
Abstract
Wood cell walls when contacted with humid atmosphere or an aqueous solution containing electrolytes or polymers undergo the phenomenon of swelling. In this work, experimental data were produced to quantify the effects of the adsorption water and solutes, which were introduced in the material by equilibration with a solution used as osmotic reservoir. For this reason, different environmental setups have been developed, allowing the control of temperature, water chemical potential, and ionic strength during the sorption process. The aim of this paper is to describe three experimental setups, focused on different levels: at the nanometric scale, small-angle scattering at controlled humidity; at the mesoscopic scale, environmental scanning electron microscopy; and at the macroscopic scale, tensile stage involving immersion of samples in solutions. Applicability and efficiency of the three setups are described. Moreover, it was shown how the combination of the results obtained via the three methodologies can be compared to expectations from a general Equation of State (EOS approach), where wood swelling with water and salt solutions is presented as the dependence of the distance between adjacent cellulose fibrils on the osmotic pressure. The total pressure calculated takes into account chemical, colloidal and mechanical terms in the force balance of the wood cell wall.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wood is a heterogeneous and anisotropic nano-composite (Fratzl and Weinkamer 2007). At nanometric scale, it is organized into stiff crystalline cellulose fibrils (50%) of a typical thickness of 2.5 nm, and parallel to each other, with a typical spacing distance between 1 and 4 nm. Fibrils are embedded in a softer and more isotropic matrix (Altaner et al. 2006), mostly comprised of hemicelluloses (20–35%), lignin (15–30%) and water (Fengel and Wegener 2003). The nano-structure of cellulose and the distribution of the polymers of the matrix in the cellulose structure are not yet known in all cases, but are readily characterized by small-angle X-ray microtomography using compression wood (Fernandes et al. 2011). The gel between cellulose crystals is hygroscopic and swells while absorbing water (Elbaum et al. 2008). From a macroscopic point of view, wood cell walls can be described as mechanical model, based on the assumption that the matrix, if it was ideally free of any constraints, would tend to swell isotropically, but the presence of the stiff fibrils that act as rigid anisotropic elements induces significant elastic strain. This strain is strongly dependent on the winding angle of the cellulose microfibrils (Salmén 2004) that regulates tensile and compressive stress generation (Fratzl et al. 2009).
Understanding and rationalizing the process of water uptake by wood is a point of primary importance, as it implies remarkable dimensional and structural changes and variations of material’s mechanical properties. As a matter of fact, biological organisms optimize their interactions with water to obtain composition- and structure-dependent properties on which they rely to accomplish certain vital function (e.g., seed dispersal, spatial re-orientation and organ locomotion) (Fratzl and Barth 2009). These interactions include volume changes, mechanical properties changes and conformational changes and can induce passive hydro-actuated swelling or shrinkage, which the organism translates into movements or into stress generation thanks to their sensors and actuators (Elbaum et al. 2008). Considering forces in play during material deformation, the observed swelling can be understood as the balance resulting from different contributions, attractive and repulsive, acting simultaneously. Chemical and elastic mechanical terms have always been taken into account in wood chemistry, but the importance of the entropic and colloidal terms has been considered only recently by the work of Medronho et al. (2012), who showed that a good solvent to dissolve cellulose should overcome the low entropy gain in the balance of the free energy of mixing with the polymers (Medronho and Lindman 2014). This provides better chances to compare theoretical calculations with experimental observations, including the mesoscale (i.e., the colloidal scale) (Singh et al. 2015; Alves et al. 2016). All the terms are summed in the form of a constitutive master equation (Bertinetti et al. 2013) (Eq. 1):
This formulation takes into account microscopic chemical (i.e., the integral of all chemical interactions via bonds with the nearest neighbor), mesoscopic colloidal (i.e., electrostatics, van der Waals, solvation and entropic terms) as well as macroscopic mechanical contributions (Bertinetti et al. 2016). In the case of wood cell walls, van der Waals forces between crystals are negligible by two orders of magnitude. Repulsive hydration forces between cellulose crystals separated by a layer of water adsorbed of the crystals with restricted conformation, giving rise to an exponentially decaying hydration force between crystals, are taken into account (Parsegian and Zemb 2011). Configurational entropy of the matrix (Chang et al. 2012; Bertinetti et al. 2016) and chemical cross-linking between the cellulose fibrils and the matrix components (Whitney et al. 1998) are combined with the elastic energy generated by the material’s anisotropy (Fratzl et al. 2009). This approach is consistent with the multi-scale organization of natural materials into building blocks, whose components are assembled at different hierarchical levels to form composites able to obtain specific structural and mechanical properties (Weinkamer and Fratzl 2011). Moreover, it leads to the formulation of a general Equation of State (i.e, the relation between osmotic pressure and distance between adjacent cellulose crystals) with only a minimal number of parameters, all measurable independently, as already known for other tissues [see the case of multi-lamellar vesicles (LeNeveu et al. 1977), DNA (Podgornik et al. 1977) as well as generic “meat” (Poulanne and Halonen 2010)]. With respect to the general sorption models (BET, GAB, Dent, Pierce, etc.) (Volkova et al. 2012), based on experimental parametric sorption isotherms, an Equation of State has the peculiarity of taking into account structural- and compositional-specific information of the material to predict water sorption.
At the industrial scale, wood is mostly used without any impregnation (Rowell 2014). Nevertheless, the development of wood-processing technologies, where wood material is treated with several kinds of chemicals to induce changes in the chemical nature that increase its properties, led to significant results in increasing wood properties, in terms of dimensional stability, chemical and biological resistance (preservatives), durability (fire retardants), etc. These processes involve treatments with specific reactants (Kazi et al. 1998). Wood modification implies the formation of covalent bonds between the polymeric constituents of the wood cell walls and reactants, including epoxides, isocyanates, anhydrides and diols (Mathias et al. 1991). The most common processes involve either reactions of furfurylation or acetylation (Mantanis 2017), whose primary effect is a considerable increase in dimensional stability, with a sensitive decrease in the degrading effects of weathering with respect to the untreated wood, due to the substitution of the hydrophilic hydroxyl groups with hydrophobic side groups (Beckers et al. 1998).
