
Abstract
It has been suggested that interleukin-6 (IL-6) plays a key role in the pathogenesis of rheumatoid arthritis (RA), including osteoporosis not only in inflamed joints but also in the whole body. However, previous in vitro studies regarding the effects of IL-6 on osteoblast differentiation are inconsistent. The aim of this study was to examine the effects and signal transduction of IL-6 on osteoblast differentiation in MC3T3-E1 cells and primary murine calvarial osteoblasts. IL-6 and its soluble receptor significantly reduced alkaline phosphatase (ALP) activity, the expression of osteoblastic genes (Runx2, osterix, and osteocalcin), and mineralization in a dose-dependent manner, which indicates negative effects of IL-6 on osteoblast differentiation. Signal transduction studies demonstrated that IL-6 activated not only two major signaling pathways, SHP2/MEK/ERK and JAK/STAT3, but also the SHP2/PI3K/Akt2 signaling pathway. The negative effect of IL-6 on osteoblast differentiation was restored by inhibition of MEK as well as PI3K, while it was enhanced by inhibition of STAT3. Knockdown of MEK2 and Akt2 transfected with siRNA enhanced ALP activity and gene expression of Runx2. These results indicate that IL-6 negatively regulates osteoblast differentiation through SHP2/MEK2/ERK and SHP2/PI3K/Akt2 pathways, while affecting it positively through JAK/STAT3. Inhibition of MEK2 and Akt2 signaling in osteoblasts might be of potential use in the treatment of osteoporosis in RA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation-mediated bone loss is a major feature of various bone diseases, including rheumatoid arthritis (RA). Interleukin-6 (IL-6) contributes to the development of arthritis and is present at high concentrations in the serum and synovial fluid of patients with RA [1–4]. Soluble IL-6 receptor (sIL-6R) is also elevated in the serum and synovial fluid of RA patients [5, 6], and IL-6 exerts its action by binding either to its membrane-bound receptor (mIL-6R) or to sIL-6R. Moreover, IL-6 is closely associated with the expression of receptor activator of NF-κB ligand (RANKL) in osteoblasts [7]. That is to say, IL-6 acts indirectly on osteoclastogenesis by stimulating the release of RANKL by cells within bone tissues such as osteoblasts [8]. It can unquestionably be said that IL-6 plays a major role in the pathogenesis of RA [9–12], including osteoporosis not only in inflamed joints but also in the whole body.
There have been several studies on the effect of IL-6 on bone turnover in animal models. In IL-6 knock-out mice, microstructure abnormalities in cortical bones and delayed fracture healing were observed [13, 14], in spite of the evident normal phenotype [15]. Also, bone loss after estrogen depletion was mitigated in IL-6-deficient mice, while a high level of IL-6 and bone loss are seen in wild-type mice [13]. Moreover, IL-6-overexpressed-transgenic mice develop osteopenia and defective ossification, in which the activity of mature osteoblasts is significantly decreased [16]. All these findings, together with studies on human RA patients [17, 18], indicate that IL-6 plays a major role in bone turnover and is an important regulator of bone homeostasis.
Recently, several biological agents have been introduced for the treatment of RA and have demonstrated not only potent anti-inflammatory effects but also inhibitory effects on joint destruction. Among these biological agents, tocilizumab, an anti-IL-6 receptor antibody, has been reported to increase serum bone formation markers in RA patients [19], suggesting that IL-6 has a negative effect on osteoblast differentiation. However, previous reports regarding the effects of IL-6 on osteoblast differentiation in vitro have been inconsistent [20]. IL-6 has been shown to decrease the expression of differentiation markers in osteoblasts [21, 22] and to inhibit bone formation [23], while it has been shown to induce osteoblast differentiation [24, 25].
Binding of IL-6 with sIL-6R or mIL-6R leads to subsequent homodimerization of the signal-transducing molecule gp130, followed by activation of two major intracellular signaling pathways, Janus protein tyrosine kinase (JAK)/signal transducer and activator of transcription factors (STAT) 3, or Src-homology domain 2 containing protein-tyrosine phosphatase (SHP2)/mitogen-activated protein kinase-extracellular signal–regulated kinase kinase (MEK)/mitogen-activated protein kinase (MAPK), also called extracellular signal-regulated kinase (ERK) [26]. There have been many reports in which the effects of IL-6 on JAK/STAT3 and SHP2/ERK signal transduction pathways have been studied in osteoblasts, though it is still controversial whether differentiation is enhanced by IL-6 [9, 20]. SHP2 can also form a tertiary complex with the scaffolding proteins Gab1/2 and the p85 subunit of phosphatidylinositol-3-kinase (PI3K) [27], which leads to activation of the Akt pathway. Several papers have so far reported that the PI3K/Akt pathway triggered by IL-6 plays important roles in various cells [28–32], but no reports have been published regarding the effect of IL-6 on this pathway in osteoblasts.
