
Abstract
Src homology-2 domain-containing phosphatase 2 (SHP2) is a ubiquitously expressed phosphatase that is vital for skeletal development and maintenance of chondrocytes, osteoblasts, and osteoclasts. Study of SHP2 function in small animal models has led to insights in phenotypes observed in SHP2-mutant human disease, such as Noonan syndrome. In recent years, allosteric SHP2 inhibitors have been developed to specifically target the protein in neoplastic processes. These inhibitors are highly specific and have great potential for disease modulation in cancer and other pathologies, including bone disorders. In this review, we discuss the importance of SHP2 and related signaling pathways (e.g., Ras/MEK/ERK, JAK/STAT, PI3K/Akt) in skeletal development. We review rodent models of pathologic processes caused by germline mutations that activate SHP2 enzymatic activity, with a focus on the skeletal phenotype seen in these patients. Finally, we discuss SHP2 inhibitors in development and their potential for disease modulation in these genetic diseases, particularly as it relates to the skeleton.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ubiquitously expressed Src Homology-2 domain-containing phosphatase 2 (SHP2), encoded by the PTPN11 gene, participates in the development and physiological regulation of tissues derived from all three germ layers [1, 2]. Dysregulation of protein tyrosine phosphorylation, including SHP2, is a major contributor to many diseases, including cancer, autoimmune disorders, chronic respiratory diseases, and diabetes. The skeletal system is especially sensitive to alterations in SHP2 activity, as evidenced by well-documented phenotypes associated with mutant SHP2 proteins. With multiple allosteric SHP2 inhibitors currently in early clinical trials for cancer treatments, SHP2 inhibition may also be beneficial for treatment of skeletal diseases. In this review, we summarize the importance of SHP2 in controlling skeletal development and homeostasis, as well as direct SHP2-mediated signaling pathways that regulate osteoblasts and osteoclasts (Fig. 1). We will consider lessons learned from alterations in SHP2 activity in human diseases, murine models, and through targeted inhibition.

Major SHP2-mediated signaling pathways regulating osteoblasts and osteoclasts
SHP2 Structure and Function
Structure
The structure of SHP2 is tightly connected to its biochemical mechanism of action. SHP2 is a non-membrane associated tyrosine phosphatase containing two N-terminal Src Homology 2 (SH2) domains, a central phosphatase domain, and C-terminal regulatory tyrosine residues [3]. In the inactive conformation, the N-terminal SH2 domain obstructs and distorts the catalytic site [3, 4] (Fig. 2a). Binding of the SHP2 SH2 domains to phospho-tyrosine residues induces a conformational change that removes the steric hindrance to unfold the protein and expose the active site [5]. Maximal phosphatase activity is achieved when both SH2 domains bind two neighboring phospho-tyrosine residues on large scaffolds and activated receptors [6]. SHP2 interacts in this way with IGF1R and its scaffolds to enhance osteogenic precursor proliferation and differentiation [7, 8]. This interaction has also been observed in response to a diverse array of receptors, ligands, and scaffolds critical to bone development and maintenance of osteoblasts and osteoclasts, including FGFR1 [9], PDGFR [10, 11], ErbB [12], DDR2 [13], cytokines [8], IL-6 [14, 15], M-CSF [7, 16], integrin [17], IGF-1 [18, 19], insulin [20, 21], RANK [22], FRS-2 [2, 23], SHC [24], GAB1 [8], GAB2 [25, 26], IFN-ɣ [27], BMP-2 [28], TGF-β [28], OSM [29], LIF [29], and E2 [30]. Such diversity of interactions underlies a critical role for SHP2 in mediating growth factor and cytokine signaling (for review of other protein tyrosine phosphatases and their skeletal signaling, we refer the reader to [31]).

Mechanism of SHP2 activation. a Interaction between SHP2 SH2 domains and phospho-tyrosine residues ( ) induces a conformational change exposing the catalytic site and alleviating auto-inhibition. b Putative mechanisms of SHP2-mediated RAS activation
) induces a conformational change exposing the catalytic site and alleviating auto-inhibition. b Putative mechanisms of SHP2-mediated RAS activation
Function
The principal effect of SHP2 activation is to promote Ras signaling. Various mechanisms of SHP2-dependent Ras activation have been proposed [2, 32]. Recently, a model has emerged describing Ras-GDP as a direct substrate of SHP2 phosphatase [33, 34]. In this model, SHP2-mediated dephosphorylation of RAS Y32 relieves an inhibitory feedback mechanism to promote RAS-GTP cycling and enhanced pathway activation [34] (Fig. 2b). While Ras cycling occurs in the presence of Y32 phosphorylation, dephosphorylation of this residue accelerates the cycling rate. Thus, SHP2-mediated dephosphorylation of Ras provides a highly dynamic method of regulating Ras signaling. SHP2 may also potentiate Ras signaling through its association with the GRB2/SOS RasGEF complex. This non-catalytic scaffolding mechanism of action is supported by studies of phosphatase-dead SHP2 mutants that continue to influence intracellular signaling [32, 35,36,37].
While most studies have focused on SHP2-mediated ERK1/2 activation, SHP2 activation is also important for signaling through the collateral MAPK pathways, p38 [38], JNK [39], and ERK5 [40]. Regulation of NF-κB by SHP2 can proceed through both Ras-dependent [41, 42] and Ras-independent [43] mechanisms. Further, there is evidence for Ras-independent SHP2-mediated control of PI3K/AKT [44], JAK2/STAT3 [45], JAK2/STAT5 [46], SMAD [28], and NFAT [22] signaling.
Skeletal development is a highly dynamic process dependent upon the function of anabolic osteoblasts and catabolic osteoclasts. SHP2 is expressed in osteoblasts and their osteoprogenitors, and a detailed review of targeted disruption of SHP2 function in these cell types is provided below. The role of Ras-MAPK signaling in osteoblast development and function remains controversial [47,48,49], as interruption of Ras/MAPK signaling can either enhance or repress osteoblast function [50]. However, multiple studies have shown that MAPK pathway inhibition promotes osteogenic differentiation and function of MC3T3-E1 cells [51, 52]. Similarly, Ras-hyperactivation reduced bone mineral content in multiple models, most notably in children with neurofibromatosis type 1 [53]. Given the divergent data available from model organisms, high-quality clinical studies are needed to examine the secondary effects of pharmacologic Ras/MAPK inhibition on osteoblastic differentiation and function. Encouragingly, MAPK inhibition with selumetinib enhanced bone mineral density in a patient with neurofibromatosis, although it is unclear if this is due to the effects of pathway inhibition on the function of osteoblasts, osteoclasts, or both cell types [54].
The catalytic regulatory cells of bone, osteoclasts, are derived from hematopoietic stem cells (HSCs) through the myeloid lineage. Pharmacologic and genetic SHP2 inhibition lead to increased bone mineral density by reducing osteoclast number and function [22]. The effects of SHP2 inhibition on bone catabolism result from its vital functions during osteoclast differentiation, from regulation of HSC survival and myeloid differentiation to mature osteoclast bone resorption. SHP2 is essential for murine HSC maintenance by promoting c-Kit expression [55, 56]. In mice, inducible ablation of Ptpn11 led to a significant reduction in bone marrow (BM) HSCs, myeloid progenitors, and peripheral leukocytes [55, 57]. Reduction in the HSC population was accompanied by departure from quiescence and increased apoptosis in Ptpn11-ablated BM [55]. The effects of SHP2 knockdown on adult HSCs were independently corroborated in isolated human BM-derived cells. SHP2 knockdown in vitro in human CD34+ cells led to significantly increased apoptosis, diminished proliferative response to growth factor stimulation, and reduced myeloid differentiation [46]. In addition to regulating myeloid and osteoclastic differentiation, SHP2 is known to regulate osteoclast function through its control of RANK-induced NFATc1 expression, a key regulatory transcription factor for osteoclastic resorption genes [22]. As reviewed below, pharmacologic or genetic inhibition of SHP2 dramatically reduces osteoclast number and function.
SHP2 in Human Skeletal Pathology
A variety of human skeletal pathologies have been linked to SHP2. The most well-characterized have been genetic disorders that arise from germline mutation of the PTPN11 gene. SHP2 is also a bona fide oncogene in leukemia, with a growing body of evidence implicating its role in solid tumors as well. Here, we consider the skeletal pathologies associated with genetic syndromes and neoplasms driven by dysregulated SHP2 (Table 1).
Germline Mutation of PTPN11
Noonan Syndrome
Noonan syndrome (NS; OMIM #163950) is an autosomal dominant genetic syndrome with an estimated incidence of 1 in 1000–2500 births. Bony dysmorphisms, including short stature, pectus carinatum, pectus excavatum, kyphosis, and scoliosis, are common. Typical facies display hypertelorism, ptosis, low-set ears, and a broad or webbed neck. Significant clinical features, such as congenital heart defects, hypertrophic cardiomyopathy, intellectual disability, and myeloid leukemia, are also common [58,59,60]. One of the most prevailing phenotypes of NS is short stature. In a study of 73 adults with NS, > 50% women and 38% of men were below the 3rd percentile for height [61]. Growth hormone insensitivity and a concomitant reduced serum IGF-1 are typical in NS patients [62, 63].