Historically, swelling of cellulose induced by various salts has been studied for a long time, and salts swelling power was for the first time related to the degree of hydration of their cations by Von Weimarn in 1912 (Rahim and Huque 1966). Later studies that started to classify salts effect according to the lyotropic series were performed on cellulose (Heymann and McKillop 1939) and its derivatives, such as methylcellulose (Heymann et al. 1937). Within this framework, the theoretical knowledge regarding wood swelling with water has been later extended to the case of swelling after impregnation of wood with solutions containing electrolytes. By means of TEM micrographs, AgNO3 has been proved to impregnate wood at the microfibrillar level and increase the swelling by softening the cell walls (Wallström and Lindberg 2000).
Several salts have recently been experimentally investigated (Barbetta et al. 2017b), exploring the Hofmeister series, which defines ions as structure making or structure breaking, according to the strength of their interactions with water (Baer and Mundy 2013). A particular custom-built tensile stage allowed the measurement of dimensional changes, in terms of axial strain, occurring in the wood material fully immersed in water and in different salt solutions, while kept in iso-stress and iso-osmolar conditions. Results showed that all salts induce a positive swelling and that the maximum swelling can be obtained by using a salt composed of a chaotropic ion combined with a kosmotropic one (e.g, NaI, CaBr2). In contact initially with water then later with concentrated salt solutions, wood materials undergo two different processes: first water flows out because of the difference in osmotic pressure between the external solution, but later ions diffuse and adsorb within the wood cell walls, inducing an important re-swelling. In the case of so-called antagonistic salts, i.e., chaotropic anion associated with kosmotropic cation, according to the definition given by Onuki et al. (2011), the chaotropic species easily loses the first hydration shell. As a consequence, they appear as “ejected” from bulk water and adsorb on the hemicelluloses structure. This creates a net electrical field that drives fully hydrated kosmotropes within the matrix + fibrils structure and causes swelling. In terms of the Equation of State, a colloidal electrostatic term due to the presence of salts can be added to the force balance.
The aim of the present work is to combine complementary multi-scale techniques to compare swelling observed at different scales for compression wood and to compare those to the generic Equation of State for impregnated and non-impregnated spruce compression wood, in which swelling is presented in the form of variation of the distance between adjacent crystalline cellulose fibrils as a function of the osmotic pressure. For this purpose, environmental setups allowing the control of several parameters, such as temperature, water chemical potential (experimentally: relative humidity RH), and ionic strength of the solution used, were developed and tested in order to be able to follow the changes occurring in the material at different scales, taking advantage of three different techniques, whose applicability, experimental protocols, advantages and disadvantages of these techniques are described in the present work:
-
at the nanometric scale, small-angle X-ray scattering (SAXS) allows the evaluation of absolute distance (nm) between adjacent cellulose fibrils of samples pre-treated with different salt solutions and in conditions of maximum swelling (i.e, they are fully immersed in solution);
-
at the microscopic level, environmental electron scanning microscopy (ESEM) is used to measure longitudinal and transversal elongation of pre-treated samples, during dehydration and rehydration cycles at different temperatures;
-
at the macroscopically observable scale, via a mechanical tensile stage (TS) it is possible to monitor in situ relative dimensional changes, in terms of axial strain, during impregnation of the material with different salt solutions.
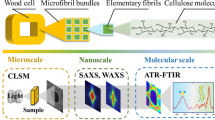
A schematic representation of the three techniques used at different scales, including a highlight on the sample dimensional changes analyzed during each experiment, and a sketch of the typical output graphs, is shown in Fig. 1.

Schematic representation of the three different techniques used, at the different scales: at the nm scale, SAXS (on top) quantifies the absolute center-to-center distance between adjacent cellulose crystals; in the µm range, ESEM (in the middle) quantifies materials’ reactions to humidity and temperature changes; at micrometer level, TS (bottom) follows in situ dimensional relative changes of samples during solution uptake
ESEM studies on spruce wood fibers were performed by the group of Burgert (Eder et al. 2008) with the aim of following in situ microtensile tests of fracture strength and behavior of various tissue types, in their hydrated state. Actually, one of the most relevant concerns of wood industry is the knowledge and optimization of the drying process, in terms of improved drying time, cost and quality (Elustondo et al. 2010). In this sense, the differential shrinkage due to material anisotropy and specific properties of its components (i.e, propensity to collapse and the propensity to induce drying stress) is studied. Observed differences showed the primary influence of cell geometry, material porosity and cell-wall thickness in structural stabilization of the material. Less drastic conditions were analyzed by Bernabei and Salvatici (2016), who studied swelling and shrinkage deformations of the wood material constituent elements (i.e., cell wall, cell lumen, fractures and parenchymatic rays) during heating, in a wide range of temperature (20–400 °C), where no cracking due to thermal treatment was detected. Moreover, determination of the dependence of porosity and shrinkage on drying, studied by the group of Perré (Redman et al. 2016), showed the effects of fibers geometry on the anisotropic swelling to explain collapse and structural degradation occurring during drying.
The investigation of the swelling of wood cell walls in contact with humid atmosphere and aqueous solutions containing electrolytes with the three devices mentioned above aims to obtain consistent results at the three scales, to be interpreted within the frame of a multi-scale approach. This can give an overall picture of the topic, including the analysis of dimensional changes at the supramolecular scale, in terms of absolute distances, upon water and solution uptake by wood (SAXS), study of the dynamics and thermodynamics aspects of impregnation (TS), and quantification of the different sensitivity of pre-treated material to environmental conditions (ESEM).
Finally, the knowledge of the experimental Equation of State can be further expanded to the case of materials impregnated with swelling agents (e.g, NaI).
Materials and methods
Slices from spruce (Picea abies) compression wood, with a thickness of 100 μm, were cut along the longitudinal direction of the tree, using a Leica RM2255 microtome, and were directly stored in a freezer (− 20 °C), so that their natural conditions were kept. Samples were unfrozen in a fridge at 4 °C the day before the beginning of the measurements. The choice of compression wood was made consistent with the previous works, because its microfibrillar angle (i.e., the spiral angle of the cellulose fibrils winding around the central lumen) can reach ca. 45°, and it is higher than normal wood (Gorisek and Torelli 1999). As the tensile stage measurements are related to the strain in the direction parallel to the fibrils orientations (see “Tensile stage (TS)” section), a higher angle implies a more sensitive response of the registered values to swelling. The measurement of the microfibrillar angle of the samples was performed via X-ray diffraction experiments, as described in the literature (Bertinetti et al. 2013, supporting information), with a Nanostar instrument (Bruker AXS). The scattered intensity was obtained from 2D patterns and plotted versus the azimuthal angle, and the center of the Gaussian curve representing the MFA distribution was taken as the reference value for the calculations.
