
Abstract
Bone is a highly dynamic tissue, and the constant actions of bone-forming and bone-resorbing cells are responsible for attaining peak bone mass, maintaining bone mass in the adults, and the subsequent bone loss with aging and menopause, as well as skeletal complications of diseases and drug side-effects. It is now accepted that the generation and activity of bone-forming osteoblasts and bone-resorbing osteoclasts is modulated by osteocytes, osteoblast-derived cells embedded in the bone matrix. The interaction among bone cells occurs through direct contact and via secreted molecules. In addition to the regulation of bone cell function, molecules released by these cells are also able to reach the circulation and have effects in other tissues and organs in healthy individuals. Moreover, bone cell products have also been associated with the establishment or progression of diseases, including cancer and muscle weakness. In this review, we will discuss the role of bone as an endocrine organ, and the effect of selected, osteoblast-, osteocyte-, and osteoclast-secreted molecules on other tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The shape and mass of the skeleton are dictated by the actions of osteoblasts and osteoclasts, which generation and function are regulated by osteocyte-produced signaling molecules [1, 2]. Dis-regulation on the pattern of gene expression and protein synthesis and secretion by these cells lead to abnormalities in bone acquisition during growth, in peak bone mass, and in the maintenance of bone mass with aging. Further, changes in bone mass and shape also alter bone strength.
Bone cell generation and function are regulated in a paracrine manner by cytokines produced locally by other cells in the bone marrow and by systemic factors such as hormones produced by endocrine glands [2]. Conversely, in addition to the bone intrinsic functions of osteoblasts, osteocytes, and osteoclasts, it is now recognized that bone cells are able to secrete molecules that have paracrine and endocrine functions, that is, can alter cellular responses of bone marrow cells and of cells in distant tissues and organs. Besides osteoclasts, osteoblasts, and osteocytes, other cells present in the bone microenvironment are known to exhibit paracrine roles and even to secrete molecules that can affect other organs. These cells include osteomacs, cells present in the blood vessels walls (for example, endothelial cells and smooth muscle cells), nerve cells, and cells present in the bone marrow, including bone cell progenitors, not described in this manuscript (for reviews on these factors, see [3,4,5,6]).
Bone-derived factors have been associated with several non-skeletal disorders, such as altered adipocytes and β-cell function, insulin metabolism regulation, fertility, food intake control, angiogenesis, immune system, and phosphate metabolism [7,8,9]. Interestingly, the RANKL/RANK system has been shown to participate in the immune response and in cancer initiation, progression, and metastasis (including bone metastasis), in particular, for breast and prostate cancer [10,11,12]. Bone cell-derived factors are also involved in other skeletal and non-skeletal pathologies, such as van Buchem disease, X-linked hypophosphatemia, autosomal hypophosphatemic rickets, McCune–Albright syndrome, and tumor-induced osteomalacia [13,14,15].
In this review, we will describe the molecules produced by osteoblasts, osteocytes, and osteoclasts that have been identified as endocrine factors, focusing on a few examples for each cell type.
Osteoblasts: Professional Secretory Cells
Osteoblasts, originated from progenitor cells present in the bone marrow, are the cells responsible for bone formation [2]. These cells are cuboidal with large nuclei located close to the basal membrane, enlarged Golgi apparatus, and extensive endoplasmic reticulum, consistent with their function as producers of extracellular matrix proteins and paracrine/endocrine factors.
The major function of osteoblasts is to produce the organic components to the bone extracellular matrix [2]. About 90% of the organic compartment is type I collagen, with smaller amounts of type III and V collagen. The remaining 10% are non-collagenous proteins (NCPs) with a vital role in regulating collagen formation and matrix mineralization, such as matrix extracellular phosphoglycoprotein (MEPE), pyrophosphate, matrix Gla protein (MGP), bone δ-carboxyglutamic acid-containing protein (osteocalcin, BGLAP), proteolipids, and bone alkaline phosphatase. Also, osteoblasts produce cell attachment proteins and small integrin-binding ligand with N‐linked glycoproteins (SIBLING proteins), including fibronectin, osteopontin, sialoprotein 1 (SSP1), osteonectin, and several other bone sialoproteins.
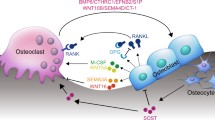
Osteoblasts secrete a wide range of different molecules with paracrine actions including receptor activator of nuclear factor kappa B ligand (RANKL/TNFSF11), monocyte colony-stimulating factor (M‐CSF‐1), osteoprotegerin (OPG/TNFRSF11B), Wnt gene family 5A (WNT5A), and WNT16, which regulate osteoclastogenesis [2, 16, 17]. RANKL and M‐CSF are molecules that bind cognate receptors (RANK and CSFR) on osteoclast precursors and facilitate their differentiation, whereas OPG functions as a RANKL decoy receptor, controlling RANK/RANKL interaction and osteoclast differentiation. Other direct interactions between osteoblasts and osteoclasts involve cell surface proteins, such as Semaphorin 3A (SEMA3A), Ephrin B4 (EPHB4), and FAS Ligand (FASL) [17]. Osteoblasts also secrete vascular endothelial growth factor (VEGF) that regulates skeletal development, osteoblast and osteoclast differentiation, bone repair, and angiogenesis [18, 19]. More recent studies described the axon guidance cue SLIT3 as a bone cell-derived angiogenic factor [20]. SLIT3 was described in 2018 as both an osteoblast-derived molecule required for fracture repair and bone healing [20] and as a osteoclast-derived factor coupling bone resorption and bone formation [21, 22]. However, in a side-by-side study it was shown that SLIT3 expression was negligible in osteoclasts compared to its expression in osteoblasts [23]. Further, deletion of Slit3 in cathepsin K-expressing osteoclasts did not alter the mouse skeletal or muscular phenotype, whereas SLIT3 global knockouts or mice with deletion of the gene in osterix-expressing cells (osteoblast precursors) exhibit vascular alterations [20, 23]. In addition to auto- and paracrine regulation of bone cell function, osteoblasts secrete molecules with endocrine action such as osteocalcin and lipocalin-2 (LCN2), which are the focus of this review, and fibroblast growth factor 23 (FGF23), reviewed elsewhere [7,8,9, 24].