Dominant activating mutations of PTPN11 are the most common cause of NS [64]. While a variety of PTPN11 mutations in NS have been described, they tend to occur in the N-terminal SH2 domain, destabilizing the auto-inhibited conformation and leading to chronic low-level activation of SHP2 [18, 58, 65, 66]. NS mutations show growth factor-independent Ras activation and chronic activation of MAPK [67]. Chronic Ras activation in a mouse model of NS with mutant SHP2 reduced serum IGF-1 levels and impaired linear growth through direct effects on chondrocyte differentiation and growth plate size [68]. SHP2 is also an important regulator of calcium oscillation in response to growth factor stimulation in primary murine fibroblasts and cardiomyocytes. SHP2-mediated control of calcium signaling alters NFAT activity and has important implications for the cardiac and skeletal phenotypes of NS [69]. Thus, the dysregulation of SHP2 activity in NS leads to skeletal phenotypes through both endocrine effects and local defects in bone cells.
Noonan Syndrome with Multiple Lentigines
Noonan Syndrome with Multiple Lentigines (NSML; OMIM #151100) was originally named LEOPARD syndrome for a clinical constellation of multiple Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, growth Retardation, and Deafness. The condition is rare, with about 200 cases reported [70]. Hypertelorism and other facial dysmorphisms are common features of NSML. Hypertrophic cardiomyopathy is the most common and life-threatening cardiac defect in these patients [71]. Lentigines, similar to café-au-lait spots, appear in childhood and increase through puberty, numbering into the thousands [70]. Patients display short stature with 25% of cases below the 3rd percentile. Sternal defects are also common in NSML, with one study demonstrating the presence of pectus carinatum or pectus excavatum in half of the NSML patients [71].
Over 85% of NSML patients have a germline heterozygous missense mutation of PTPN11 thought to produce a dominant negative SHP2 mutant [72]. The mutations described in NSML occur in the catalytic phosphatase site. Biochemical assays confirm that these mutations abrogate phosphatase activity [72]. A variety of features, including short stature, skeletal dysmorphisms, cardiac defects, and increased incidence of cancer, are shared among patients with NS and NSML. How the activating PTPN11 mutations of NS and the inactivating PTPN11 mutations of NSML give rise to an overlapping clinical picture remain unknown.
Metachrondromatosis
Metachondromatosis (OMIM #156250) is a rare inherited autosomal dominant tumor syndrome [73]. Patients develop multiple cartilage-capped bony outgrowths, exostoses, and ectopic intracortical cartilage, termed endochondromas [74]. At least 50% of metachondromatosis patients inherit one defective copy of PTPN11. Consistent with classical inherited tumor syndromes of tumor suppressors, development of metachondromatosis is dependent on a second-hit or loss of the functional copy of SHP2 in a chondrocytic precursor [75]. Loss of SHP2 in paracrine regulators of chondrocytes likely contributes to the pathology of metachondromatosis [76]. The discovery of the role of SHP2 in the pathogenesis of metachondromatosis has been further facilitated through multiple murine models (reviewed below). These studies have greatly enhanced our understanding of this previously orphan disease and suggest novel therapeutic strategies [77, 78].
Wildtype PTPN11
Neoplasia
In contrast to the hematological malignancies associated with NS, epithelial neoplasms typically express wildtype SHP2 [79, 80]. Although mutation and amplification of SHP2 in solid tumors are rare, cancers that activate Ras through RTK-amplification [81] or NF1 loss [82] rely on SHP2 function. Wildtype SHP2 is emerging as a critical node that can be targeted in a variety of solid tumors dependent on Ras activation [81,82,83]. SHP2 is also thought to play a critical role in the observed resistance to pharmacologic inhibitors of carcinomas with oncogenes in the Ras pathway [83]. While this remains an area of active research, studies have demonstrated a role for SHP2 in the intrinsic resistance of BRAF-mutant colon cancer [84] and acquired resistance in KRas-mutant pancreatic and lung cancers [85, 86]. Our understanding of the role of wildtype SHP2 in neoplastic disease is rapidly expanding, as targeted agents facilitate more rigorous study. For a more complete consideration of the role of SHP2 in these processes, the reader is referred to recent reviews [87,88,89]. Future studies are needed to better understand the role of wildtype SHP2 in the pathogenesis of non-neoplastic bone disease.
Mouse Models of SHP2 Mutants
Mouse models have contributed significantly to our understanding of the importance of SHP2 in bone development and pathology. Here, we discuss the skeletal findings of murine models with alteration of SHP2 in mesodermal tissues, comprising the mesenchymal and hematopoietic systems. Recent reviews summarizing the extra-skeletal findings of these models are also available [18, 90]. We begin with whole-body knockout and progress through time- and tissue-specific knockouts. SHP2 knockouts of all the major cell types encompassed in bone development have been characterized, including mesenchymal stem cells, hypertrophic chondrocytes, and osteoclast precursors. The scope of this work reflects the significance of SHP2 in healthy bone development and homeostasis (Figs. 3, 4).

Studies of SHP2 knockout and targeted inhibition in osteoblast development

Studies of SHP2 knockout and targeted inhibition in osteoclast development
Whole Organism
The first murine knockout of Shp2 demonstrated that homozygous deletion of residues 46–110 resulted in death by gestational day 10. Shp2 null embryos displayed notable deficiencies in gastrulation, mesodermal patterning, and anterior–posterior axis formation [91, 92]. Primary Shp2-mutant fibroblasts showed significantly reduced ERK1/2 activation by FGF, a critical embryonic growth factor [91]. Chimeric SHP2-mutant embryos demonstrated the requirement of SHP2 for limb development [9]. The progress zone of budding limbs required SHP2, phenocopying FGFR1 mutants characterized earlier and suggesting the importance of SHP2 in transmitting the cytoplasmic signal from the activated FGF receptor. These early knockout studies demonstrated that ubiquitously expressed SHP2 was active in growth factor signaling and required for embryonic development.
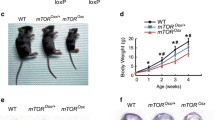
While chimeric analyses enabled study of SHP2 deletion post-gestational day 10, it was impossible to observe the effects of SHP2 knockout in adult tissues derived from embryonic structures that required wildtype SHP2. Thus, subsequent work developed multiple inducible SHP2 mutants, including a mutant with loxP-flanking of the catalytic phosphatase on exon 11 [93] and loxP-flanking of exon 4, which deletes residues 111–176 and introduces a frame-shift and premature stop codon [94].
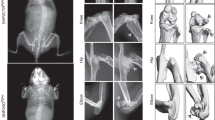
Ubiquitous expression of inducible ert2-Cre was used to model global Shp2 knockout in the adult mouse. Global Shp2 knockout in 6–8-week-old mice resulted in rapid weight-loss and early mortality, with 50% mortality at 4 weeks post-tamoxifen-mediated induction [16]. Adult Shp2 knockout mice were notable for hematopoietic and skeletal abnormalities. Mice displayed substantial reductions in common lymphoid progenitors and mature B and T cells. As early as two weeks post-induction, adult Shp2-mutant mice developed skeletal abnormalities, including kyphosis and scoliosis, characteristic of NS and NSML. Increased bone mineral density of the vertebrae, ribs, and humeral and femoral metaphyses were noted by x-ray and μCT analyses and confirmed histologically. Disorganization of cartilaginous growth plates of Shp2-mutant adult mice was extensive and likely contributed to the skeletal abnormalities of these growing mice. The osteopetrotic phenotype is at least partially due to a notable reduction in osteoclastogenesis in adult Shp2-deficient mice. These studies provided an important framework for understanding the roles of SHP2 in adult skeletal physiology and contributed important tools for interrogation of SHP2 deletion in the specific tissues that develop into and regulate the skeletal system.
Mesenchymal-Specific
The paired-related homeobox gene-1 (Prx1) promoter is utilized by mesenchymal tissues and was used to knock out Shp2 in osteocytic, adipogenic, and cartilaginous cell types [95]. These Prx1-induced Shp2 knockout mice showed absence of subcutaneous fat, reduced mature osteocytes, and increased chondrocytes, particularly hypertrophic chondrocytes. Molecular analyses indicated osteoblastic differentiation was interrupted at a late stage, as no differences in early osteoblast markers Col1a1, Runx2, Osterix or Atf4 were noted between knockout and wildtype samples, but a dramatic reduction in the mature marker, Osteocalcin, was observed. These alterations of mature mesenchymal cell types led to dramatic phenotypic skeletal changes, including drastically shortened forelimbs and hindlimbs and chest deformities characteristic of defects in endochondral ossification. Furthermore, calvarial bone defects indicated impaired intramembranous ossification. The skeletons of the Prx1-induced Shp2 knockout mice displayed reduced cortical mineral density, likely resulting from impaired osteoblast function.
The fate of the osteochondroprogenitor cell is tightly controlled by the transcription factor Sox9 [96]. Prx1-induced SHP2 deletion led to increased phosphorylation and SUMOylation of Sox9, leading to enhanced chondrogenesis [97]. Mice displayed increase cartilage mass at normal and ectopic sites, including the bone cortex and marrow. Appendicular ossification was concomitantly decreased. Studies of SHP2 knockout in mesenchymal precursor tissues display skewing toward chondrocytic differentiation over osteoblastic differentiation. These studies are remarkably consistent with the work in NS models in which activating SHP2 mutations display reductions in chondrocyte differentiation, especially hypertrophic chondrocytes [68]. These independent lines of inquiry demonstrate the necessity of SHP2 function for osteoblastic differentiation.