Solutions were prepared with salts (supplied by Alfa Aesar) just before the experiments and at room temperature, by adding weighed quantities of salts to defined volumes of Milli-Q water. A concentration of 15.2 osm was chosen for all the solutions, consistent with the previous experiments (Barbetta et al. 2017b), in which the concentration of a saturated NaCl solution (15.2 osm), which produces 37.7 MPa osmotic pressure (OP), was chosen as reference, and the concentrations of the other solutions were chosen to produce the same OP, within the limit of the experimental error. For all the experiments, unfrozen dry samples were immersed in water overnight. In the case of the experiments with salt impregnation, wood samples were immersed in the electrolytic solution for a period of ca. 2 h and then washed with water to avoid crystallization and deposition of the salt on the sample. In the case of the measurements carried out with TS, the immersion and washing steps were performed in situ while measuring the strain. This allowed us to evaluate the rate of irreversibility of the process.
SAXS
Small-angle X-ray scattering experiments were performed at the µ-spot beam line at BESSY II synchrotron radiation facilities (Berlin). The setup consists of a 7T wavelength shifter as source for the primary beam, a toroidal mirror to focus the beam horizontally and vertically, a double-crystal monochromator equipped with three pairs of crystals, zone plates and mirrors as second and third focusing options, slits, filters and pinholes at different positions, the experimental hutch with goniometer, sample scanner and a 2D MarMosaic 225 detector to collect the scattered signal. The wavelength used was 0.689 Å (E = 18 keV). Beam size on sample was set by a 50-µm pinhole. Sample–detector distance was set to 840 mm and calibrated with a crystalline silver behenate standard. A typical measuring time of 120 s for each point was chosen, and several measures of the same sample were performed in different spots. Pre-treated compression wood slices were measured in fully hydrated conditions by means of a custom-built metallic frame (Fig. 2), which allows them to be immersed during the measurement in a water layer (0.2 mm), sufficiently thin to avoid any noise in the signal.

Custom-built frame to measure samples in fully hydrated conditions, while immersed in a 0.2-mm water layer. λ = 0.689 Å, D = 840 mm
Data analysis was performed according to the studies of Jakob et al. (1996): in their work, they proved that scattering intensities derived from cellulose fibrils of the cell-wall layer have cylindrical symmetry around the longitudinal axis of the stem (Fig. 3). Therefore, spherical coordinates (q, φ) should be used to integrate the scattering intensity (Eq. 2)
in which the solid angle Ω, decomposed into the two Eulerian angles φ and χ, is independent from χ, the angle measured in the plane perpendicular to the symmetry axis.

From the collected pattern (a) to the integrated intensity signal fitted with Bessel functions (b): pattern as it appears after application of cylindrical symmetry (b) and after selective masking to isolate sample signal (c)
The mathematical derivation of the spherical average of the intensity, \(\tilde{I}\left( q \right)\), as described in the literature, is reported in Eq. 3:
\(\tilde{I}(q)\) description can be simplified as follows (Eq. 4):
where bgr indicates the background, R is taken as 1.25 nm for all the specimen, and J 1 is the Bessel function of the first kind. The average distance between cellulose crystals was then obtained by subtracting this function from the experimental data and by fitting the obtained curve with a Gaussian function close to its maximum (Barbetta et al. 2017b).
ESEM
In situ wetting and drying experiments were performed using a FEI Quanta 200 Environmental scanning electron microscope (Eindhoven, Netherlands), equipped with a field electron gun (sketched in Fig. 4). Specific attention was paid to the sample preparation, the use of a Peltier stage, the choice of the experimental conditions and the image processing.

Schematic representation of ESEM chamber: sample is fixed via a copper-holding system to a Peltier stage, which allows to control the temperature. A tunable water vapor source allows the user to control the humidity in the chamber
Several 2–5 mm × 2–5 mm large pieces of wood were cut from the stored slices. Samples were immersed for a period of 2 h in the salt solutions and then washed for at least 30 min in Milli-Q water at 4 °C.
Once ready, the sample was inserted in between the aluminum sample holder and a copper flat washer to avoid twisting and deformation during the hydration and dehydration experiments. The sample holder is put on the Peltier stage that allows the user to regulate the sample temperature with an acceptable precision (± 0.1 °C) in the temperature range from − 10 to + 60 °C. The maximum water vapor that can be incorporated in the ESEM chamber reaches 2400 Pa. According to the liquid/gas water equilibrium, a 100% relative humidity value can be reached in the temperature range from 0 to 20 °C.
A key point for the success of the experiments is the control of the initial pumping sequence. A purge cycle was necessary before each analysis, allowing the air initially present in the ESEM chamber to be replaced by water vapor, in order to obtain a pure water vapor atmosphere. Part of the volume of the air in the ESEM chamber was pumped out without dehydrating the sample. (RH was kept close to 100%.) The gas was then replaced by water vapor. Successive purges led to 99.5% water vapor in the chamber.
As the studied material turned out to be very sensitive to electron beam damages, particular attention was put to the choice of electron beam conditions. Experiments were performed with various accelerating voltages, between 5 and 30 keV, beam intensities and frame times (generally: 3 s), depending on the pressure of the chamber, to limit the sample exposure to the electron beam, and to reduce electron beam damages while recording images with a high signal–noise ratio (Turkulin et al. 2004a, b).
Once the sample was introduced into the ESEM chamber under wet conditions (99.5% RH) at T = 5 °C, the temperature was kept constant. Three different zones of each sample were observed: low magnification images (image size 1024 × 880 pixels) were recorded every 2 min on the first zone, for a total time of 16 min (Position 1). One low magnification image was recorded at t = 12 min on the second zone (1024 × 880 pixels, Position 2), and another low magnification image was recorded at t = 13 min on the third zone (4096 × 3520 pixels, Position 3). Then, water vapor pressure in the chamber was tuned to decrease RH to 90%, and the image sequences were recorded again. This protocol was repeated at 76, 50 and 25% RH. Afterward, the ramp was repeated in the opposite direction. After all the images had been recorded at T = 5 °C, the same cycle was performed at 10, 15 and 20 °C.