Osteocalcin
Osteocalcin is the most abundant non-collagenous protein of the bone extracellular matrix produced by osteoblasts and its deletion in osteocalcin-deficient mice (Ocn−/− mice) results in a high bone mass phenotype due to an increase in bone formation without impairing bone resorption [25]. In addition, osteocalcin acts as an endocrine hormone on multiple organs, including adipose tissue, liver, muscle, pancreas, testis, and brain (Fig. 1) [7,8,9]. Before secretion by osteoblasts, osteocalcin is posttranslationally modified to γ-carboxylated osteocalcin (GlaOCN). The acidic environment generated by osteoclasts is responsible to decarboxylation of GlaOCN into undercarboxylated osteocalcin (GluOCN), decreasing its affinity for hydroxyapatite and helping osteocalcin reach the circulation [8]. GluOCN is the bioactive osteocalcin form with action as an endocrine hormone.

Communication of bone cells with other tissues. Schematic representation of the effects of osteoblasts, osteocytes, and osteoclasts in other cells and tissues, highlighting the molecules described in the text. Parts of the figure were drawn using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). RANKL, receptor activator of nuclear factor kappa B ligand; PGE2, prostaglandin E2, FGF23, fibroblast growth factor 23; OCN, osteocalcin; LCN2, lipocalin-2; PDGF-BB, platelet-derived growth factor BB; PTH, parathyroid hormone; NT, neurotransmitters
GluOCN functions as a hormone, regulating β-cell function and adipocyte gene expression [7, 26]. Thus, Ocn−/− mice are abnormally fat due to increased fat mass, adipocyte number, and serum triglyceride levels. In addition, Ocn−/− exhibit higher blood glucose and lower insulin serum levels compared to wild-type mice. Further, mice lacking osteocalcin specifically in osteoblasts show decreased β-cell proliferation, glucose intolerance, and insulin secretion and resistance, all actions that result from lack of activation of the GPRC6A receptor [27, 28]. Additional studies showed that the effect of administration of undercarboxylated osteocalcin on serum insulin levels are mediated by secretion of the glucagon-like peptide-1 (GLP-1) from the gut [26].
The role of osteocalcin on the regulation of glucose homeostasis was also put in evidence in in vitro studies showing that low concentrations of osteocalcin (0.03 to 0.3 ng/ml) regulate cell proliferation and insulin expression in β-cells and adipocytes. In addition, higher concentrations of osteocalcin (10 to 30 ng/ml) increased adiponectin, Pgc1, and Ucp1 expression in adipocytes. These in vitro observations were confirmed in 8-week-old wild-type mice, which received osteocalcin by delivering pumps. These studies showed that osteocalcin is able to regulate insulin secretion, insulin sensitivity, and fat mass in vivo [29].
In liver, osteocalcin was shown to prevent lipid accumulation by partially restoring insulin sensitivity and glucose tolerance in mice fed with a high-fat diet and receiving daily osteocalcin injections [29]. In addition, high-fat diet-fed mice showed increased liver expression of the gene encoding for TNFα, which was normalized after osteocalcin injections.
Osteocalcin also increased energy expenditure and enhanced uptake and utilization of glucose in skeletal muscle, protecting from diet-induced obesity [30]. Moreover, osteocalcin increased the expression of the Nrf1 and Mcad genes implicated in energy consumption and enhanced mitochondrial activity in muscle [27].
A recent study in animal models proposed that OCN attenuates the endothelial dysfunction in atherogenic abdominal aorta seen in controls, by a mechanism independent of the GPRC6A receptor [31]. But no positive or negative impact of osteocalcin was observed in healthy aortic rings. Further, the association between osteocalcin levels and vascular function in humans is inconsistent, as recently reviewed by Tacey et al. [32].
GPRC6A has been proposed as an osteocalcin receptor in Leydig cells, in which osteocalcin promotes testosterone synthesis and is required for full fertility in male mice [33]. This conclusion is based on studies showing that testis size and weight and the size of epididymis and seminal vesicles, as well as sperm count are significantly decreased in Ocn−/− mice. However, no changes in testosterone levels were observed in men after 2 years of zoledronic acid therapy, which results in substantial reduction in osteocalcin levels, arguing against a biologically significant role for osteocalcin in the regulation of testosterone in adult men [34].
Another potential target for osteocalcin actions has been proposed to be the brain [35]. Thus, the passive behavior observed in Ocn−/− mice suggested a role of osteocalcin in central nervous system. Consistent with this hypothesis, osteocalcin, which is able to cross the blood–brain barrier, inhibits the synthesis of monoamine neurotransmitters by reaching neurons of the brainstem, midbrain, and hippocampus. Further, osteocalcin intracerebroventricular infusions corrected the neurotransmitter deficiency, normalized tryptophan hydroxylase 2 (Tph2), tyrosine hydroxylase (Th), glutamate decarboxylase 1 and 2 (Gad1/Gad2) expression, corrected anxiety and depression, and partially improved the spatial learning in Ocn−/− mice.