Chondrocyte-Specific
Given its role in determining the fate of the osteochondroprogenitor, it is not surprising that knock out of SHP2 has been pursued in progressively more differentiated cell types. To date, only one recent study has reported SHP2 deletion in Bglap+ mouse bone cells, which leads to osteopenia, failure of bone cell maturation, and enhanced osteoclast activity [98]. However, many studies have targeted SHP2 deletion in mature chondrocytes and are discussed below.
Shp2 deletion in chondrocytes delayed terminal differentiation in vitro, prolonging the hypertrophic stage [76]. Tamoxifen-inducible deletion of Shp2 driven by the Col2α1 promoter of early chondrocytes produced osteopenia, kyphoscoliosis, chest wall deformities, and lesions reminiscent of metachondromatosis in adult mice [74, 77, 99, 100], similar to those reported in Cathepsin K-driven loss of Shp2 in chondrocyte precursors [78]. Seven weeks post-induction, the mice displayed disorganized and enlarged vertebral growth plates with accompanying ectopic ossification and endochondromas [76]. Importantly, tamoxifen-induced Shp2 deletion was incomplete, and the endochondral lesions were composed of Shp2 mutant and wildtype chondrocytes, indicating the importance of paracrine effects of Shp2-mutant cells in promoting cartilage disorganization. Shp2 knockout of Fsp1-positive, bone-lining fibroblasts was sufficient to induce cartilaginous outgrowths of the metacarpals, phalanges, fibula, tibia, and femur, confirming the role of Shp2 in paracrine chondrocyte regulation. While FGF2 protein expression was increased by Shp2 deletion in the developing rib and paw, activation of Erk was noticeably impaired [74]. The role of SHP2 in chondrocyte differentiation involves a MAPK-dependent regulation of Indian hedgehog [78]. Shp2 function is also important in orofacial cartilage, as its depletion leads to mandibular condyle deformity [101]. In addition, development of primary cilia, a critical signaling organelle, was disrupted in the absence of Shp2. Taken together, Shp2 mutation in early chondrocytes promotes cartilaginous expansion and recapitulates the phenotype of metachondromatosis.
While it was clear that SHP2 is vital for proper cartilage differentiation, the dramatic effects of its knockout in early chondrocytes prevented researchers from assessing its role in the later stages of endochondral ossification. Col10α1 is not produced until the final stages of endochondral ossification when hypertrophic chondrocytes undergo terminal differentiation and secrete this matrix component for mineralization. Col10α1-driven Shp2 knockout in hypertrophic chondrocytes did not produce metachondromatosis, confirming the importance of Shp2-loss in a proliferative progenitor population for the pathology of this disease [100]. Shp2 knockout in hypertrophic chondrocytes resulted in significant osteopenia, promoted maintenance of chondrocytic master transcriptional regulator Sox9, and reduced expression of osteogenic genes. It is possible that SHP2 controls the trans-differentiation of a small pool of hypertrophic chondrocytes to an osteogenic cell type. Lineage tracing studies demonstrated that Col10α1-expressing cells downregulated Col2α1 and upregulated osteogenic genes, Ibsp, Mmp13, Runx2 and Ctnnb1, and that this switch was impaired with Shp2 deletion. While the phenomenon of osteogenic trans-differentiation remains controversial, it is an enticing prospect given the role of SHP2 in regulating SOX9 expression and ultimate cell fate of the osteochondrogenic precursor cell [97].
Hematopoietic-Specific
Early chimeric studies indicated a critical role for SHP2 in hematopoiesis and regulation of myeloid cell development [102]. Inducible Shp2 knockout in murine HSCs utilizing Mx1-Cre confirmed the essential role of Shp2 in HSC maintenance and survival [57]. Loss of Shp2 led to BM aplasia and reduced lifespan to a median of 5 weeks. HSCs lacking Shp2 displayed impaired Erk activation after stem cell factor stimulation and increased Noxa-mediated apoptosis. Restoration of Erk signaling by expression of mutant Kras rescued survival in Shp2-deficient HSCs. Similar results were obtained when SHP2 function was studied in human CD34+ HSCs [46, 103]. Knockdown reduced CD34+ cell proliferation, survival, and colony-forming units and inhibited myeloid differentiation [46]. Thus, SHP2 is necessary for HSC maintenance by promoting ERK signaling.
Myeloid-specific Shp2 knockout mouse models have facilitated precise studies that show SHP2 functions at multiple points in myeloid differentiation to promote osteoclast development, a critical element of skeletal physiology. The Lysozyme M (LysM) promoter has been extensively used to drive specific knockout in monocytes, macrophages, and osteoclasts [104,105,106]. LysM-driven Shp2 knockout resulted in mild osteopetrosis [78]. Cathepsin K (Ctsk) promoter-driven Shp2 deletion produced osteopetrosis, in addition to scoliosis and metaphyseal exostoses. Transplant of Ctsk-driven Shp2 knockout BM into lethally irradiated recipient mice confirmed that the osteopetrosis arose from Shp2 knockout in osteoclasts. The metaphyseal exostoses, however, arose from a previously unknown population of Ctsk-expressing chondroprogenitors and phenotypically recapitulate the observation of SHP2 loss in metachondromatosis. The osteopetrosis caused by Shp2 deletion in osteoclasts was later characterized in greater detail [22]. Shp2 knockout in these cells led to a 42.8% increase in bone volume to trabecular volume ratio, increased trabeculae number, and decreased trabecular spacing. Mice had fewer osteoclasts, while in vitro cultured BM produced fewer osteoclasts when stimulated with M-CSF and RANKL. Shp2-deficient pre-osteoclasts demonstrated impaired RANK-dependent preosteoclast fusion. The knockout pre-osteoclasts failed to upregulate Nfatc1, an essential osteoclast transcription factor that drives expression of the fusion protein, DC-STAMP. Analysis of the few Shp2-deficient osteoclasts that did develop demonstrated functional impairment with reduced pit resorption. Together, these studies showed that SHP2 is essential for osteoclast development and activity and further demonstrate its importance in skeletal development and homeostasis.
Targeted Modulation of Shp2
Given the causal role of SHP2 in genetic syndromes and neoplastic disease, it is not surprising that it has long been the subject of intense pharmacologic discovery programs. However, developing targeted tyrosine phosphatase inhibitors has proven more challenging than targeting the tyrosine kinase domain [107]. Targeting the human phosphatase active site has been difficult, partially due to its highly conserved nature [108, 109]. Another difficulty arises from the strongly positive nature of the catalytic site, which is necessary for interacting with the phosphate group [108, 110]. Strong polar charges of potential active site inhibitors reduce their bioavailability and clinical development [109, 110]. Despite these challenges, a variety of SHP2 competitive inhibitors have been described, although none has progressed to clinical studies. Recently, a new class of allosteric SHP2 inhibitors has emerged and progressed rapidly into clinical trials. Here, we consider the inhibitors that have been used in vivo and in clinical studies, with an emphasis on their application in musculoskeletal and other non-neoplastic pathologies (Table 2). For reviews focusing on SHP2 inhibition in cancer, the reader is referred to other sources [87, 90].
Competitive Inhibitors
NSC-87877
The first competitive SHP2 inhibitor to be reported showed similar activity against SHP1 and SHP2. The specificity of NSC-87877 continues to be of concern, as a recent report demonstrated that NSC-87877 failed to prevent PDGF-BB or EGF-induced MEK1/2 or ERK1/2 activation, which is highly dependent on SHP2 [10]. This study underlies the inherent difficulty of understanding protein function with small molecule inhibitors. The best studies of function should combine genetic and pharmacological methodologies.
The effect of NSC-87877 on osteoclast development has been reported [22]. Differentiation of osteoclasts from BM precursor cells in the presence of NSC-87877 was significantly reduced. The authors demonstrated a reduction in the total number of osteoclasts and in expression of osteoclastic differentiation markers, including Trap, Cathepsin K, DC-Stamp, and Nfatc1, with Shp2 inhibition. As NFATc1 is thought to be the master transcription factor of osteoclastogenesis, the authors conclude that SHP2-mediated inhibition of osteoclast differentiation is predominantly the result of decreased NFATc1 expression. Consistent with this conclusion, overexpression of NFATc1 rescued the effect of NSC-87877 treatment on osteoclast differentiation. Functionally, osteoclasts cultured in the presence of NSC-87877 or with SHP2 knockout showed defective pit resorption. Interestingly, removal of NSC-87877 led to a rebound in osteoclast numbers, suggesting Shp2 inhibition prevented the maturation of osteoclasts but did not kill the progenitor population. This observation has profound implications for targeted inhibition of SHP2 in skeletal pathologies. To date, anti-osteoclastic therapies, such as bisphosphonates, produce an irreversible reduction in osteoclast numbers [111]. Thus, SHP2 inhibition may provide a novel strategy for inhibiting osteoclastogenesis in a reversible manner.