Image processing was performed using Fiji software (Schindelin et al. 2012): at first, stack alignment was processed, and a semiautomatic tracking of two clear details on the image series was performed using the TrackMate plug-in implemented in the software. From the extracted coordinate dataset, the distances between the two details were calculated for the image series (Fig. 5). In the case of Position 1, sample overexposure led to remarkable beam damaging effects, while problems with analysis of Position 3 concerned an overproducing of tracking points by the plug-in due to the high resolution. Therefore, most reliable results were obtained with the analysis of Position 2.

Collected images for a wood sample after impregnation cycle with NaI, at two different scales: on the left (a), ×33 magnification gives an overall view of the central zone of the sample, from which the three described positions were chosen; on the right (b), ×500 magnification, used for sample analysis. Two different points, which could be recognized for the whole duration of the experiment, were chosen as references to calculate sample’s dimensional changes during temperature and humidity cycles
Tensile stage (TS)
In situ length changes of the sample during impregnation with different salt solutions were performed via a custom-built tensile stage setup (sketched in Fig. 6), which has already been described in the literature (Barbetta et al. 2017b).

In the tensile stage, the sample is fixed in between two clamps and fully immersed in solution. Main components of the setup are: a load cell (in bold on the left) to control and measure axial tensile forces, and a linear motor (on the right) to control and measure the strain
The setup is made of three main components: (1) the sample-holding system consists of two stainless-steel “L”-shaped arms, with clamps on the sides facing each other. The sample slices (typically: 100 or 150 μm thick, 2 cm long, 3 mm wide) can be fixed between the clamps by screwing the two plates of each clamp. A metal cuvette of 25 mL volume can fit in this stage, so that the sample is fully immersed in solution during the entire experiment. (2) A PI M-126.DG1 linear motor stage is connected to one of the two arms and allows it to be driven along one axis, with a maximum distance between the two clamps of 3.5 cm. (3) A 0.5-N load cell (Honeywell 31E Low) measures and controls the axial force along the same axis. The load cell was mounted on the fixed arm to avoid noise due to the movements.
A custom-written software allows the motor to drive the arm either to control the strain (motor control mode) or to regulate the force (force control mode). Values of motor position and force are logged by the software as a function of time.
Once clamped, samples were immersed in Milli-Q water and stretched by moving the motor until buckling was removed. This can be noticed by a net linear increase in the force registered by the load cell with increasing distances. Force was calibrated in this condition, and samples were then equilibrated overnight under 0 N force. In that time, an axial elongation of 3% of its length was observed. The length of the sample after this time was considered as the reference distance for the strain calculations. Measurements consisted in cyclical immersions of the sample in water and solutions, for periods of 2 h and for a maximum number of five cycles. Experiments were performed in iso-stress conditions (i.e, imposing a force of 0 N with the load cell), by recording the variation of the motor position as a response of the different salt solutions. To quantify the effects produced by each solution, it was necessary to work in iso-osmotic conditions: a 6.1-M NaCl solution, which produces a 37.7-MPa osmotic pressure, was taken as reference, as in the previous work by Barbetta et al. (2017b), and the NaI and NaBr solutions were prepared with the same osmolarity (15.2 Osm). For this, values of osmotic coefficients were taken from literature and fitted to obtain the required concentration. After the experiments were finished, sample strains were plotted as a function of the number of cycles, for each solution used.
Measure at macroscopic scale
The particularly modified tensile stage with humidity control environmental cell was used (Bertinetti et al. 2015). This cell allows the measurement of dimensional changes occurring in dry samples: the sealed chamber is kept at constant temperature of 25 °C via a Huber cooling bath circulation thermostat, while humidity was controlled with a Setaram WetSys humidity generator, working with a 200-mL/min flow, and measured with a Sensirion Sht75 digital humidity and temperature sensor, placed in the vicinity of the sample.
Results and discussion
Results
Figure 7 summarizes the impregnation and dehydration cycle for the case of the two samples, with and without impregnation with electrolytes. In this figure, cellulose crystals are represented as black hexagons, coated by a thin layer of matrix, shown in gray. At this point of the authors’ knowledge, it is not possible to evaluate single contributions of the individual components of the swelling matrix, which are therefore represented as indistinguishable. Chaotropic I− is sketched in red, while kosmotropic Na+ is sketched in green. Dry samples were immersed in water until equilibration (step A). Sample length at this state was taken as reference. Some of the samples were immersed in salt solution and then washed (step C1); a positive strain was registered for all samples after this step. The same dehydration process was then performed for both pre-treated (step C2) and non-pre-treated samples (B), and strain was registered.

Top view of a schematic representation of wood cell-wall material: hexagonal and parallel crystalline cellulose fibrils (black) surrounded by a thin layer of a matrix of hemicelluloses and lignin (gray). Dry samples were immersed in water until equilibration (A), and inter-crystalline distance was measured via SAXS technique. The strain measured in this condition was taken as the reference for strain calculation. Then, samples were either dehydrated (B) or further treated (C1) by impregnation with NaI solution (I− ions in red, Na+ ions in green) before dehydration (C2)
At first, preliminary considerations should be done to explain what the strains observed with the tensile stage and the ESEM measurements refer to. As already discussed, the results obtained from experiments performed with the tensile stage are evaluated by considering the swelling in terms of axial strain, εx. To convert this value into swelling between crystals, one should take into account that the fibrils are winding around the central lumen with a certain angle, the so-called microfibrillar angle (MFA) (Weinkamer and Fratzl 2011). In the case of the compression wood specimen used, this is around 45°. The peculiarity of experiments performed with ESEM is that data analysis allowed us to take into account strain in both transversal and in longitudinal directions, as the elongation was intended as distance in between recognizable points, and this could be separated into the two contributions εx, in parallel direction with respect to fiber orientation, and εy, in the perpendicular tangential direction. At first, the relationship between εx and εy at different relative humidity values is reported in Fig. 8.