Lipocalin-2
LCN2 is a ubiquitously expressed protein that has been associated with processes, such as cellular differentiation, inflammation, and cancer, and it is known to induce survival/apoptotic signals [36]. In bone, at the local level, osteoblast-derived LCN2 negatively alters bone development and reduces trabecular number and bone mass by affecting growth plate development and osteoblast differentiation [37]. Further, it increases bone resorption by enhancing osteoclast activity, as demonstrated in transgenic mice overexpressing LCN2 in bone. In addition, an endocrine function of LCN2 has been proposed, by which the molecule regulates energy homeostasis, increasing insulin secretion and sensitivity, and glucose tolerance [38]. LCN2 deletion in osteoblastic cells (Lcn2osb−/− mice), but not in adipocytes, results in decreased glucose tolerance, insulin sensitivity and secretion, and increase in food intake. On the other hand, Lcn2fat−/− mice lacking LCN2 in adipocytes did not exhibit differences in these parameters. LCN2 decreases food intake acting on hypothalamus (Fig. 1) by activating MC4R-dependent anorexigenic signaling function, as demonstrated following LCN2 administration to Mc4r−/− mice [39]. Osteocalcin expression and activity were not affected suggesting a direct effect of LCN2 on pancreatic islets. However, the role of the bone on body weight remains unclear. In addition, previous studies reported that LCN2 deficiency in global Lcn2−/− mice improved systemic insulin sensitivity but does not affect food intake [40, 41].
In addition to the effects described for osteoblastic LCN2 and consistent with an outside-in effect of LCN2, kidney-produced LCN2 increases FGF23 synthesis by osteocytes in response to inflammation and in chronic kidney disease (CKD) [42, 43].
Osteocytes: Sending Signals from within the Bone
Osteocytes originate from osteoblasts once they become completely surrounded by mineralized bone matrix [2, 44]. Because of their localization within the bone matrix and the extensive network of projections that allow the communication among osteocytes and with cells on the bone surface, it has been long thought that osteocytes are responsible of sensing mechanical signals, triggering bone formation and/or resorption, depending on the magnitude of the mechanical signals or whether bones are loaded or not [45]. It is now accepted that osteocytes are also the target of hormonal and pharmacological stimuli and that in turn they release factors that alter the formation and activity of osteoblasts and osteoclasts [44]. The main products of osteocytes affecting bone remodeling are Sost/sclerostin, a potent inhibitor of bone formation, and RANKL/OPG, pro- and anti-osteoclastogenic cytokines indispensable for normal bone resorption [45]. In this section, we will describe the components of the osteocyte secretome that have effects on distal tissues and organs.
Mechanical stimulation of osteocytic cells leads to the release of small molecules, namely prostaglandin E2 (PGE2), adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NAD+), and nitric oxide (NO) [45, 46]. In vivo studies showed that these molecules are involved in the anabolic response to mechanical loading. Interestingly, studies have shown that skeletal muscle from aged humans (79 ± 2-year-old) exhibit reduced expression of prostaglandin receptors, compared to younger individuals (25 ± 1-year-old), raising the possibility that reduced response to osteocyte-derived prostaglandins is involved in the ameliorated response to exercise in the elderly [47]. However, the effect of small molecules released by osteocytes as a consequence of mechanical stimulation in cells other than osteoblasts and osteoclasts has not been tested.
Osteocytes are the main source of the receptor activator of Nfκb ligand (RANKL) for osteoclast differentiation and bone resorption [48, 49]. In addition to its osteoclastogenic actions, RANKL has been shown to reduce skeletal muscle mass and strength in mice overexpressing human RANKL [50]. Thus, mature skeletal muscle cells express RANK, the RANKL receptor, and its activation regulates Ca2 + storage, function, and phenotype in denervated skeletal muscles [51]. In particular, skeletal muscle deletion of RANK using the muscle creatine kinase-Cre increases muscle fatigability, results in altered fiber-type composition in both control and denervated muscles and reduces the sarcoendoplasmic reticulum Ca2 + -ATPase (SERCA) activity in the fast twitched extensor digitorum longus, EDL, muscle. Further, muscle cells secrete the RANKL decoy receptor osteoprotegerin (OPG) in vitro, and administration of OPG-Fc, a long-lasting OPG derivate, improves skeletal muscle integrity and function and reduces skeletal muscle inflammatory cell infiltration in the mdx mouse model of muscular dystrophy [52]. In addition, elevated RANKL levels are associated with skeletal muscle atrophy in a model of non-metastatic ovarian cancer, whereas anti-RANKL treatment reduced myotube atrophy and the decrease in skeletal muscle mass and strength in this animal model [12].
More recently, a novel RANKL receptor, leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4, also known as GPR48) has been described [53, 54]. LGR4 competes with RANK for RANKL binding, suppressing downstream signaling [53]. As expected, deletion of LGR4 results in increased osteoclasts and bone resorption. Other studies showed that absence of LGR4 increases lipid oxidation-related gene expression and reduces glucose transporter type 4 (Glut4) levels thereby regulating energy expenditure in skeletal muscle [55]. Yet, whether LGR4 mediates, at least in part, the effects of RANKL in skeletal muscle is not known. It should be noted, however, that the source of RANKL was not determined in these studies and, even though it has been proposed the osteocytic RANKL is the main regulator of osteoclastogenesis [48, 49], the possibility that other cells are the source of skeletal muscle-affecting RANKL, such as T cells [56, 57], cannot be ruled out.
Another function ascribed to osteocytes is the regulation of mineral metabolism (recently reviewed in [58]). As discussed elsewhere in the special issue [24] fibroblast growth factor 23 (FGF23), a phosphaturic factor, is secreted by osteocytes in response to increased blood phosphate and 1,25(OH)2 vitamin D [59, 60]. First identified as the cause of autosomal recessive hypophosphatemic rickets [61], FGF23 activates FGF receptor/Klotho1 complex in the kidney, resulting in increased urinary phosphate excretion (phosphaturia) (Fig. 1).
