssiedu/nemesis/. Access date 5 Apr 2016
France SC, Rosel PE, Agenbroad JE, Mullineaux LS, Kocher TD (1996) DNA sequence variation of mitochondrial large-subunit rRNA provides support for a two subclass organization of the Anthozoa (Cnidaria). Mol Mar Biol Biotechnol 5:77–86
Freeman, AS and Byers, JE (2006) Divergent induced responses to an invasive predator in marine mussel populations. Science 313:831–833
Frey MA, Simard N, Robichaud DD, Martin JL, Therriault TW (2014) Fouling around: vessel sea-chests as a vector for the introduction and spread of aquatic invasive species. Manag Biol Invasions 5:21–30
Fukui Y (1988) Comparative studies on the life history of the Grapsid crabs (Crustacea, Brachyura) inhabiting intertidal cobble and boulder shores. Publ Seto Mar Biol Lab 33:121–162
Geller JB, Darling JA, Carlton JT (2010) Genetic perspectives on marine biological invasions. Ann Rev Mar Sci 2:367–393
Gollasch S (1999) The Asian decapod Hemigrapsus penicillatus (de Haan, 1835) (Grapsidae, Decapoda) introduced in European waters: status quo and future perspective. Helgolander Meeresuntersuchungen 52:359–366
Gotelli NJ, Colwell RK (2001) Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett 4:379–391
Griffen BD, Byers JE (2006) Partitioning mechanisms of predator interference in different habitats. Oecologia 146:608–614
Grosberg R, Cunningham CW (2000) Genetic structure in the sea from populations to communities. In: Bertness M, Gaines S, Hay M (eds) Marine Community Ecology. Sinauer Associates, Sunderland, pp 61–84
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9(5):1322–1332
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Klassen G (2012) Biological Synopsis of the Asian Shore Crab. Hemigrapsus sanguineus Canadian Manuscript Report of Fisheries Aquatic Sciences 2978:43
Kolbe JJ, Larson A, Losos JB, de Queiroz K (2008) Admixture determines genetic diversity and population differentiation in the biological invasion of a lizard species. Biol Lett 4:434–437
Kraemer GP, Sellberg M, Gordon A, Main J (2007) Eight-year record of Hemigrapsus sanguineus (Asian shore crab) invasion in western Long Island Sound estuary. Northeast Nat 14:207–224
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evolut 17:386–391
Lejeusne, C, Saunier, A, Petit, N, Beguer, M, Otani, M, Carlton, JT, Rico, C and Green, AJ (2014) High genetic diversity and absence of founder effects in a worldwide aquatic invader. Sci Rep 4. doi:10.1038/srep05808
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Micu D, Nita V, Todorova V (2010) First record of the Japanese shore crab Hemigrapsus sanguineus (de Haan, 1835) (Brachyura: Grapsoidea: Varunidae) from the Black Sea. Aquat Invasions 5:S1–S4
Miller AW, Minton MS, Ruiz GM (2011) Geographic limitations and regional differences in ships’ ballast water management to reduce marine invasions in the contiguous United States. Bioscience 61:880–887
Minton MS, Verling E, Miller AW, Ruiz GM (2005) Reducing propagule supply and coastal invasions via ships: effects of emerging strategies. Front Ecol Environ 3:304–308
Miura O, Torchin ME, Kuris, A M, Hechinger RF, Chiba S (2006) Introduced cryptic species of parasites exhibit different invasion pathways. Proc Natl Acad Sci 103:19818–19823
Moore WS (1995) Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 1:718–726
Muirhead JR, Minton MS, Miller AW, Ruiz GM (2015) Projected effects of the Panama Canal expansion on shipping traffic and biological invasions. Divers Distrib 21:75–87
National Ballast Information Clearinghouse (NBIC) (2016) NBIC Online Database, Electronic publication, Smithsonian Environmental Research Center and United States Coast Guard. doi:10.5479/data.serc.nbic, http://www.invasionssiedu/nbic/searchhtml. Accessed 25 Mar 2016
O’Connor NJ (2014) Invasion dynamics on a temperate rocky shore: from early invasion to establishment of a marine invader. Biol Invasions 16:73–87
Ogura H, Miyajima Y, Kitazawa T (2010) Changes in ecosystem services of SATOUMI in Chiba prefecture. Rep Chiba Biodivers Cent 2:141–156
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Poux C, Gothland M, Holl AC, Spilmont N, Arnaud JF (2015) Highly polymorphic microsatellite markers in two species, the invasive shore crabs Hemigrapsus sanguineus and Hemigrapsus takanoi (Decapoda, Varunidae). Conserv Genet Resour 7:569–572
Pringle JM, Blakeslee, A M, Byers JE, Roman J (2011) Asymmetric dispersal allows an upstream region to control population structure throughout a species’ range. Proc Natl Acad Sci 108:15288–15293
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rius M, Turon X, Bernardi G, Volckaert FA, Viard F (2015) Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol Invasions 17:869–885