Elongation in the two directions, parallel and perpendicular with respect to fiber orientation (see Fig. 5), measured at 5 °C with ESEM, is reported for the case of sample immersed in water (blue), NaBr solution (black) and NaI solution (red)
The trend, even at low temperatures, shows a linear correspondence in the cases of water and NaBr, a salt which does not induce remarkable swelling on wood (εx = 0.3% after several cycles of impregnation, as measured with tensile stage). On the other hand, the fitted curve for the case of NaI (εx = 3% with tensile stage) is based on a nonlinear exponential growth curve, and the strain on the transversal direction εy leads to values more than 10% higher than the longitudinal ones at low humidity, implying the fact that the microfibrillar angle increases with swelling, as reported in the literature. It is also pointed out that fibers might detach, and eventually twist, during the temperature and relative humidity cycles. This affects the measurement of the εy and might lead to an overestimation of these values. For this reason, and in order to compare the results with the ones obtained via tensile stage experiments, focus was put on the analysis of εx.
In the ESEM experiment, strain was calculated by taking the initial point (wet condition) as reference. This means that it was not possible to report the results in a way in which they were comparable for the different salts without the support of other techniques. This experimental difficulty was solved by taking the maximum swelling measured with TS as starting point for data evaluation (raw data are shown in Fig. 9a). For all the three samples, a remarkable shrinkage is registered with decreasing relative humidity. Samples pre-treated with NaI, inducing high swelling, undergo slightly higher-dimensional changes (εx(100 %RH→25 %RH) = − 3.4 ± 0.6%) if compared to the samples that were simply immersed in water (εx(100 %RH→25 %RH) = − 2.5 ± 0.5%) and the ones pre-treated with NaBr, whose impregnation induces negligible swelling (εx(100 %RH→25 %RH) = − 3.1 ± 0.4%).

Strain results as a function of relative humidity measured at 20 °C with ESEM (a) and at 25 °C with TS (b). Samples were untreated (blue), or pre-treated with NaBr solution (black) and NaI solution (red)
Results obtained for the untreated sample were compared with the ones obtained via the modified tensile stage (Fig. 9b), in which untreated samples, previously immersed in water (step A in Fig. 7), were clamped into an isolated chamber, with the possibility of controlling the relative humidity thanks to a humidity generator. In this case, compression wood foils, of the same thickness as the ones used in the described setups, undergo a strain εx of ca. 6.7 ± 0.4% when environmental humidity is driven from dry to water-saturated vapor. Data comparison shows a sensitive difference of ca. 2.5% strain between the results obtained with the ESEM (∆εx = 2.5% ca.) and TS (∆εx = 5% ca.) for the step between 100% RH and 25% RH.
This discrepancy may have different origins. At first, image deformation as a function of the acceleration voltage leads to an error of ca. 0.5%. Moreover, it should be considered what can be called an “individual effect”: data analysis of tensile stage experiments takes into account the final effects of all the fibers of the sample and therefore gives an averaged result of their strain, while on the other hand ESEM analysis of εx takes into account one single fiber per time. In this sense, fibers behavior could vary from case to case. It is pointed out that, despite this fact, there is no evidence of registered individual values of εx (TS) higher than εx (ESEM): this ensured that the difference in the result is not due to an incorrect choice of representative samples. A third aspect that should be considered is a temperature difference of ca. 5 °C between the operative conditions in the ESEM-controlled chamber (20 °C) and the environmental temperature of the fume hood, where the TS experiments were performed (ca. 25 °C). Figure 10 shows trends for the case of the three samples analyzed via microscopy, as a function of the chamber temperature.

Temperature-dependent sample shrinkage measured with ESEM at constant 25% relative humidity, for samples immersed in water (blue), NaBr solution (black) and NaI solution (red). The blue dashed line represents the curve of water uptake predicted by the theoretical model
In all cases, a slight temperature-dependent decrease in the swelling with decreasing relative humidity was observed. For the case of the non-pre-treated sample, strain varies by 0.6% over a temperature range of 15 °C. This could be attributed to an increase in the configuration entropy of the matrix constituted by water, hemicelluloses and lignin, as confirmed by implementing the theoretical model of Bertinetti et al. (2016) to predict water uptake, obtained by writing the master EOS in the form described by Eq. 1 with calculations at different temperatures. Derivation is computed using as input parameter a chemical cross-link enthalpy per contact point of 2.5 K B T in water. In the model, hydration force and the entropy of mixing of the matrix are the two dominant contributions to swelling. The mixing entropy, which depends on temperature, is evaluated assuming that polymers have infinite chain, according to the Flory expression for the partial entropy of mixing, and assuming for the interaction parameter χ at low volume fraction a value of 0.5 (Bertinetti et al. 2016). Theoretical and experimental results show a negligible dependence of the strain on temperature. Again, it can be assumed that the temperature difference between the two different setups contributes to the difference in strain, but not even this aspect itself could explain all the observed difference.
The most delicate step in the ESEM analysis concerns the sample-holding system: specimens’ displacement and twisting are limited by the presence of the copper ring, but not avoided at all. On the other hand, a too strong holding (e.g., sticking) would inhibit sample swelling. Thanks to the automatic alignment plug-in from Fiji software, one can overcome displacement, but eventual partial sample bending and consequent detaching from the stage could not be controlled, and not even evaluated, as the image focus, which could give indications of bending, must be tuned for each step according to temperature and relative humidity conditions. In this regard, it was calculated that a lifting with an angle of 10° would imply a loss of 1.5% on the measurement of εx, while an angle of 12.5° gives an error of − 2.5% to be added to the deformation. It is therefore reasonable that this small and undetectable movement of the samples in the Peltier stage could be the main source of error. In this sense, at the current state of the art, experiments performed with the tensile stage in controlled humidity conditions could give more reliable results.
Vapor pressure paradox
The term “vapor pressure paradox” refers to a particularity of water uptake by phospholipids (Rand and Parsegian 1989): lipid samples in a 100% humidity water vapor atmosphere do not take up the same amount of water as when they are in direct contact with liquid water, but less (Jendrasiak and Mendible 1976). Therefore, the water layer between a bilayer is thicker in the second case (Tristram-Nagle et al. 1998). This is a consequence of the fact that hydrating from the vapor creates an interface different from the case of samples immersed in solution (Podgornik and Parsegian 1997): this imposes an extra surface tension that inhibits fluctuations responsible for a repulsive entropic force, which increases spacing.