Osteocytes also secrete several molecules involved in the activation and inhibition of the Wnt signaling pathway, with potential impact outside of bone cells [1]. These include Dikkopf1 (Dkk1) and soluble frizzled-related protein 1 (Sfrp1), both with systemic effects. However, whether osteocytic Dkk1 and/or Sfrp1 have a systemic role is not clear. On the other hand, sclerostin, the product of the Sost gene and a very potent inhibitor of Wnt signaling and bone formation, produced mainly by osteocytes, has been shown to have systemic effects (Fig. 1). First identified in individuals with sclerosteosis and van Buchem disease, in which its reduction/absence results in high bone mass [62], was originally described as an osteocyte-produced BMP antagonist [63], later found to be a potent inhibitor of the Wnt signaling pathway [64, 65]. Due to its potent bone formation inhibitory effects, sclerostin was targeted for anabolic treatments and neutralizing antibodies were generated [66]. Studies in animals demonstrated the effectiveness of the neutralizing antibodies in increasing bone mass and strength and preventing deleterious effects of surgical and pharmacological approaches, as well as of traumatic events [67,68,69,70,71]. The anti-sclerostin antibody was tested in humans and demonstrated high effectiveness [72,73,74] and was approved by the FDA in 2019 for the treatment of patients at high risk of bone fractures [75]. However, there is evidence of increased cardiovascular events in individuals treated with anti-sclerostin, suggesting sclerostin might have an effect on the vasculature. Indeed, evidence of reduced tissue deterioration has been shown in the aorta of mice-treated recombinant sclerostin or transgenic for Sost, whereas human samples from aortic aneurysm showed reduced sclerostin levels [76]. However, it is not clear whether osteocyte-derived versus locally produced sclerostin are the cause of this cardiovascular pathology.
Osteoclasts: Secretion Versus Release of Matrix-Embedded Signaling Molecules
Osteoclasts are unique cells with the ability of bone resorption and therefore essential to remodeling and maintain bone integrity. They have a short lifespan but are highly active cells. Located on trabecular and endosteal bone surfaces, as well as on the periosteal surface during bone development and growth, these cells derive from hematopoietic stem cells differentiated by stimulation of growth factors and cytokines such as M-CSF and RANKL secreted by osteoblast and osteocytes, among other cells [77]. Differentiation gives rise to immature mononuclear cells that merge to form large, multinucleated osteoclasts with high movement and migration capacity [78].
Mature osteoclasts are polarized cells with a basolateral domain in contact to vascular stream and a resorptive domain facing bone surface, where the ruffled border is located [2]. The structure of these cells matches their physiological functions; they are responsible of bone resorption, protein transport, and waste products processing, but also secretion of molecules and proteins. For the resorption process, the osteoclasts attachment to bone matrix is facilitated by podosomes action, anchoring the cells in the sealing zone and therefore giving place to the resorption pit, a compartment where protons, chloride ions, and matrix-degrading enzymes like cathepsin K and tartrate-resistant acid phosphatase (TRAP) are secreted through the ruffled border. Due to low pH, enzyme activation occurs, enhancing mineral and proteins release from bone matrix [79].
During bone resorption osteoclasts release from bone matrix different growth factors such as transforming growth factor β (TGF-β) and insulin-like growth type 1 (IGF-1), cytokines, and factors that stimulate bone formation [80, 81]. These released factors, such as bone morphogenetic protein 6 (BMP6), collagen triple helix repeat-containing 1 (CTHRC1), sphingosine-1-phosphate (S1P), Wnt family member 10B (WNT10B), semaphorin-4D (SEMA4D), and cardiotrophin-1 (CT-1) can carry out paracrine actions, while other factors can act on vascular tissue, including platelet-derived growth factor (PDGF).
Osteoclast-Released Factors with Paracrine Actions—Clastokines.
Studies demonstrated that, in addition to their traditional role in bone resorption and as cells able to release factors stored within the bone matrix [2], osteoclasts participate in the immune response. The localization and origin of the cells were a clue for this novel osteoclast role. Thus, on one hand osteoclasts are located at the same place where hematopoiesis occurs and on the other, similar to macrophages, they differentiate from cells of the myeloid lineage. These facts were key for investigators to find the role of osteoclasts on immunity: they suppress T cell response to the activators α-CD3/CD28. Osteoclast and T cell interaction could be cell contact independent, via the secretion of chemokines by osteoclasts. Osteoclasts were also shown to participate in the immunosuppressive microenvironment by releasing molecules, like death ligand 1 (PD-L1), CD200, and galectin-9 (Fig. 1) [82]. Another study has demonstrated that osteoclasts interact with T cells by mechanisms similar to those used by antigen-presenting cells [83]. Thus, osteoclast membranes express major histocompatibility complex (MHC) I and II molecules, which are needed for antigen presentation. Osteoclasts also express costimulatory molecules CD80, CD86, and CD40 and are able to release cytokines IL-10, TGF-β, IL-6, and TNF-α, required to activate T cells [84]. Recent studies and, in particular, genomic analyses have provided the molecular basis for the role of osteoclasts in the immune response, which is mediated by the secretion of cytokines, recently named “clastokines” [85]. Further, microarray transcriptomics revealed that mature human osteoclasts are transcriptionally closer to dendritic cells than to monocytes [82].
PDGF-BB
Platelet-derived growth factor (PDGF) is a factor secreted by preosteoclast involved in the maintenance of bone homeostasis and vessel formation during bone modeling and remodeling. Deletion of the Pdgfb gene in TRAP + preosteoclasts caused low cortical and trabecular bone mass and reduced angiogenesis in young healthy mice [86].