NSC-87877 has been used to inhibit SHP2 in a variety of non-skeletal models. This SHP2 inhibitor reduced ERK activation and viability of breast cancer cell lines [112]. SHP2 was required for survival of proliferating cardiac progenitor cells and for upregulation of cardiac proteins, such as myosin heavy chain and the cardiac master transcriptional regulator, Nkx2.5, which was inhibited upon treatment with NSC-87877 [23]. The effect of NSC-87877 was specific to its inhibition of SHP2 by rescue with constitutively active SHP2, an important control given the similar potency of this compound in inhibiting SHP1. Treatment with NSC-87877 ameliorated TGF-β-induced dermal- and bleomycin-induced pulmonary fibrosis [45]. The role of SHP2 in hippocampal synaptic plasticity and memory was examined in a model of contextual fear conditioning [113]. Long-term potentiation (LTP) of glutamatergic synapses results from increased post-synaptic expression of AMPA receptors and has been shown to be Ras-dependent [114, 115]. Fear conditioning with foot shock recruited SHP2 and the AMPA subunit GluA1 to spines, indicating LTP [113]. Importantly, NSC-87877 treatment prevented GluA1 recruitment.
PHPS1
The next competitive inhibitor to become available was PHPS1 [116]. PHPS1 demonstrated reduced potency but greater SHP2 specificity compared to NSC-87877. PHPS1 was used to inhibit SHP2 activation in the murine MC3T3-E1 pre-osteoblast cell line and primary murine calvarial osteoblasts [14]. Markers of osteoblast differentiation, including Runx2, osterix, osteocalcin and alkaline phosphatase, increased with SHP2 inhibition in MC3T3-E1 cells. Extracellular matrix mineralization by MC3T3-E1 cells increased with SHP2 inhibition. PHPS1 partially inhibited the IL-6-dependent activation of ERK and AKT; however, complete AKT or ERK inhibition had no effect on MC3T3 mineralization. The authors conclude that SHP2 inhibition prevents the negative effects of IL-6 on osteoblast differentiation, but it is difficult to explain how partial inhibition by PHPS1 has such a dramatic effect that was not observed with specific MEK or AKT inhibition.
The use of PHPS1 for SHP2 inhibition in extra-skeletal systems has focused on modulation of the immune response and application to cancer. SHP2 was involved in the recovery of the endothelial barrier after thrombin-mediated inflammation by direct phosphatase action on β-catenin [117]. SHP2 inhibition with PHPS1 was similar to SHP2 silencing in delaying endothelial barrier recovery after thrombin treatment. SHP2 inhibition with PHPS1 or conditional Shp2 knockout enhanced IL-4-directed M2 polarization, which may play a role in promoting the pathogenesis of bleomycin-induced pulmonary fibrosis [45, 118]. Similarly, treatment with PHPS1 attenuated the immune response and skewed macrophages toward an M2 phenotype when activated by bacterial infection with Haemophilus influenzae [119]. SHP2 activation of the M1 phenotype was independent of ERK and, instead, required NF-kB. SHP2 may also be involved in mediating the pro-inflammatory response to cigarette smoke that results in chronic obstructive pulmonary disease [120]. Treatment with PHPS1 reduced cigarette smoke-induced IL-8 and inflammatory infiltration. The role of SHP2 in regulating signaling by the estrogen receptor (ER) in breast cancer cell lines was examined, and SHP2 mRNA and protein expression were found to be increased by treatment with estradiol [121]. Further, PHPS1-mediated SHP2 inhibition delayed tumor formation in vivo, and PHPS1 and NSC-87877 suppressed estradiol-dependent transcription of Cyclin D1 and proliferation.
A recent study examined the role of SHP2 in the pathophysiology of high cholesterol-induced atherosclerosis [122]. LDL receptor-deficient mice were fed a high cholesterol diet for 4 weeks. During the final week of the high cholesterol diet, the mice received daily intraperitoneal injection with PHPS1 or vehicle. PHPS1 treatment significantly reduced the total number, size, and smooth muscle cell content of atherosclerotic plaques. In vitro studies with vascular smooth muscle cells (VSMCs) revealed that PHPS1 treatment reduced ERK phosphorylation and VSMC proliferation induced by oxidized LDL. However, PHPS1 did not reduce the proliferation of inactivated VSMCs. Thus, activated VSMCs, which drive the pathology of atherosclerosis, may be selectively sensitive to SHP2 inhibition.
II-B08
II-B08 was developed from a screen designed to find bidentate SHP2 competitive inhibitors with affinity for the conserved PTP active site and the less conserved flanking residues, which are also important for substrate binding [123]. II-B08 inhibited long-term ERK activation approximately 60 min after EGF treatment in HEK293. Early EGF-dependent ERK activation, however, was not affected by SHP2 inhibition with II-B08 [124]. The lack of early EGF-dependent ERK activation and suppression of PGDFRβ phosphorylation, which is upstream of and not thought to be regulated by SHP2 [10], raise concerns regarding the specificity of II-B08.
II-B08 was used to interrogate the role of SHP2 in mast cell function and mast cell proliferative diseases. II-B08 inhibited SCF-induced chemotaxis of BM-derived mast cells [125] and enhanced apoptosis following growth factor withdrawal in mast cells, indicating the importance of SHP2 in regulating the activity of apoptotic effectors, such as Bim, downstream of survival signals [83, 126]. II-B08 reduced Kit+ leukemia cell viability and colony formation in vitro [127] and reduced proliferation and increased survival in vivo [128]. II-B08 inhibited GM-CSF-dependent proliferation of BM mononuclear cells expressing wildtype or NS-mutant SHP2 [124]. As demonstrated by these studies, II-B08 has primarily been used to inhibit SHP2 in models of RTK- and SHP2-driven cancers.
11a-1
The compound, 11a-1, was developed through a structure-guided and fragment-based library approach to improve on the published inhibitor, II-B08 [108]. The SHP2 IC50 of 11a-1 is 0.20 µM, with at least fivefold selectivity over 20 related protein tyrosine phosphatases. Treatment with 11a-1 ameliorated TGF-β-induced dermal and bleomycin-induced pulmonary fibrosis [45]. The role of SHP2 in the pathogenesis of rheumatoid arthritis was also examined [129]. SHP2 inhibition with 11a-1 reduced severity of arthritis, as indicated by reductions in inflammation, bone and cartilage erosion, and ankle swelling in a serum transfer model of rheumatoid arthritis. In vitro, 11a-1 treatment reduced migration and TNF- or IL-1β-induced upregulation of matrix metalloproteinases by rheumatoid arthritis fibroblast-like synoviocytes. Systemic lupus erythematosus (SLE) is another common autoinflammatory pathology that may benefit from SHP2 inhibition [130]. SHP2 inhibition with 11a-1 increased survival, ameliorated the common SLE-related renal pathology crescentic glomerulonephritis, and reduced skin lesions in an SLE murine model.
GS-493
The most potent and selective competitive SHP2 inhibitor reported to date is GS-493 [131]. While GS-493 showed enhanced SHP2 selectively in phosphatase assays, follow-up studies demonstrated its ability to directly dephosphorylate PDGFRβ, raising concerns of off-target effects that have continually plagued the competitive SHP2 inhibitors [10].
GS-493 was used to probe the role of SHP2 in a musculoskeletal mesodermal population [132]. Proliferation of postnatal myogenic progenitor cells was inhibited by SHP2 knockdown or targeted inhibition by GS-493 in vivo. The effect of SHP2 inhibition was thought to be due to the dramatic inhibition of phosphorylated ERK, as proliferation was rescued by constitutively active MEK. To date, there have been no reports assessing GS-493 in osteocytic populations. The development of GS-493 has provided a competitive SHP2 inhibitor with dramatically improved potency and selectivity. It has been used extensively in cancer research [84, 85, 131, 133] and will likely continue to facilitate a variety of new studies.
Allosteric Inhibitors
Recently a novel class of SHP2 allosteric inhibitors has been developed [81]. These compounds were developed utilizing a screen that selected small molecules with efficacy against full-length SHP2 activated by a biphosphoryl peptide but excluded compounds with efficacy against the catalytic subunit of SHP2. This simple but effective experimental design identified a class of inhibitors with allosteric activity. Structural studies demonstrated that the allosteric site is at the interface of SHP2 N and C terminals and that the interaction with the inhibitor stabilizes the closed, inactive conformation of SHP2. The structural work predicted the importance of two residues, T253 and Q257, which interacted with the inhibitor to stabilize the closed confirmation. Indeed, mutation of these residues had little effect on SHP2 phosphatase activity but reduced the potency of the inhibitor by 1000-fold, confirming the allosteric nature of the inhibitor. In comparison to the active site inhibitors, published studies with multiple allosteric inhibitors, SHP099 [81], TNO-155 [134], and RMC4550 [82], demonstrated their striking specificity for SHP2 over other human phosphatases.
Despite their recent introduction, the allosteric SHP2 inhibitors have already been extensively studied in cancer [87, 135]. The speed with which these agents have entered early-stage clinical trials is a testament to the promise they hold for treating cancer patients. These studies have led to an important observation on the potency of these inhibitors in targeting mutant SHP2. As described above, the activating mutation of NS predisposes individuals to leukemias. These mutations tend to destabilize the closed conformation of SHP2, in direct opposition to the action of the allosteric SHP2 inhibitors. Investigation revealed that some of the common mutant SHP2 proteins in NS are resistant to allosteric inhibition [136]. However, allosteric inhibitors targeting mutant leukemogenic SHP2 mutants have already been developed [137]. The application of these agents to human genetic disease continues to be explored [138].