In the case of wood, wetting tests were performed with the tensile stage on dry compression wood samples: de-frozen slices were equilibrated overnight in iso-stress mode in liquid water, under constant 0 N force condition. During that time, an increase in the axial strain εx of ca. 3% was registered. This value appears significantly lower than the ones measured with increasing and decreasing humidity, showing an opposite behavior with respect to the case of vapor pressure paradox.
Experimental Equation of State
Raw data analyzed in the previous section can be further elaborated and implemented in the form of an experimental Equation of State (EOS). The EOS quantifies the relation occurring between the osmotic pressure OP and the center-to-center distance between adjacent cellulose crystals (d). The points of maximum swelling of the EOS curve (i.e., at 100% RH, and expressed in terms of absolute distances) were calculated via small-angle X-ray scattering experiments in fully hydrated conditions, as described in “SAXS” section, for the sample pre-treated with NaI and for the one simply immersed in water, which was used as a reference. Data analysis gave values of 4.25 and 4.04 nm, respectively. The other humidity-dependent points of the curve were obtained from ESEM results. As these results were expressed in terms of percentage of strain with respect to the starting point of the experiment itself, in order to correlate SAXS and ESEM results and therefore relate the point in the EOS at 100% humidity to the others, the strain εx measured with ESEM must be at first re-scaled into an inter-crystalline strain, εD. This was possible considering that fibers are oriented with an angle of 45°, as typical for spruce compression wood (Barbetta et al. 2017b). From that, values of d (expressed in nm) at each humidity step can be obtained by calculations from εD. It must be pointed out that for the case of the sample pre-treated with a salt, the microfibrillar angle is also slightly depending on swelling (as reported in Fig. 8).
Results of data elaboration, reported in Fig. 11, qualitatively show the higher sensitivity to humidity changes of the sample treated with the swelling agent, NaI (red points).

Compression wood Equation of State for samples immersed in water (blue) and impregnated with NaI solution, reported as osmotic pressure as a function of inter-crystalline distance. Point of maximum swelling was measured with SAXS experiment in fully hydrated conditions. Empty points refer to humidity-controlled TS measurements, the filled ones to ESEM. The shaded areas represent the error committed with ESEM measurement, due to uncontrollable bending of the sample, to a maximum of 2.5%, indicated by the lines. Black line refers to theoretical sorption behavior, in its lower limit where all the contributions opposing to swelling are multiplied by a factor of 1.5 and the ones favoring swelling divided by the same factor
For the case of untreated samples (i.e., wood simply immersed in water), experimental results are presented, obtained via desorption ESEM measurement (blue-filled points) and with the humidity-controlled TS described with respect to Fig. 9b (blue empty points). In both red and blue curves, the shaded areas from the points, leading to the continuous lines of the same colors, represent the incertitude due to the ESEM experimental setup problems. In particular, the continuous lines correspond to a 2.5% uncontrollable error.
Results can be compared with the calculated results obtained from modeling (Bertinetti et al. 2016; Barbetta et al. 2017a). The black line represents the theoretically predicted sorption behavior at its lower limit, which was calculated summing up all the contributions in the force balance, and multiplying all the attractive terms by a factor of 1.5 and dividing all the repulsive ones by 1.5. The limits take into account possible inaccuracies and variability of the literature data used in the model, and the fact that the accuracy of the model itself could be further refined, by considering the interactions between the matrix components, which is neglected at the current state, as the matrix is considered as an homogeneous medium, and by taking into account the topology of the lignin network. The force balance included the enthalpy of chemical binding between cellulose fibrils and the matrix, the hydration repelling cellulose crystals in the water medium, the configurational entropy of the matrix, van der Waals force between crystals, and the elastic energy generated by anisotropic swelling. Further implementations of the model could include an electrostatic term in the calculation that takes into account the contribution to swelling given by the presence of electrolytes according to their nature and concentration.
As shown in the figure, a reasonable consistency between the experimental and the calculated curve can be observed for the case of untreated wood.
An alternative way to present the same results, more often used in materials science, is in the form of a sorption isotherm, in which material’s moisture content is reported as a function of the relative humidity. The conversion is made considering the geometrical model, which is described in Fig. 7, and the correlation between the inter-crystalline distance and the moisture content is given in Supplementary information. Results reported in the form of MC vs. RH for the case of the untreated sample via TS in controlled humidity conditions (Fig. 9b) are shown in Supplementary information. The results look consistent, within the limits of the experimental errors, with respect to recent literature data concerning spruce sorption analysis (Hill et al. 2009; Murata et al. 2013).
Conclusion
Figure 12 resumes the humidity-dependent axial strain changes registered during a dehydration experiment for spruce compression wood foil, with both a macroscopic tensile stage device, and by means of ESEM, a technique giving information at the microscopic scale. In this work, it was attempted to establish an experimental protocol, consisting of a multi-scale combination of different techniques, to define the response of wood to environmental condition changes, in terms of an experimental Equation of State. In particular, the goal of testing different experimental procedures was to extend the knowledge concerning wood impregnation with water or solution containing electrolytes, and the subsequent swelling, in humid or dry atmosphere, at different temperatures, or in contact with water or aqueous solutions containing salts. Result discrepancies were explored and discussed.

Compression wood Equation of State for samples immersed in water, reported as osmotic pressure as a function of axial strain. Empty points refer to humidity-controlled TS measurements, the filled ones to ESEM. The shaded areas represent the error committed with ESEM measurement, due to uncontrollable bending of the sample, to a maximum of 2.5%, indicated by the lines
It was shown how SAXS data from synchrotron measurements of pre-treated samples immersed in solution allow the evaluation of the absolute distance between cellulose crystals in fully hydrated solutions (nanometric scale). At the current state of present setups, this method was the only one that gives us the possibility to investigate the sample while immersed in solution in “static conditions” (i.e., without alterations of any environmental parameter). Spruce compression wood slices of a defined thickness of 100 μm were measured to explore the effects of salt impregnation, and registered values of inter-crystalline distance of 4.04 nm for the untreated sample and of 4.25 nm for the sample pre-treated with NaI. The analysis could be furthermore implemented as a function of osmotic pressure, both in “static” and in “dynamic” modes, via a closed chamber that allows the control of relative humidity.