PDGFs are important serum factors that stimulate smooth muscle cell migration and proliferation and a role of PDGF-BB in the development of neointimal hyperplasia after joint injury and in atherosclerosis acting as vascular aging–inducing factor has been proposed [87]. In aged mice under metabolic stress, preosteoclasts exhibit increased PDGF-BB secretion and arterial stiffening and low bone mass compared to healthy mice, suggesting that high PDGF-BB negatively regulates osteoblasts differentiation and contributes to vascular aging in a paracrine manner [88].
Summary and Conclusion
Mounting evidence supports the role of bone cells not only in the regulation of bone mass and strength but also in controlling bone homeostasis. In this manuscript, we discussed how gene mutations in bone-released factors can affect other organs and cause a wide range of diseases. These interactions shed light to a side of the field that is not usually analyzed and highlights the need to pay attention to bone tissue physiology and functionalities affecting others biological process.
Although some studies are still controversial, osteocalcin, FGF23, and sclerostin stand out as bone-produced endocrine factors. Yet, further investigation is needed to tease out the contribution of bone release versus local production for some of those molecules. The challenge for the future will be to identify and find the specific functions of bone-released factors and understand the reciprocal interaction between bone and other organs in favor of a better understanding of skeletal and non-skeletal diseases.
References
Plotkin LI, Bruzzaniti A (2019) Molecular signaling in bone cells: Regulation of cell differentiation and survival. Adv Protein Chem Struct Biol. https://doi.org/10.1016/bs.apcsb.2019.01.002
Bellido T, Plotkin LI, Bruzzaniti A (2019) Bone Cells. In: Burr DB, Allen MR (eds) Basic and Applied Bone Biology, 2nd edn. Elsevier, London Wall
Weivoda MM, Bradley EW (2023) Macrophages and Bone Remodeling. J Bone Miner Res 38(3):359–369. https://doi.org/10.1002/jbmr.4773
Alvarez MB, Xu L, Childress PJ, Maupin KA, Mohamad SF, Chitteti BR, Himes E, Olivos DJ 3rd, Cheng YH, Conway SJ, Srour EF, Kacena MA (2018) Megakaryocyte and Osteoblast Interactions Modulate Bone Mass and Hematopoiesis. Stem Cells Dev 27(10):671–682. https://doi.org/10.1089/scd.2017.0178
Cheng X, Zhang M, Xie Y, Xu Y, Du R, Wu B, Guan Z, Wang W, Sun W, Xu T, Zhu S, Wu L, Wang X, Shi H, Sun B, Zhang Y (2023) Bone marrow-derived mesenchymal stem cells accelerate angiogenesis in pregnant experimentally induced deep venous thrombosis rat model via up-regulation of pro-angiogenic secretogranin II. Int Immunopharmacol 118:110025. https://doi.org/10.1016/j.intimp.2023.110025
Assefa F (2023) The role of sensory and sympathetic nerves in craniofacial bone regeneration. Neuropeptides 99:102328. https://doi.org/10.1016/j.npep.2023.102328
Ferron M, Lacombe J (2014) Regulation of energy metabolism by the skeleton: osteocalcin and beyond. Arch Biochem Biophys 561:137–146. https://doi.org/10.1016/j.abb.2014.05.022
Mizokami A, Kawakubo-Yasukochi T, Hirata M (2017) Osteocalcin and its endocrine functions. Biochem Pharmacol 132:1–8. https://doi.org/10.1016/j.bcp.2017.02.001
Oldknow KJ, MacRae VE, Farquharson C (2015) Endocrine role of bone: recent and emerging perspectives beyond osteocalcin. J Endocrinol 225(1):R1-19. https://doi.org/10.1530/JOE-14-0584
Casimiro S, Vilhais G, Gomes I, Costa L (2021) The Roadmap of RANKL/RANK Pathway in Cancer. Cells. https://doi.org/10.3390/cells10081978
Infante M, Fabi A, Cognetti F, Gorini S, Caprio M, Fabbri A (2019) RANKL/RANK/OPG system beyond bone remodeling: involvement in breast cancer and clinical perspectives. J Exp Clin Cancer Res 38(1):12. https://doi.org/10.1186/s13046-018-1001-2
Pin F, Jones AJ, Huot JR, Narasimhan A, Zimmers TA, Bonewald LF, Bonetto A (2022) RANKL Blockade Reduces Cachexia and Bone Loss Induced by Non-Metastatic Ovarian Cancer in Mice. J Bone Miner Res 37(3):381–396. https://doi.org/10.1002/jbmr.4480
Athonvarangkul D, Insogna KL (2021) New Therapies for Hypophosphatemia-Related to FGF23 Excess. Calcif Tissue Int 108(1):143–157. https://doi.org/10.1007/s00223-020-00705-3
Takashi Y, Kawanami D, Fukumoto S (2021) FGF23 and Hypophosphatemic Rickets/Osteomalacia. Curr Osteoporos Rep 19(6):669–675. https://doi.org/10.1007/s11914-021-00709-4
Costa AG, Bilezikian JP (2012) Sclerostin: therapeutic horizons based upon its actions. Curr Osteoporos Rep 10(1):64–72. https://doi.org/10.1007/s11914-011-0089-5
Han Y, You X, Xing W, Zhang Z, Zou W (2018) Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res 6:16. https://doi.org/10.1038/s41413-018-0019-6
Kim JM, Lin C, Stavre Z, Greenblatt MB, Shim JH (2020) Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells. https://doi.org/10.3390/cells9092073
Coultas L, Chawengsaksophak K, Rossant J (2005) Endothelial cells and VEGF in vascular development. Nature 438(7070):937–945. https://doi.org/10.1038/nature04479