To date, evaluation of the utility of SHP2 allosteric inhibitors in non-cancer pathologies has been limited. The allosteric agent, SHP099, was used to probe the importance of SHP2 in insulin signaling, a critical pathway in skeletal development [139,140,141]. Inhibition of SHP2 delayed insulin receptor endocytosis and improved insulin sensitivity in mice [139]. One group reported efficacy of SHP099 in two in vivo models of liver fibrosis [142]. SHP2 inhibition reduced liver fibrosis through its activity in platelet-derived growth factor signaling. Importantly, the dose of SHP099 used in vivo was much lower than that reported for cancer studies, an important consideration for pathologies that may require long-term treatment. Despite the long-held known role for SHP2 in bone development and maintenance, the effect of SHP2 inhibition on bone in vivo has been understudied, to date. However, a recent report demonstrates the potential for this new class of targeted inhibitor to modulate musculoskeletal pathology [143]. In this report, investigators showed that SHP2 is highly expressed in intervertebral discs where it acts in the role described above to inhibit chondrocytic differentiation and gene expression. Injection of SHP2 inhibitor, SHP099, into the intervertebral disc promoted cartilaginous protein expression and had a therapeutic effect in a rat model of degenerative disc disease.
Conclusion
A significant effort has been made to understand the importance of SHP2 in development and disease through a global, concerted effort combining genetic and pharmacologic methodologies. Together, these studies demonstrate the important role of SHP2 in skeletal development and maintenance through actions on osteoblasts and osteoclasts. Osteoblasts arise from mesenchymal cells with the potential to differentiate into alternative mesenchymal tissues, including adipocytes and chondrocytes. The lack of SHP2 activity in mesenchymal tissues, either by genetic dysfunction or targeted inhibition, prevents osteoblastic in favor of chondrocytic differentiation. These findings align with the known role of ERK1/2 in osteoblastic differentiation through phosphorylation of RUNX2 [48, 144, 145]. Failure of SHP2-mutant mesenchymal cells to activate ERK1/2 may prevent activation of RUNX2 and osteoblast differentiation, and promote Sox9 expression and stability, resulting in chondrocyte differentiation (Fig. 5).

SHP2 regulates differentiation of osteoblasts and osteoclasts
These studies also established an important role for SHP2 in osteoclast development. SHP2 is necessary for hematopoietic stem cell renewal, survival, and myeloid differentiation. It is required for expression and activity [93] of the osteoclastogenic master transcriptional regulator, NFATc1. In addition to its regulation of ERK1/2 activation by M-CSF and RANKL, there is evidence of a role for SHP2 in controlling growth factor-mediated calcium oscillations, a key regulator of NFATc1 activity [69]. Future studies should also investigate the potential role of nuclear SHP2 in osteoblast, osteoclast, chondrocyte, and osteochondroprogenitor maintenance and survival. Nuclear SHP2 has been shown to be involved in regulating STAT5 signaling, as well as cell proliferation [146]. Interactions between YAP1 and SHP2 have also been shown to promote translocation of SHP2 to the nucleus, leading to activation of the Wnt pathway in tumors from patients with non-small cell lung cancer [147]. As Wnt signaling is also an important signaling pathway in bone maintenance, nuclear SHP2 may be an interesting target of further study in bone cells.
SHP2 is essential for proper function of chondrocytes, osteoblasts, and osteoclasts, all the cell types regulating endochondral ossification and skeletal maintenance. Targeted loss of SHP2 in tissues that promote endochondral ossification, such as Bglap+ bone cells and early and late hypertrophic chondrocytes, produces significant osteopenia. In contrast, SHP2 knockout in osteoclasts results in osteopetrosis. While SHP2 functions in promoting both bone formation and resorption, it is important to note that, in murine models with ubiquitous SHP2 knockout, the mice developed rapid and progressive osteopetrosis, indicating a predominant effect on osteoclasts and the importance of tightly regulated balance for bone homeostasis.
Insights gained from study of SHP2 function in model organisms help to explain the phenotypes seen in SHP2-mutant human disease [148]. Germline mutations in SHP2 are among the most common rasopathies. The clinical syndromes demonstrate a spectrum of manifestations that result from aberrations in SHP2-mediated signaling. While this spectrum is broad, many of the prominent features relate to the role of SHP2 in mesenchymal development and skeletal homeostasis. These features have dramatic consequences for the patients born with these conditions, which may require surgical intervention and lifelong neoplastic surveillance.
Highly specific SHP2 inhibitors have been developed and are now in clinical trials. While these agents were initially developed for neoplastic conditions, they may represent a new tool in modulating SHP2-mediated genetic disease. Recent clinical successes have demonstrated that targeted agents can be useful in genetic syndromes, such as neurofibromatosis and achondroplasia. Decades of research and development have provided deep mechanistic understanding of the pathophysiology of clinical syndromes that result from SHP2 mutation and small molecules capable of modulating its function. Additionally, many of the major signaling pathways influenced by alterations in SHP2 activity are involved in multiple pathological conditions. Therefore, SHP2 inhibitors may have clinical benefit in other diseases, not just cancer, including diabetes [149, 150], autoimmune disease [130], chronic respiratory diseases [151], and musculoskeletal disorders [148]. It is important to be mindful of what was learned in the model systems and genetic syndromes as these agents move forward in clinical development. This foundation will help evaluate for adverse effects and expand therapeutic applications.
References
Ahmad S, Banville D, Zhao Z, Fischer EH, Shen SH (1993) A widely expressed human protein-tyrosine phosphatase containing src homology 2 domains. Proc Natl Acad Sci U S A 90:2197–2201. https://doi.org/10.1073/pnas.90.6.2197
Neel BG, Gu H, Pao L (2003) The ’Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293. https://doi.org/10.1016/S0968-0004(03)00091-4
Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92:441–450. https://doi.org/10.1016/s0092-8674(00)80938-1
Yu Z-H, Xu J, Walls CD, Chen L, Zhang S, Zhang R et al (2013) Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem 288:10472–10482. https://doi.org/10.1074/jbc.M113.450023
Ran H, Tsutsumi R, Araki T, Neel BG (2016) Sticking it to cancer with molecular glue for SHP2. Cancer Cell 30:194–196. https://doi.org/10.1016/j.ccell.2016.07.010
Cunnick JM, Mei L, Doupnik CA, Wu J (2001) Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem 276:24380–24387. https://doi.org/10.1074/jbc.M010275200
Yao Z, Darowski K, St-Denis N, Wong V, Offensperger F, Villedieu A et al (2017) A Global Analysis of the receptor tyrosine kinase-protein phosphatase interactome. Mol Cell 65:347–360. https://doi.org/10.1016/j.molcel.2016.12.004
Fukada T, Yoshida Y, Nishida K, Ohtani T, Shirogane T, Hibi M et al (1999) Signaling through Gp130: toward a general scenario of cytokine action. Growth Factors 17:81–91. https://doi.org/10.3109/08977199909103518
Saxton TM, Ciruna BG, Holmyard D, Kulkarni S, Harpal K, Rossant J et al (2000) The SH2 tyrosine phosphatase shp2 is required for mammalian limb development. Nat Genet 24:420–423. https://doi.org/10.1038/74279
Tsutsumi R, Ran H, Neel BG (2018) Off-target inhibition by active site-targeting SHP2 inhibitors. FEBS Open Bio 8:1405–1411. https://doi.org/10.1002/2211-5463.12493
Yin H, Huang J, Cao X, Wang Z-X, Cao J, Hu Y et al (2018) Inhibition of Src homology 2 domain-containing protein tyrosine phosphatase-2 facilitates cd31hiendomucinhi blood vessel and bone formation in ovariectomized mice. Cell Physiol Biochem 50:1068–1083. https://doi.org/10.1159/000494531
Tanowitz M, Si J, Yu DH, Feng GS, Mei L (1999) Regulation of neuregulin-mediated acetylcholine receptor synthesis by protein tyrosine phosphatase SHP2. J Neurosci 19:9426–9435
Iwai LK, Payne LS, Luczynski MT, Chang F, Xu H, Clinton RW et al (2013) Phosphoproteomics of collagen receptor networks reveals SHP-2 phosphorylation downstream of wild-type DDR2 and its lung cancer mutants. Biochem J 454:501–513. https://doi.org/10.1042/BJ20121750
Kaneshiro S, Ebina K, Shi K, Higuchi C, Hirao M, Okamoto M et al (2014) IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Metab 32:378–392. https://doi.org/10.1007/s00774-013-0514-1
Sims NA, Jenkins BJ, Quinn JMW, Nakamura A, Glatt M, Gillespie MT et al (2004) Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest 113:379–389. https://doi.org/10.1172/JCI19872
Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, Wilkinson JE et al (2011) Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech 4:228–239. https://doi.org/10.1242/dmm.006130
Nishida K, Hirano T (2003) The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci 94:1029–1033. https://doi.org/10.1111/j.1349-7006.2003.tb01396.x
Tajan M, Pernin-Grandjean J, Beton N, Gennero I, Capilla F, Neel BG et al (2018) Noonan syndrome-causing SHP2 mutants impair ERK-dependent chondrocyte differentiation during endochondral bone growth. Hum Mol Genet 27:2276–2289. https://doi.org/10.1093/hmg/ddy133
Kapur S, Mohan S, Baylink DJ, Lau K-HW (2005) Fluid shear stress synergizes with insulin-like growth factor-I (IGF-I) on osteoblast proliferation through integrin-dependent activation of IGF-I mitogenic signaling pathway. J Biol Chem 280:20163–20170. https://doi.org/10.1074/jbc.M501460200
Ergun-Longmire B, Wajnrajch MP (2000) Growth and Growth Disorders. In: Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K et al (eds) Feingold KR. Endotext. MDText.com, Inc, South Dartmouth (MA)
Tajan M, Batut A, Cadoudal T, Deleruyelle S, Le Gonidec S, Saint Laurent C et al (2014) LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc Natl Acad Sci U S A 111:E4494-4503. https://doi.org/10.1073/pnas.1406107111
Zhou Y, Mohan A, Moore DC, Lin L, Zhou FL, Cao J et al (2015) SHP2 regulates osteoclastogenesis by promoting preosteoclast fusion. FASEB J 29:1635–1645. https://doi.org/10.1096/fj.14-260844
Langdon YG, Goetz SC, Berg AE, Swanik JT, Conlon FL (2007) SHP-2 is required for the maintenance of cardiac progenitors. Development 134:4119–4130. https://doi.org/10.1242/dev.009290
Lecoq-Lafon C, Verdier F, Fichelson S, Chrétien S, Gisselbrecht S, Lacombe C et al (1999) Erythropoietin induces the tyrosine phosphorylation of GAB1 and its association with SHC, SHP2, SHIP, and phosphatidylinositol 3-kinase. Blood 93:2578–2585
Gu H, Pratt JC, Burakoff SJ, Neel BG (1998) Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell 2:729–740. https://doi.org/10.1016/s1097-2765(00)80288-9
Gu H, Neel BG (2003) The “Gab” in signal transduction. Trends Cell Biol 13:122–130. https://doi.org/10.1016/s0962-8924(03)00002-3
Cunningham CC, Corr EM, McCarthy GM, Dunne A (2016) Intra-articular basic calcium phosphate and monosodium urate crystals inhibit anti-osteoclastogenic cytokine signalling. Osteoarthr Cartil 24:2141–2152. https://doi.org/10.1016/j.joca.2016.07.001
Wang L, Huang J, Moore DC, Song Y, Ehrlich MG, Yang W (2019) SHP2 regulates intramembranous ossification by modifying the TGFβ and BMP2 signaling pathway. Bone 120:327–335. https://doi.org/10.1016/j.bone.2018.11.014
Persson E, Souza PPC, Floriano-Marcelino T, Conaway HH, Henning P, Lerner UH (2019) Activation of Shc1 allows oncostatin M to induce RANKL and osteoclast formation more effectively than leukemia inhibitory factor. Front Immunol 10:1164. https://doi.org/10.3389/fimmu.2019.01164
Park H-J, Gholam-Zadeh M, Yoon S-Y, Suh J-H, Choi H-S (2021) Estrogen decreases cytoskeletal organization by forming an ERα/SHP2/c-Src complex in osteoclasts to protect against ovariectomy-induced bone loss in mice. Antioxidants 10:619. https://doi.org/10.3390/antiox10040619
Yang H, Wang L, Shigley C, Yang W (2022) Protein tyrosine phosphatases in skeletal development and diseases. Bone Res 10:10. https://doi.org/10.1038/s41413-021-00181-x
Dance M, Montagner A, Salles J-P, Yart A, Raynal P (2008) The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 20:453–459. https://doi.org/10.1016/j.cellsig.2007.10.002
Bunda S, Heir P, Srikumar T, Cook JD, Burrell K, Kano Y et al (2014) Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci U S A 111:E3785-3794. https://doi.org/10.1073/pnas.1406559111
Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y et al (2015) Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 6:8859. https://doi.org/10.1038/ncomms9859
Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG (1994) Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A 91:7335–7339. https://doi.org/10.1073/pnas.91.15.7335
Li W, Nishimura R, Kashishian A, Batzer AG, Kim WJ, Cooper JA et al (1994) A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol 14:509–517. https://doi.org/10.1128/mcb.14.1.509-517.1994
Vogel W, Ullrich A (1996) Multiple in vivo phosphorylated tyrosine phosphatase SHP-2 engages binding to Grb2 via tyrosine 584. Cell Growth Differ 7:1589–1597
Chong ZZ, Lin S-H, Kang J-Q, Maiese K (2003) The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol Neurobiol 23:561–578. https://doi.org/10.1023/a:1025158314016
Chan G, Kalaitzidis D, Neel BG (2008) The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 27:179–192. https://doi.org/10.1007/s10555-008-9126-y
Nakaoka Y, Shioyama W, Kunimoto S, Arita Y, Higuchi K, Yamamoto K et al (2010) SHP2 mediates gp130-dependent cardiomyocyte hypertrophy via negative regulation of skeletal alpha-actin gene. J Mol Cell Cardiol 49:157–164. https://doi.org/10.1016/j.yjmcc.2010.03.001
Kang HJ, Chung D-H, Sung CO, Yoo SH, Yu E, Kim N et al (2017) SHP2 is induced by the HBx-NF-κB pathway and contributes to fibrosis during human early hepatocellular carcinoma development. Oncotarget 8:27263–27276. https://doi.org/10.18632/oncotarget.15930
Yart A, Laffargue M, Mayeux P, Chretien S, Peres C, Tonks N et al (2001) A critical role for phosphoinositide 3-kinase upstream of Gab1 and SHP2 in the activation of Ras and mitogen-activated protein kinases by epidermal growth factor. J Biol Chem 276:8856–8864. https://doi.org/10.1074/jbc.M006966200
Liu W, Yu W-M, Zhang J, Chan RJ, Loh ML, Zhang Z et al (2017) Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 31:1415–1422. https://doi.org/10.1038/leu.2016.326
Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R et al (2002) Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 22:4062–4072. https://doi.org/10.1128/MCB.22.12.4062-4072.2002
Zehender A, Huang J, Györfi A-H, Matei A-E, Trinh-Minh T, Xu X et al (2018) The tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat Commun 9:3259. https://doi.org/10.1038/s41467-018-05768-3
Li L, Modi H, McDonald T, Rossi J, Yee J-K, Bhatia R (2011) A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood 118:1504–1515. https://doi.org/10.1182/blood-2010-06-288910
Schindeler A, Little DG (2006) Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res 21:1331–1338. https://doi.org/10.1359/jbmr.060603
Ge C, Xiao G, Jiang D, Franceschi RT (2007) Critical role of the extracellular signal–regulated kinase–MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol 176:709–718. https://doi.org/10.1083/jcb.200610046
Majidinia M, Sadeghpour A, Yousefi B (2018) The roles of signaling pathways in bone repair and regeneration. J Cell Physiol 233:2937–2948. https://doi.org/10.1002/jcp.26042
Weng T, Mao F, Wang Y, Sun Q, Li R, Yang G et al (2010) Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. J Cell Sci 123:682–689. https://doi.org/10.1242/jcs.058396
Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H et al (2002) Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res 17:1785–1794. https://doi.org/10.1359/jbmr.2002.17.10.1785
Nakayama K, Tamura Y, Suzawa M, Harada S-I, Fukumoto S, Kato M et al (2003) Receptor tyrosine kinases inhibit bone morphogenetic protein-Smad responsive promoter activity and differentiation of murine MC3T3-E1 osteoblast-like cells. J Bone Miner Res 18:827–835. https://doi.org/10.1359/jbmr.2003.18.5.827
Armstrong L, Jett K, Birch P, Kendler DL, McKay H, Tsang E et al (2013) The generalized bone phenotype in children with neurofibromatosis 1: a sibling matched case-control study. Am J Med Genet A 161A:1654–1661. https://doi.org/10.1002/ajmg.a.36001
Ma Y, Gross AM, Dombi E, Pemov A, Choi K, Chaney K et al (2020) A molecular basis for neurofibroma-associated skeletal manifestations in NF1. Genet Med 22:1786–1793. https://doi.org/10.1038/s41436-020-0885-3
Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W et al (2011) Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood 117:5350–5361. https://doi.org/10.1182/blood-2011-01-333476
Kan C, Yang F, Wang S (2018) SHP2-mediated signal networks in stem cell homeostasis and dysfunction. Stem Cells Int 2018:e8351374. https://doi.org/10.1155/2018/8351374
Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S et al (2011) Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood 117:4253–4261. https://doi.org/10.1182/blood-2010-11-319517
Niihori T, Aoki Y, Ohashi H, Kurosawa K, Kondoh T, Ishikiriyama S et al (2005) Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J Hum Genet 50:192–202. https://doi.org/10.1007/s10038-005-0239-7
Tartaglia M, Gelb BD, Zenker M (2011) Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 25:161–179. https://doi.org/10.1016/j.beem.2010.09.002
Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME et al (2010) Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126:746–759. https://doi.org/10.1542/peds.2009-3207
Noonan JA, Raaijmakers R, Hall BD (2003) Adult height in Noonan syndrome. Am J Med Genet 123A:68–71. https://doi.org/10.1002/ajmg.a.20502