ESEM techniques allowed us to observe the sample at microscopic scale, to quantify the sensitivity of differently pre-treated materials to environmental condition changes (humidity, temperature). Reported results show that ionic specific effect on swelling properties of wood, already described in a previous work, was qualitatively confirmed. The sample pre-treated with a swelling-inducing salt NaI showed a higher sensitivity to relative humidity changes (3.40% axial strain difference from 100% RH to 25% RH) compared to the one pre-treated with a non-swelling-inducing salt NaBr (3.13%) and to the untreated sample (2.45%). Temperature effects are limited to a maximum strain difference of 0.8% in a ∆T of 15 °C and might be due to an increase in the matrix configurational entropy. To make results more reliable with the state-of-the-art data, the sample-holding system should be implemented to avoid uncontrollable errors due to moving and bending of the sample. For the untreated sample, a difference on the axial strain of ca. 2.5% was registered with respect to the results obtained via a custom-built tensile stage, designed to follow in situ relative swelling of the material during drying, which gave comparable results with respect to literature.
By means of tensile stage measurement, the relative swelling of the material during the water/solution sorption was also quantified, and it was shown that with respect to an untreated sample immersed in water, whose strain in wet conditions was taken as reference (i.e., εx = 0%), impregnation with NaI induces a sensitively higher axial strain εx of 3%.
At the current state of development, the techniques show good potential for applications in the field, but the setups could be furthermore improved in order to have more consistent results.
As a continuation of the previous works (Barbetta et al. 2017a, b), it was intended to extend the study on the ion-specific effects, based on the Hofmeister series, from the case of the analysis of the effects of impregnation on the material to the case of the response of already treated samples to changes in the environmental conditions. For this reason, results obtained with the different techniques for the untreated wood samples have been combined in the form of an experimental Equation of State that quantifies the link between osmotic pressure and wood swelling, reported in terms of distance between cellulose crystal, and compared to the one predicted from modeling. The resulting picture shows consistent trends for the two cases that converge at high humidity, while a tendency to diverge is detected at lower humidity points. Both ESEM and TS experimental setups, as explained, and model input parameters of the calculations could be furthermore implemented to obtain more accurate results, for example recent investigations of the individual forces contributing to swelling given by the single components of the swellable matrix (Barbetta et al. 2017a).
The complexity of wood in terms of composition and molecular and hierarchical structure has challenged scientists belonging to different communities for a long time. A comprehensive understanding of the tissues requires the description of the materials at multiple length scales and needs an underlying molecular model. With this contribution, a combination of experimental techniques and a physicochemical model is proposed, which allow for describing the changes at the nanometric/molecular levels accompanying wood swelling. Despite the results not being fully consistent at all the considered scales, it was shown how to reconcile different languages and conventions used in different communities to obtain a common description of the material.
References
Altaner C, Apperley DC, Jarvis MC (2006) Spatial relationships between polymers in Sitka spruce: proton spin-diffusion studies. Holzforschung 60:665–673
Alves L, Medronho B, Antunes FE, Lindman B (2016) Dissolution state of cellulose in aqueous system- 2 acidic solvents. Carbohydr Polym 20(151):707–715
Baer MD, Mundy CJ (2013) An ab initio approach to understanding the specific ion effect. Faraday Discuss 160:89–101
Barbetta A, Bertinetti L, Zemb T (2017a) Composition dependent Equation of State of cellulose based plant tissues in the presence of electrolytes. Colloids Surf A Physicochem Eng Asp 532:314–322. https://doi.org/10.1016/j.colsurfa.2017.04.075
Barbetta A, Fratzl P, Zemb T, Bertinetti L (2017b) Impregnation and swelling of wood with salts: ion specific kinetics and thermodynamics effects. Adv Mater Interfaces 4:1600437. https://doi.org/10.1002/admi.201600437
Beckers EPJ, de Meijer M, Militz H, Stevens M (1998) Performance of finishes on wood that is chemically modified by acetylation. J Coat Technol 70(878):59–67
Bernabei M, Salvatici MC (2016) In situ ESEM observations of spruce wood (Picea abies Karst.) during heat treatment. Wood Sci Technol 50:715–726
Bertinetti L, Fischer FD, Fratzl P (2013) Physicochemical basis for water-actuated movement and stress generation in nonliving plant tissues. Phys Rev Lett 11:238001
Bertinetti L, Masic A, Schuetz R, Barbetta A, Seidt B, Wagermaier W, Fratzl P (2015) Osmotically driven tensile stress in collagen-based mineralized tissues. J Mech Behav Biomed Mater 52:14–21
Bertinetti L, Fratzl P, Zemb T (2016) Chemical, colloidal and mechanical contributions to the state of water in wood cell walls. New J Phys 18:083048
Chang S, Quignard F, Di Renzo F, Clair B (2012) Solvent polarity and internal stresses control the swelling behavior of green wood during dehydration in organic solution. BioResources 7:2418–2430
Eder M, Stanzl-Tschegg S, Burgert I (2008) The fracture behaviour of single wood fibers is governed by geometrical constraints: in situ ESEM studies on three fiber types. Wood Sci Technol 42:679–689
Elbaum R, Gorb S, Fratzl P (2008) Structures in the cell wall that enable hygroscopic movement of wheat awns. J Struct Biol 164:101–107
Elustondo DM, Oliveira L, Avramidis S (2010) New methodology to optimize sorting in wood drying. Maderas Cienc Tecnol 12(2):79–91
Fengel D, Wegener G (2003) Wood–chemistry, ultrastructure, reactions. Verlag Kessel, Remagen, pp 26–59, 227–238, 296–315