Hu K, Olsen BR (2016) Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest 126(2):509–526. https://doi.org/10.1172/JCI82585
Xu R, Yallowitz A, Qin A, Wu Z, Shin DY, Kim JM, Debnath S, Ji G, Bostrom MP, Yang X, Zhang C, Dong H, Kermani P, Lalani S, Li N, Liu Y, Poulos MG, Wach A, Zhang Y, Inoue K, Di Lorenzo A, Zhao B, Butler JM, Shim JH, Glimcher LH, Greenblatt MB (2018) Targeting skeletal endothelium to ameliorate bone loss. Nat Med 24(6):823–833. https://doi.org/10.1038/s41591-018-0020-z
Kim BJ, Lee YS, Lee SY, Baek WY, Choi YJ, Moon SA, Lee SH, Kim JE, Chang EJ, Kim EY, Yoon J, Kim SW, Ryu SH, Lee SK, Lorenzo JA, Ahn SH, Kim H, Lee KU, Kim GS, Koh JM (2018) Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J Clin Invest 128(4):1429–1441. https://doi.org/10.1172/JCI91086
Koh JM (2018) Osteoclast-derived SLIT3 is a coupling factor linking bone resorption to bone formation. BMB Rep 51(6):263–264. https://doi.org/10.5483/bmbrep.2018.51.6.109
Li N, Inoue K, Sun J, Niu Y, Lalani S, Yallowitz A, Yang X, Zhang C, Shen R, Zhao B, Xu R, Greenblatt MB (2020) Osteoclasts are not a source of SLIT3. Bone Res 8:11. https://doi.org/10.1038/s41413-020-0086-3
Clinkenbeard EL (2023) Regulation of expression and secretion of Fibroblast Growth Factor 23. Calcif Tissue Int
Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, Bradley A, Karsenty G (1996) Increased bone formation in osteocalcin-deficient mice. Nature 382:448–452
Mizokami A, Yasutake Y, Gao J, Matsuda M, Takahashi I, Takeuchi H, Hirata M (2013) Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS ONE 8(2):e57375. https://doi.org/10.1371/journal.pone.0057375
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130(3):456–469. https://doi.org/10.1016/j.cell.2007.05.047
Pi M, Chen L, Huang MZ, Zhu W, Ringhofer B, Luo J, Christenson L, Li B, Zhang J, Jackson PD, Faber P, Brunden KR, Harrington JJ, Quarles LD (2008) GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS ONE 3(12):e3858. https://doi.org/10.1371/journal.pone.0003858
Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A 105(13):5266–5270. https://doi.org/10.1073/pnas.0711119105
Ferron M, McKee MD, Levine RL, Ducy P, Karsenty G (2012) Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone 50(2):568–575. https://doi.org/10.1016/j.bone.2011.04.017
Qaradakhi T, Gadanec LK, Tacey AB, Hare DL, Buxton BF, Apostolopoulos V, Levinger I, Zulli A (2019) The Effect of Recombinant Undercarboxylated Osteocalcin on Endothelial Dysfunction. Calcif Tissue Int 105(5):546–556. https://doi.org/10.1007/s00223-019-00600-6
Tacey A, Hayes A, Zulli A, Levinger I (2021) Osteocalcin and vascular function: is there a cross-talk? Mol Metab 49:101205. https://doi.org/10.1016/j.molmet.2021.101205
Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, Hermo L, Suarez S, Roth BL, Ducy P, Karsenty G (2011) Endocrine regulation of male fertility by the skeleton. Cell 144(5):796–809. https://doi.org/10.1016/j.cell.2011.02.004
Bolland MJ, Grey A, Horne AM, Reid IR (2013) Testosterone levels following decreases in serum osteocalcin. Calcif Tissue Int 93(2):133–136. https://doi.org/10.1007/s00223-013-9730-x
Oury F, Khrimian L, Denny CA, Gardin A, Chamouni A, Goeden N, Huang YY, Lee H, Srinivas P, Gao XB, Suyama S, Langer T, Mann JJ, Horvath TL, Bonnin A, Karsenty G (2013) Maternal and offspring pools of osteocalcin influence brain development and functions. Cell 155(1):228–241
Flower DR (1994) The lipocalin protein family: a role in cell regulation. FEBS Lett 354(1):7–11. https://doi.org/10.1016/0014-5793(94)01078-1
Costa D, Lazzarini E, Canciani B, Giuliani A, Spano R, Marozzi K, Manescu A, Cancedda R, Tavella S (2013) Altered bone development and turnover in transgenic mice over-expressing lipocalin-2 in bone. J Cell Physiol 228(11):2210–2221. https://doi.org/10.1002/jcp.24391
Jaberi SA, Cohen A, D’Souza C, Abdulrazzaq YM, Ojha S, Bastaki S, Adeghate EA (2021) Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed Pharmacother 142:112002. https://doi.org/10.1016/j.biopha.2021.112002
Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J, Lanzano P, Deng L, Leibel RL, Rubin M, Nicholas T, Chung W, Zeltser LM, Williams KW, Pessin JE, Kousteni S (2017) MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature. https://doi.org/10.1038/nature21697
Law IK, Xu A, Lam KS, Berger T, Mak TW, Vanhoutte PM, Liu JT, Sweeney G, Zhou M, Yang B, Wang Y (2010) Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes 59(4):872–882. https://doi.org/10.2337/db09-1541
Guo H, Jin D, Zhang Y, Wright W, Bazuine M, Brockman DA, Bernlohr DA, Chen X (2010) Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes 59(6):1376–1385. https://doi.org/10.2337/db09-1735
Courbon G, Francis C, Gerber C, Neuburg S, Wang X, Lynch E, Isakova T, Babitt JL, Wolf M, Martin A, David V (2021) Lipocalin 2 stimulates bone fibroblast growth factor 23 production in chronic kidney disease. Bone Res 9(1):35. https://doi.org/10.1038/s41413-021-00154-0
Courbon G, David V (2022) Lipocalin-2: a novel link between the injured kidney and the bone. Curr Opin Nephrol Hypertens 31(4):312–319. https://doi.org/10.1097/MNH.0000000000000804