Binder G, Neuer K, Ranke MB, Wittekindt NE (2005) PTPN11 mutations are associated with mild growth hormone resistance in individuals with Noonan syndrome. J Clin Endocrinol Metab 90:5377–5381. https://doi.org/10.1210/jc.2005-0995
Limal J-M, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S et al (2006) Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab 91:300–306. https://doi.org/10.1210/jc.2005-0983
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H et al (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465–468. https://doi.org/10.1038/ng772
Keilhack H, David FS, McGregor M, Cantley LC, Neel BG (2005) Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem 280:30984–30993. https://doi.org/10.1074/jbc.M504699200
Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V et al (2006) Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 78:279–290
Fragale A, Tartaglia M, Wu J, Gelb BD (2004) Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat 23:267–277. https://doi.org/10.1002/humu.20005
De Rocca S-Nédélec A, Edouard T, Tréguer K, Tajan M, Araki T, Dance M et al (2012) Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci U S A 109:4257–4262. https://doi.org/10.1073/pnas.1119803109
Uhlén P, Burch PM, Zito CI, Estrada M, Ehrlich BE, Bennett AM (2006) Gain-of-function/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. Proc Natl Acad Sci U S A 103:2160–2165. https://doi.org/10.1073/pnas.0510876103
Sarkozy A, Digilio MC, Dallapiccola B (2008) Leopard syndrome. Orphanet J Rare Dis 3:13. https://doi.org/10.1186/1750-1172-3-13
Digilio MC, Sarkozy A, de Zorzi A, Pacileo G, Limongelli G, Mingarelli R et al (2006) LEOPARD syndrome: clinical diagnosis in the first year of life. Am J Med Genet A 140:740–746. https://doi.org/10.1002/ajmg.a.31156
Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 281:6785–6792. https://doi.org/10.1074/jbc.M513068200
Sobreira NLM, Cirulli ET, Avramopoulos D, Wohler E, Oswald GL, Stevens EL et al (2010) Whole-genome sequencing of a single proband together with linkage analysis identifies a mendelian disease gene. PLoS Genet 6:e1000991. https://doi.org/10.1371/journal.pgen.1000991
Kim HKW, Feng G-S, Chen D, King PD, Kamiya N (2014) Targeted disruption of Shp2 in chondrocytes leads to metachondromatosis with multiple cartilaginous protrusions. J Bone Miner Res 29:761–769. https://doi.org/10.1002/jbmr.2062
Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafé L, Superti-Furga A et al (2011) Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet 7:e1002050. https://doi.org/10.1371/journal.pgen.1002050
Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML (2014) SHP2 Regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLOS Genet 10:e1004364. https://doi.org/10.1371/journal.pgen.1004364
Yang W, Neel BG (2013) From an orphan disease to a generalized molecular mechanism. Rare Dis 1:e26657. https://doi.org/10.4161/rdis.26657
Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q et al (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499:491–495. https://doi.org/10.1038/nature12396
Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K et al (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 64:8816–8820. https://doi.org/10.1158/0008-5472.CAN-04-1923
Martinelli S, Carta C, Flex E, Binni F, Cordisco EL, Moretti S et al (2006) Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet Cytogenet 166:124–129. https://doi.org/10.1016/j.cancergencyto.2005.10.003
Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG et al (2016) Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535:148–152. https://doi.org/10.1038/nature18621
Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D et al (2018) RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20:1064–1073. https://doi.org/10.1038/s41556-018-0169-1
Sun B, Jensen NR, Chung D, Yang M, LaRue AC, Cheung HW et al (2019) Synergistic effects of SHP2 and PI3K pathway inhibitors in GAB2-overexpressing ovarian cancer. Am J Cancer Res 9:145–159
Prahallad A, Heynen GJJE, Germano G, Willems SM, Evers B, Vecchione L et al (2015) PTPN11 Is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep 12:1978–1985. https://doi.org/10.1016/j.celrep.2015.08.037
Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D et al (2018) Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med 24:954–960. https://doi.org/10.1038/s41591-018-0024-8
Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ et al (2018) SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov 8:1237–1249. https://doi.org/10.1158/2159-8290.CD-18-0444
Kerr DL, Haderk F, Bivona TG (2021) Allosteric SHP2 inhibitors in cancer: targeting the intersection of RAS, resistance, and the immune microenvironment. Curr Opin Chem Biol 62:1–12. https://doi.org/10.1016/j.cbpa.2020.11.007
Mohi MG, Neel BG (2007) The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev 17:23–30. https://doi.org/10.1016/j.gde.2006.12.011
Shen D, Chen W, Zhu J, Wu G, Shen R, Xi M et al (2020) Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur J Med Chem 190:112117. https://doi.org/10.1016/j.ejmech.2020.112117
Tajan M, de Rocca SA, Valet P, Edouard T, Yart A (2015) SHP2 sails from physiology to pathology. Eur J Med Genet 58:509–525. https://doi.org/10.1016/j.ejmg.2015.08.005
Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F et al (1997) Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16:2352–2364. https://doi.org/10.1093/emboj/16.9.2352
Saxton TM, Pawson T (1999) Morphogenetic movements at gastrulation require the SH2 tyrosine phosphatase Shp2. Proc Natl Acad Sci U S A 96:3790–3795. https://doi.org/10.1073/pnas.96.7.3790
Fornaro M, Burch PM, Yang W, Zhang L, Hamilton CE, Kim JH et al (2006) SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J Cell Biol 175:87–97. https://doi.org/10.1083/jcb.200602029
Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T et al (2004) Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell 13:341–355. https://doi.org/10.1016/s1097-2765(04)00050-4
Lapinski PE, Meyer MF, Feng G-S, Kamiya N, King PD (2013) Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis Model Mech 6:1448–1458. https://doi.org/10.1242/dmm.012849
de Crombrugghe B, Lefebvre V, Behringer RR, Bi W, Murakami S, Huang W (2000) Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol 19:389–394. https://doi.org/10.1016/s0945-053x(00)00094-9
Zuo C, Wang L, Kamalesh RM, Bowen ME, Moore DC, Dooner MS et al (2018) SHP2 regulates skeletal cell fate by modifying SOX9 expression and transcriptional activity. Bone Res 6:12. https://doi.org/10.1038/s41413-018-0013-z
Wang L, Yang H, Huang J, Pei S, Wang L, Feng JQ et al (2021) Targeted Ptpn11 deletion in mice reveals the essential role of SHP2 in osteoblast differentiation and skeletal homeostasis. Bone Res 9:6. https://doi.org/10.1038/s41413-020-00129-7
Kim HKW, Aruwajoye O, Sucato D, Richards BS, Feng G-S, Chen D et al (2013) Induction of SHP2 deficiency in chondrocytes causes severe scoliosis and kyphosis in mice. Spine 38:E1307-1312. https://doi.org/10.1097/BRS.0b013e3182a3d370
Wang L, Huang J, Moore DC, Zuo C, Wu Q, Xie L et al (2017) SHP2 regulates the osteogenic fate of growth plate hypertrophic chondrocytes. Sci Rep 7:12699. https://doi.org/10.1038/s41598-017-12767-9
Kamiya N, Shen J, Noda K, Kitami M, Feng G-S, Chen D et al (2015) SHP2-deficiency in chondrocytes deforms orofacial cartilage and ciliogenesis in mice. J Bone Miner Res 30:2028–2032. https://doi.org/10.1002/jbmr.2541
Qu CK, Nguyen S, Chen J, Feng GS (2001) Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood 97:911–914. https://doi.org/10.1182/blood.v97.4.911
Broxmeyer HE, Etienne-Julan M, Gotoh A, Braun SE, Lu L, Cooper S et al (2013) Hematopoietic colony formation from human growth factor-dependent TF1 cells and human cord blood myeloid progenitor cells depends on SHP2 phosphatase function. Stem Cells Dev 22:998–1006. https://doi.org/10.1089/scd.2012.0478
Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277. https://doi.org/10.1023/a:1008942828960
Kim H-J, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ et al (2006) Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest 116:2152–2160. https://doi.org/10.1172/JCI28084
Yang C, McCoy K, Davis JL, Schmidt-Supprian M, Sasaki Y, Faccio R et al (2010) NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS ONE 5:e15383. https://doi.org/10.1371/journal.pone.0015383
Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J (2010) Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr Pharm Des 16:1843–1862. https://doi.org/10.2174/138161210791209027
Zeng L-F, Zhang R-Y, Yu Z-H, Li S, Wu L, Gunawan AM et al (2014) Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem 57:6594–6609. https://doi.org/10.1021/jm5006176
He R, Zeng L-F, He Y, Zhang S, Zhang Z-Y (2013) Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J 280:731–750. https://doi.org/10.1111/j.1742-4658.2012.08718.x
Garcia Fortanet J, Chen CH-T, Chen Y-NP, Chen Z, Deng Z, Firestone B et al (2016) Allosteric inhibition of SHP2: identification of a potent, selective, and orally efficacious phosphatase inhibitor. J Med Chem 59:7773–7782. https://doi.org/10.1021/acs.jmedchem.6b00680