Fernandes AN, Thomas LH, Altaner CM, Callow P, Firsyth VT, Apperley DC, Kennedy CK, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. PNAS 108(47):E1195–E1203
Fratzl F, Barth FG (2009) Biomaterial systems for mechanosensing and actuation. Nature 462:442–448
Fratzl P, Weinkamer R (2007) Nature’s hierarchical materials. Prog Mater Sci 52:1263–1334
Fratzl P, Elbaum R, Burgert I (2009) Cellulose fibrils direct plant organ movements. Faraday Discuss 139:275–282
Gorisek Z, Torelli N (1999) Microfibril angle in juvenile, adult and compression wood of spruce and silver fir. Phyton 39(3):129–132
Heymann E, McKillop GC (1939) Studies on the lyotropic series. III The adsorption of salts on cellulose. J Phys Chem 45(2):195–203
Heymann E, Bleakley HG, Docking AR (1937) Studies on the lyotropic series. I The adsorption of salts on methylcellulose. J Phys Chem 42(3):353–368
Hill CAS, Norton A, Newman G (2009) The water vapor sorption behavior of natural fibers. J Appl Polym Sci 112:1524–1537
Jakob HF, Tschegg SE, Fratzl P (1996) Hydration dependence of the wood-cell wall structure in Picea abies. A small-angle X-ray scattering study. Marcomolecules 29:8435–8440
Jendrasiak GL, Mendible JC (1976) The effect of the phase transition on the hydration and electrical conductivity of phospholipids. Biochim Biophys Acta 424:133–148
Kazi KMF, Jollez P, Chornet E (1998) Preimpregnation: an important step for biomass refining processes. Biomass Bioenergy 15(2):125–141
LeNeveu DM, Rand RP, Parsegian VA, Gingell D (1977) Measurement and modification of forces between lecithin bilayers. Biophys J 18:209–230
Mantanis GI (2017) Chemical modification of wood by acetylation or furfurylation: a review of the present scaled-up technologies. BioResources 12(2):4478–4489
Mathias LJ, Lee S, Wright JR, Warren SC (1991) Improvement of wood properties by impregnation with multifunctional monomers. J Appl Polym Sci 42:55–67
Medronho B, Lindman B (2014) Competing forces during cellulose dissolution: from solvents to mechanisms. COCIS 19(1):32–40
Medronho B, Romano A, Graça Miguel M, Stigsson L, Lindman B (2012) Rationalizing cellulose (in)solubility: reviewing basic physicochemical aspects and role of hydrophilic interactions. Cellulose 19:581–587
Murata K, Watanabe Y, Nakano T (2013) Effect of thermal treatment on fracture properties and adsorption properties of spruce wood. Materials 6:4186–4197
Onuki A, Okamoto R, Araki T (2011) Phase transitions in soft matter induced by selective solvation. Bull Chem Soc Jpn 84(6):569–587
Parsegian VA, Zemb T (2011) Hydration forces: observations, explanations, expectations, questions. COCIS 16:618–624
Podgornik R, Parsegian VA (1997) On a possible microscopic mechanism underlying the vapor pressure paradox. Biophys J 72:942–952
Podgornik R, Strey HH, Parsegian VA (1977) Colloidal DNA. Biophys J 18:209–230
Poulanne E, Halonen M (2010) Theoretical aspects of water-holding in meat. Meat Sci 86:151–165
Rahim KA, Huque MM (1966) Swelling of cellulose in salt solutions and its relation to the radii of ions. J Text I 57(8):T374–T376
Rand RP, Parsegian VA (1989) Hydration forces between phospholipid bilayers. Biochim Biophys Acta 988:351–376
Redman AL, Bailleres H, Turner I, Perré P (2016) Characterisation of wood-water relationships and transverse anatomy and their relationship to drying degrade. Wood Sci Technol 50:739–757
Rowell RM (2014) Acetylation of wood—a review. Int J Lignocellulosic Prod 1:1–27
Salmén L (2004) Micromechanical understanding of the cell-wall structure. Biologies 327:873–880
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saafeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682
Singh P, Duarte H, Alves L, Antunes F, Le Moigne N, Dormanns J, Duchemin B, Staiger MP, Medronho B (2015) From cellulose dissolution and regeneration to added value applications—synergism between molecular understanding and material development. Cellulose—fundamental aspects and current trends. InTech Chapter 1, pp 1–44
Tristram-Nagle S, Petrache HI, Suter RM, Nagle JF (1998) Effect of substrate roughness on D spacing supports theoretical resolution of vapor pressure paradox. Biophys J 74:1421–1427
Turkulin H, Holzer L, Richter K (2004a) Application of the ESEM technique in wood research. Part I. Optimization of imaging parameters and working conditions. Wood Fiber Sci 37(4):552–564
Turkulin H, Holzer L, Richter K (2004b) Application of the ESEM technique in wood research. Part II. Comparison of operational models. Wood Fiber Sci 37(4):565–573
Volkova N, Ibrahim V, Hatti-Kaul R, Wadsö L (2012) Water sorption isotherms of Kraft lignin and its composites. Carbohydr Polym 87:1817–1821
Wallström L, Lindberg KAH (2000) The diffusion, size and location of added silver grains in the cell walls of Swedish pine, Pinus sylvestris. Wood Sci Technol 34:403–415
Weinkamer R, Fratzl P (2011) Mechanical adaptation of biological materials—the examples of bone and wood. Mater Sci Eng 31:1161–1173
Whitney SEC, Brigham JE, Darke AH, Grant Reid JS, Gidley MJ (1998) Structural aspects of the interaction of mannan-based polysaccharides with bacterial cellulose. Carbohydr Res 307:299–309
Acknowledgements
The authors thank Klaus Bienert and Marco Bott, Max Planck Institute of Colloids and Interfaces, for technical support and development of the in situ mechanical testing devices, Stefan Siegel and Chenghao Li for their support during the X-ray measurements at the μ-spot beam line (BESSY II, Helmholtz–Zentrum für Materialien und Energie, Berlin, Germany). The excellence laboratory LABEX “Chemisyst” ANR 2011-05 is acknowledged for the French–German Ph.D. Stipendium. TZ acknowledges the Humboldt foundation for support. The financial support by DFG through Leibniz Award to PF is also acknowledged. This work was fully undertaken within the scope of the French–German agreement L.I.A. RECYCLING between CNRS/INC and MPG/MPIKG. The authors thank Helmuth Möhwald for numerous helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Barbetta, A., Bertinetti, L., Lautru, J. et al. Nano-, meso- and macro-swelling characterization of impregnated compression wood cell walls. Wood Sci Technol 52, 421–443 (2018). https://doi.org/10.1007/s00226-017-0978-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-017-0978-6