Plotkin LI, Bellido T (2016) Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol 12(10):593–605. https://doi.org/10.1038/nrendo.2016.71
Dallas SL, Prideaux M, Bonewald LF (2013) The osteocyte: an endocrine cell … and more. Endocr Rev 34(5):658–690
Rochefort GY, Pallu S, Benhamou CL (2010) Osteocyte: the unrecognized side of bone tissue. Osteoporos Int. https://doi.org/10.1007/s00198-010-1194-5
Liu SZ, Jemiolo B, Lavin KM, Lester BE, Trappe SW (1985) Trappe TA (2016) Prostaglandin E2/cyclooxygenase pathway in human skeletal muscle: influence of muscle fiber type and age. J Appl Physiol 120(5):546–551. https://doi.org/10.1152/japplphysiol.00396.2015
Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, Bonewald L, Manolagas SC, O’Brien CA (2015) Osteocytes, not Osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 10(9):e0138189. https://doi.org/10.1371/journal.pone.0138189
Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O’Brien CA (2014) Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone 66:146–154. https://doi.org/10.1016/j.bone.2014.06.006
Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S (2019) RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest 129(8):3214–3223. https://doi.org/10.1172/JCI125915
Dufresne SS, Dumont NA, Boulanger-Piette A, Fajardo VA, Gamu D, Kake-Guena SA, David RO, Bouchard P, Lavergne E, Penninger JM, Pape PC, Tupling AR, Frenette J (2016) Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am J Physiol Cell Physiol 310(8):C663-672. https://doi.org/10.1152/ajpcell.00285.2015
Dufresne SS, Dumont NA, Bouchard P, Lavergne E, Penninger JM, Frenette J (2015) Osteoprotegerin protects against muscular dystrophy. Am J Pathol 185(4):920–926. https://doi.org/10.1016/j.ajpath.2015.01.006
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, Huang J, Dai W, Li C, Zheng C, Xu L, Chen H, Wang J, Li D, Siwko S, Penninger JM, Ning G, Xiao J, Liu M (2016) LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med 22(5):539–546. https://doi.org/10.1038/nm.4076
Filipowska J, Kondegowda NG, Leon-Rivera N, Dhawan S, Vasavada RC (2022) LGR4, a G Protein-coupled receptor with a systemic role: from development to metabolic regulation. Front Endocrinol (Lausanne) 13:867001. https://doi.org/10.3389/fendo.2022.867001
Sun Y, Hong J, Chen M, Ke Y, Zhao S, Liu W, Ma Q, Shi J, Zou Y, Ning T, Zhang Z, Liu R, Wang J, Ning G (2015) Ablation of Lgr4 enhances energy adaptation in skeletal muscle via activation of Ampk/Sirt1/Pgc1alpha pathway. Biochem Biophys Res Commun 464(2):396–400. https://doi.org/10.1016/j.bbrc.2015.06.066
Pacifici R (2016) T cells, osteoblasts, and osteocytes: interacting lineages key for the bone anabolic and catabolic activities of parathyroid hormone. Ann N Y Acad Sci 1364(1):11–24. https://doi.org/10.1111/nyas.12969
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408(6812):600–605
Robling AG, Bonewald LF (2020) The Osteocyte: New Insights. Annu Rev Physiol 82:485–506. https://doi.org/10.1146/annurev-physiol-021119-034332
Agoro R, Ni P, Noonan ML, White KE (2020) Osteocytic FGF23 and Its kidney function. Front Endocrinol (Lausanne) 11:592. https://doi.org/10.3389/fendo.2020.00592
Ubaidus S, Li M, Sultana S, de Freitas PH, Oda K, Maeda T, Takagi R, Amizuka N (2009) FGF23 is mainly synthesized by osteocytes in the regularly distributed osteocytic lacunar canalicular system established after physiological bone remodeling. J Electron Microsc (Tokyo) 58(6):381–392. https://doi.org/10.1093/jmicro/dfp032
White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26(3):345–348
Bezooijen RL, Dijke PT, Papapoulos SE, Lowik CWGM (2005) SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev 16(3):319–327
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22(23):6267–6276
Ott SM (2005) Sclerostin and Wnt signaling–the pathway to bone strength. J Clin Endocrinol Metab 90(12):6741–6743. https://doi.org/10.1210/jc.2005-2370
Bezooijen RLV, Papapoulos ES, Hamdy AN, Lowik WC (2008) SOST/Sclerostin: An Osteocyte-Derived Inhibitor of Bone Formation that Antagoizes Canonical Wnt signaling. In: Bilezikian PJ, Raisz LG, Martin TJ (eds) Principles of Bone Biology, 3rd edn. Elsevier’s Science & Technology, San Diego, pp 139–152
Hoeppner LH, Secreto FJ, Westendorf JJ (2009) Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets 13(4):485–496. https://doi.org/10.1517/14728220902841961
Eddleston A, Marenzana M, Moore AR, Stephens P, Muzylak M, Marshall D, Robinson MK (2009) A short treatment with an antibody to sclerostin can inhibit bone loss in an ongoing model of colitis. J Bone Miner Res 24(10):1662–1671
Marenzana M, Greenslade K, Eddleston A, Okoye R, Marshall D, Moore A, Robinson MK (2011) Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthritis Rheum 63(8):2385–2395
Li X, Ominsky MS, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Dwyer D, Grisanti M, Stolina M, Kostenuik PJ, Simonet WS, Paszty C, Ke HZ (2011) Increased bone formation and bone mass induced by sclerostin antibody Is not affected by pretreatment or cotreatment with alendronate in osteopenic. Ovariectomized Rats Endocrinol 152(9):3312–3322