Reszka AA, Rodan GA (2003) Bisphosphonate mechanism of action. Curr Rheumatol Rep 5:65–74. https://doi.org/10.1007/s11926-003-0085-6
Chen L, Sung S-S, Yip MLR, Lawrence HR, Ren Y, Guida WC et al (2006) Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol 70:562–570. https://doi.org/10.1124/mol.106.025536
Zhang B, Du Y, Lu W, Yan X, Yang Q, Yang W et al (2016) Increased activity of Src homology 2 domain containing phosphotyrosine phosphatase 2 (Shp2) regulates activity-dependent AMPA receptor trafficking. J Biol Chem 291:18856–18866. https://doi.org/10.1074/jbc.M116.714501
Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. https://doi.org/10.1146/annurev.neuro.25.112701.142758
Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110:443–455. https://doi.org/10.1016/s0092-8674(02)00897-8
Hellmuth K, Grosskopf S, Lum CT, Würtele M, Röder N, von Kries JP et al (2008) Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A 105:7275–7280. https://doi.org/10.1073/pnas.0710468105
Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of β-catenin phosphorylation. Mol Biol Cell 23:4212–4225. https://doi.org/10.1091/mbc.E12-01-0038
Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y et al (1950) Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol 2014(193):2801–2811. https://doi.org/10.4049/jimmunol.1303463
Zhao L, Xia J, Li T, Zhou H, Ouyang W, Hong Z et al (2016) Shp2 deficiency impairs the inflammatory response against haemophilus influenzae by regulating macrophage polarization. J Infect Dis 214:625–633. https://doi.org/10.1093/infdis/jiw205
Li F-F, Shen J, Shen H-J, Zhang X, Cao R, Zhang Y et al (1950) Shp2 plays an important role in acute cigarette smoke-mediated lung inflammation. J Immunol 2012(189):3159–3167. https://doi.org/10.4049/jimmunol.1200197
Li S, Wang L, Zhao Q, Liu Y, He L, Xu Q et al (2014) SHP2 positively regulates tgfβ1-induced epithelial-mesenchymal transition modulated by its novel interacting protein hook1. J Biol Chem 289:34152–34160. https://doi.org/10.1074/jbc.M113.546077
Chen J, Cao Z, Guan J (2018) SHP2 inhibitor PHPS1 protects against atherosclerosis by inhibiting smooth muscle cell proliferation. BMC Cardiovasc Disord 18:72. https://doi.org/10.1186/s12872-018-0816-2
Zhang Z-Y (2002) Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol 42:209–234. https://doi.org/10.1146/annurev.pharmtox.42.083001.144616
Zhang X, He Y, Liu S, Yu Z, Jiang Z-X, Yang Z et al (2010) Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). J Med Chem 53:2482–2493. https://doi.org/10.1021/jm901645u
Sharma N, Everingham S, Ramdas B, Kapur R, Craig AWB (1950) SHP2 phosphatase promotes mast cell chemotaxis toward stem cell factor via enhancing activation of the Lyn/Vav/Rac signaling axis. J Immunol 2014(192):4859–4866. https://doi.org/10.4049/jimmunol.1301155
Sharma N, Kumar V, Everingham S, Mali RS, Kapur R, Zeng L-F et al (2012) SH2 domain-containing phosphatase 2 is a critical regulator of connective tissue mast cell survival and homeostasis in mice. Mol Cell Biol 32:2653–2663. https://doi.org/10.1128/MCB.00308-12
Sharma N, Everingham S, Zeng L-F, Zhang Z-Y, Kapur R, Craig AWB (2014) Oncogenic KIT-induced aggressive systemic mastocytosis requires SHP2/PTPN11 phosphatase for disease progression in mice. Oncotarget 5:6130–6141. https://doi.org/10.18632/oncotarget.2177
Mali RS, Ma P, Zeng L-F, Martin H, Ramdas B, He Y et al (2012) Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood 120:2669–2678. https://doi.org/10.1182/blood-2011-08-375873
Maeshima K, Stanford SM, Hammaker D, Sacchetti C, Zeng L, Ai R et al (2016) Abnormal PTPN11 enhancer methylation promotes rheumatoid arthritis fibroblast-like synoviocyte aggressiveness and joint inflammation. JCI Insight 1:e86580. https://doi.org/10.1172/jci.insight.86580
Wang J, Mizui M, Zeng L-F, Bronson R, Finnell M, Terhorst C et al (2016) Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus. J Clin Invest 126:2077–2092. https://doi.org/10.1172/JCI87037
Grosskopf S, Eckert C, Arkona C, Radetzki S, Böhm K, Heinemann U et al (2015) Selective inhibitors of the protein tyrosine phosphatase SHP2 block cellular motility and growth of cancer cells in vitro and in vivo. ChemMedChem 10:815–826. https://doi.org/10.1002/cmdc.201500015
Griger J, Schneider R, Lahmann I, Schöwel V, Keller C, Spuler S et al (2017) Loss of Ptpn11 (Shp2) drives satellite cells into quiescence. Elife 6:e21552. https://doi.org/10.7554/eLife.21552
Sun Y-J, Zhuo Z-L, Xian H-P, Chen K-Z, Yang F, Zhao X-T (2017) Shp2 regulates migratory behavior and response to EGFR-TKIs through ERK1/2 pathway activation in non-small cell lung cancer cells. Oncotarget 8:91123–91133. https://doi.org/10.18632/oncotarget.20249
LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H et al (2020) Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J Med Chem 63:13578–13594. https://doi.org/10.1021/acs.jmedchem.0c01170
Yuan X, Bu H, Zhou J, Yang C-Y, Zhang H (2020) Recent advances of SHP2 inhibitors in cancer therapy: current development and clinical application. J Med Chem 63:11368–11396. https://doi.org/10.1021/acs.jmedchem.0c00249
Sun X, Ren Y, Gunawan S, Teng P, Chen Z, Lawrence HR et al (2018) Selective inhibition of leukemia-associated SHP2E69K mutant by the allosteric SHP2 inhibitor SHP099. Leukemia 32:1246–1249. https://doi.org/10.1038/s41375-018-0020-5
Xie J, Si X, Gu S, Wang M, Shen J, Li H et al (2017) Allosteric inhibitors of SHP2 with therapeutic potential for cancer treatment. J Med Chem 60:10205–10219. https://doi.org/10.1021/acs.jmedchem.7b01520
Gripp KW, Schill L, Schoyer L, Stronach B, Bennett AM, Blaser S et al (2020) The sixth international RASopathies symposium: precision medicine-From promise to practice. Am J Med Genet A 182:597–606. https://doi.org/10.1002/ajmg.a.61434
Choi E, Kikuchi S, Gao H, Brodzik K, Nassour I, Yopp A et al (2019) Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat Commun 10:1473. https://doi.org/10.1038/s41467-019-09318-3
Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A et al (2010) Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142:296–308. https://doi.org/10.1016/j.cell.2010.06.003
Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D et al (2010) Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142:309–319. https://doi.org/10.1016/j.cell.2010.06.002
Kostallari E, Hirsova P, Prasnicka A, Verma VK, Yaqoob U, Wongjarupong N et al (2018) Hepatic stellate cell-derived platelet-derived growth factor receptor-alpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology 68:333–348. https://doi.org/10.1002/hep.29803
Wang J, Huang L, Huang Y, Jiang Y, Zhang L, Feng G et al (2021) Therapeutic effect of the injectable thermosensitive hydrogel loaded with SHP099 on intervertebral disc degeneration. Life Sci 266:118891. https://doi.org/10.1016/j.lfs.2020.118891
Celil AB, Campbell PG (2005) BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem 280:31353–31359. https://doi.org/10.1074/jbc.M503845200
Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT (2012) Interactions between extracellular signal-regulated kinase 1/2 and P38 map kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res 27:538–551. https://doi.org/10.1002/jbmr.561
Nabinger SC, Li X, Ramdas B, He Y, Zhang X, Zeng L et al (2013) The Protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia 27:398–408. https://doi.org/10.1038/leu.2012.308
Chen M-J, Wang Y-C, Wu D-W, Chen C-Y, Lee H (2019) Association of nuclear localization of SHP2 and YAP1 with unfavorable prognosis in non-small cell lung cancer. Pathol - Res Pract 215:801–806. https://doi.org/10.1016/j.prp.2019.01.027
Zhang Y, Lu W, Zhao Q, Chen J, Wang T, Ji J (2022) The role of the protein tyrosine phosphatase SHP2 in ossification. Dev Dyn 251:748–758. https://doi.org/10.1002/dvdy.449
Paccoud R, Saint-Laurent C, Piccolo E, Tajan M, Dortignac A, Pereira O et al (2021) SHP2 drives inflammation-triggered insulin resistance by reshaping tissue macrophage populations. Sci Transl Med 13:eabe2587. https://doi.org/10.1126/scitranslmed.abe2587
Yue X, Han T, Hao W, Wang M, Fu Y (2020) SHP2 knockdown ameliorates liver insulin resistance by activating IRS-2 phosphorylation through the AKT and ERK1/2 signaling pathways. FEBS Open Bio 10:2578–2587. https://doi.org/10.1002/2211-5463.12992
Chang C-J, Lin C-F, Chen B-C, Lin P-Y, Chen C-L (2022) SHP2: The protein tyrosine phosphatase involved in chronic pulmonary inflammation and fibrosis. IUBMB Life 74:131–142. https://doi.org/10.1002/iub.2559
Acknowledgements
This work was supported by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs (VA Merit Award to A.C.L., BX000333 and BX005168).
Author information
Authors and Affiliations
Contributions
All authors read and approved of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Nathaniel R. Jensen, Ryan R. Kelly, Kirsten D. Kelly, Stephanie K. Khoo, Sara J. Sidles and Amanda C. LaRue declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jensen, N.R., Kelly, R.R., Kelly, K.D. et al. From Stem to Sternum: The Role of Shp2 in the Skeleton. Calcif Tissue Int 112, 403–421 (2023). https://doi.org/10.1007/s00223-022-01042-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-022-01042-3