Roman J (2006) Diluting the founder effect: cryptic invasions expand a marine invader’s range. Proc R Soc Lond B Biol Sci 273:2453–2459
Roman J, Darling JA (2007) Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol 22:454–464
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Ruesink JL, Lenihan HS, Trimble AC, Heiman KW, Micheli F, Byers JE, Kay MC (2005) Introduction of non-native oysters: ecosystem effects and restoration implications. Ann Rev Ecol Evol Syst 36:643–689
Ruiz GM, Fofonoff, P W, Steves BP, Carlton JT (2015) Invasion history and vector dynamics in coastal marine ecosystems: A North American perspective. Aquatic Ecosyst Health Manag 18:299–311
Seebens H, Gastner M, Blasius B (2013) The risk of marine bioinvasion caused by global shipping. Ecol Lett 16:782–790
Seeley B, Sewell J, Clark PF (2015) First GB records of the invasive Asian shore crab, Hemigrapsus sanguineus from Glamorgan, Wales and Kent, England. Mar Biodiver Rec 8:e102
Steinberg MK, Krimsky LS, Epifanio CE (2008) Induction of metamorphosis in the Asian shore crab Hemigrapsus sanguineus: effects of biofilms and substratum texture. Estuaries Coasts 31:738–744
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Takada Y (1999) Influence of shade and number of boulder layers on mobile organisms on a warm temperate boulder shore. Mar Ecol Prog Ser 189:171–179
Tepolt CK, Palumbi SR (2015) Transcriptome sequencing reveals both neutral and adaptive genome dynamics in a marine invader. Mol Ecol 24:4145–4158
Torchin ME, Byers JE, Huspeni TC (2005) Differential parasitism of native and introduced snails: replacement of a parasite fauna. Biol Invasions 7:885–894
Tyrrell MC, Harris LG, Pederson J (2000) Potential impact of the introduced Asian shore crab, Hemigrapsus sanguineus. In: northern New England: Diet, feeding preferences, and overlap with the green crab (ed) Carcinus maenas Marine Bioinvasions: Proceedings of the First National Conference. MIT Sea Grant College Program, Cambridge, MA pp 208–220
Warkentine BE, Rachlin JW (2010) The first record of Palaemon macrodactylus (oriental shrimp) from the eastern coast of North America. Northeast Nat 17:91–102
Williams AB, McDermott JJ (1990) An eastern United States record for the western Indo-Pacific crab, Hemigrapsus sanguineus (Crustacea: Decapoda: Grapsidae). Proc Biol Soc Wash 103:108–109
Yamaguchi T, Tokunaga S, Aratake H (1994) Contagious infection by the rhizocephalan parasite Sacculina sp in the grapsid crab Hemigrapsus sanguineus (De Haan). Crustacean Res 23:89–119
Yoon M, Hong S-E, Nam YK, Kim DS (2011) Genetic diversity of the Asian shore crab, Hemigrapsus sanguineus, in Korea and Japan inferred from mitochondrial cytochrome c oxidase subunit I gene. Animal Cells Syst 15:243–249
Acknowledgements
We thank the following for their lab/field help: K. Bartley, A. Fowler, A. Freeman, Z. Holmes, I. Kernin, S. Kulins, M. Roy, and M. Sanderson. We thank J. McDermott for sending us two preserved 1996 samples. O. Miura was partly supported by the “Program to Disseminate Tenure Tracking System”, MEXT, Japanese Government, and facilitated by JSPS KAKENHI Grant Number 25840160. J. Urabe, W. Makino, and O. Miura were supported by the “Tohoku Ecosystem-Associated Marine Science”, MEXT. C. Keogh was supported by a NSF GRF and a JSPS Short Term Fellowship. J. Onufrey was a 2014 semi-finalist for the National Intel Talent Search Program mentored by A. Blakeslee, while she attended Wantagh High School (Wantagh, NY). We thank Long Island University-Post for their support and facilities during that mentorship. Finally, we thank two anonymous reviewers for their helpful and constructive suggestions that greatly improved our manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All international, national, and institutional guidelines for the care and use of the marine invertebrate animals in this study were followed. There were no human participants in this study.
Additional information
Communicated by T.Reusch.
The original version of this article was revised: One of the author name “Yumi Kamakura” in the authorship was incorrectly published in this original version and the same is corrected here.
Reviewed by undisclosed experts.
An erratum to this article is available at http://dx.doi.org/10.1007/s00227-017-3117-x.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Blakeslee, A.M.H., Kamakura, Y., Onufrey, J. et al. Reconstructing the Invasion History of the Asian shorecrab, Hemigrapsus sanguineus (De Haan 1835) in the Western Atlantic. Mar Biol 164, 47 (2017). https://doi.org/10.1007/s00227-017-3069-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-017-3069-1