Ominsky MS, Brown DL, Van G, Cordover D, Pacheco E, Frazier E, Cherepow L, Higgins-Garn M, Aguirre JI, Wronski TJ, Stolina M, Zhou L, Pyrah I, Boyce RW (2015) Differential temporal effects of sclerostin antibody and parathyroid hormone on cancellous and cortical bone and quantitative differences in effects on the osteoblast lineage in young intact rats. Bone 81:380–391. https://doi.org/10.1016/j.bone.2015.08.007
Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C (2010) Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res 25(5):948–959. https://doi.org/10.1002/jbmr.14
Cosman F, Crittenden DB, Grauer A (2017) Romosozumab treatment in postmenopausal osteoporosis. N Engl J Med 376(4):396–397. https://doi.org/10.1056/NEJMc1615367
Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, Maddox J, Fan M, Meisner PD, Grauer A (2017) Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. https://doi.org/10.1056/NEJMoa1708322
Padhi D, Allison M, Kivitz AJ, Gutierrez MJ, Stouch B, Wang C, Jang G (2014) Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: a randomized, double-blind, placebo-controlled study. J Clin Pharmacol 54(2):168–178. https://doi.org/10.1002/jcph.239
Markham A (2019) Romosozumab: first global approval. Drugs 79(4):471–476. https://doi.org/10.1007/s40265-019-01072-6
Krishna SM, Seto SW, Jose RJ, Li J, Morton SK, Biros E, Wang Y, Nsengiyumva V, Lindeman JH, Loots GG, Rush CM, Craig JM, Golledge J (2017) Wnt Signaling Pathway Inhibitor Sclerostin Inhibits Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis. Arterioscler Thromb Vasc Biol 37(3):553–566. https://doi.org/10.1161/ATVBAHA.116.308723
Edwards JR, Mundy GR (2011) Advances in osteoclast biology: old findings and new insights from mouse models. Nat Rev Rheumatol 7(4):235–243
Cappariello A, Maurizi A, Veeriah V, Teti A (2014) The Great Beauty of the osteoclast. Arch Biochem Biophys 558:70–78. https://doi.org/10.1016/j.abb.2014.06.017
Parvizi J (2010) Osteoclasts. In: Kim GK (ed) Parvizi J. High Yield Orthopaedics Elseier, Amsterdam
Drissi H, Sanjay A (2016) The multifaceted osteoclast; far and beyond bone resorption. J Cell Biochem 117(8):1753–1756. https://doi.org/10.1002/jcb.25560
Zaidi M, Lizneva D, Yuen T (2021) The role of PDGF-BB in the bone-vascular relationship during aging. J Clin Invest. https://doi.org/10.1172/JCI153644
Madel MB, Ibanez L, Wakkach A, de Vries TJ, Teti A, Apparailly F, Blin-Wakkach C (2019) Immune function and diversity of osteoclasts in normal and pathological conditions. Front Immunol 10:1408. https://doi.org/10.3389/fimmu.2019.01408
Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q (2010) Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 116(2):210–217. https://doi.org/10.1182/blood-2009-11-255026
Grassi F, Manferdini C, Cattini L, Piacentini A, Gabusi E, Facchini A, Lisignoli G (2011) T cell suppression by osteoclasts in vitro. J Cell Physiol 226(4):982–990. https://doi.org/10.1002/jcp.22411
Gallois A, Lachuer J, Yvert G, Wierinckx A, Brunet F, Rabourdin-Combe C, Delprat C, Jurdic P, Mazzorana M (2010) Genome-wide expression analyses establish dendritic cells as a new osteoclast precursor able to generate bone-resorbing cells more efficiently than monocytes. J Bone Miner Res 25(3):661–672. https://doi.org/10.1359/jbmr.090829
Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, Li C, Xie L, Crane J, Wan M, Zhen G, Bian Q, Yu B, Chang W, Qiu T, Pickarski M, Duong LT, Windle JJ, Luo X, Liao E, Cao X (2014) PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med 20(11):1270–1278. https://doi.org/10.1038/nm.3668
Su W, Liu G, Liu X, Zhou Y, Sun Q, Zhen G, Wang X, Hu Y, Gao P, Demehri S, Cao X, Wan M (2020) Angiogenesis stimulated by elevated PDGF-BB in subchondral bone contributes to osteoarthritis development. JCI Insight. https://doi.org/10.1172/jci.insight.135446
Santhanam L, Liu G, Jandu S, Su W, Wodu BP, Savage W, Poe A, Liu X, Alexander LM, Cao X, Wan M (2021) Skeleton-secreted PDGF-BB mediates arterial stiffening. J Clin Invest. https://doi.org/10.1172/JCI147116
Acknowledgements
The authors thank Padmini Deosthale for her help with reference management.
Funding
Partial financial support was received from the National Institutes of Health/National Institute on Aging R21-AG078861, and the Veterans Research Administration Merit Award I01BX005154 to Lilian I. Plotkin, and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Ministerio de Ciencia, Tecnología e Inovación Productiva, Argentina -PIP 11220200100085- to Lucas R. Brun.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The author declares there are no competing interests associated with this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Plotkin, L.I., Sanz, N. & Brun, L.R. Messages from the Mineral: How Bone Cells Communicate with Other Tissues. Calcif Tissue Int 113, 39–47 (2023). https://doi.org/10.1007/s00223-023-01091-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-023-01091-2