The purpose of this study was to clarify the effect of IL-6 on osteoblast differentiation in vitro, with consideration of intracellular signaling pathways in murine MC3T3-E1 osteoblastic cells and primary murine calvarial osteoblasts.
Materials and methods
Ethics statement
Prior to the study, all experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Osaka University School of Medicine.
Cell culture
MC3T3-E1 osteoblastic cells were purchased from Riken Cell Bank (Tsukuba, Japan). MC3T3-E1 cells were cultured in α-minimum essential medium (α-MEM) containing 10 % fetal bovine serum (FBS; Equitech-Bio, Kerrville, TX, USA) and 1 % penicillin and streptomycin at 37 °C in a humidified atmosphere of 5 % CO2. All media were purchased from Life Technologies Japan (Tokyo, Japan). Murine primary osteoblasts were isolated from the calvariae of 3-day-old C57BL/6 mice (Charles River Laboratories Japan, Inc, Osaka, Japan) by sequential collagenase digestion as described previously [33].
MC3T3-E1 cells and murine calvarial osteoblasts were seeded at 1 × 105 cells per well in 12-well plates. After the cells reached confluence, the medium was replaced to induce osteoblast differentiation. The differentiation medium contained 10 % FBS, 10 mM β-glycerophosphate, and 50 μg/ml ascorbic acid in the absence or presence of recombinant mouse (rm) IL-6 (R&D Systems, Inc., Minneapolis, MN, USA) (10, 50 ng/mL), and rm sIL-6R (R&D Systems) (100 ng/mL). The medium and reagents were renewed every 3 days.
To study signal transduction, the following inhibitors or vehicle (DMSO) (Sigma-Aldrich, St.Louis, MO, USA) were added to culture medium at several concentrations; MEK inhibitor (U0126; 1, 2.5, 5 μM; Cell Signaling Technology, Danvers, MA, USA), STAT3 inhibitor (V Stattic; 2.5, 5 μM; Calbiochem, La Jolla, CA, USA), PI3K inhibitor (LY294002; 1, 2.5, 5 μM; Cell Signaling Technology), and SHP2 inhibitor (PHPS1; 5, 20, 40 μM; Sigma-Aldrich). These inhibitors were added 1 h before treatment with IL-6/sIL-6R. All inhibitors were maintained until the end of the culture period at the indicated concentrations.
Alkaline phosphatase (ALP) staining and activity
MC3T3-E1 cells and murine calvarial osteoblasts were treated with or without IL-6/sIL-6R and signal pathway inhibitors after the cells reached confluence and were incubated for 6 days.
For ALP staining, after fixation with 10 % formalin, cells were washed twice with phosphate-buffered saline (PBS) (pH 7.4) and incubated with ALP substrate solution, 0.1 mg/ml naphthol AS-MX (Sigma-Aldrich), and 0.6 mg/ml fast violet B salt (Sigma-Aldrich) in 0.1 M Tris–HCl (pH 8.5) for 20 min.
To measure ALP activity, cells were washed twice with PBS and lysed in Mammalian Protein Extraction Reagent (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. ALP activity was assayed using p-nitrophenylphosphate as a substrate by an Alkaline Phosphatase Test Wako (Wako Pure Chemicals Industries, Ltd., Osaka, Japan), and the protein content was measured using the Bicinchoninic Acid Protein Assay Kit (Pierce).
Proliferation assay
MC3T3-E1 cells were cultured in 96-well plates at a concentration of 2.0 × 104 cells/cm2 in α-MEM containing 10 % FBS. Cells were incubated for 1 day, after which the medium was treated with IL-6/sIL-6R for 3 days. Cell proliferation was assessed using the Premix WST-1 Cell Proliferation Assay System (Takara Bio, Inc., Otsu, Japan) according to the manufacturer’ s instructions. We performed this assay every 24 h.
Alizarin red staining
After fixation with 10 % formalin, MC3T3-E1 cells and murine calvarial osteoblasts were washed with distilled water, and stained with alizarin red S solution (Sigma-Aldrich) (pH 6.0) for 10 min, followed by incubation in 100 mM cetylpyridinium chloride for 1 h at room temperature to dissolve and release calcium-bound alizarin red. The absorbance of the released alizarin red was then measured at 570 nm [34]. To measure the value of absorbance for alizarin red, the absorbance data were normalized by total DNA content. Total DNA was extracted using a DNeasy Blood & Tissue Kit (Qiagen, Düsseldorf, Germany).
Knockdown of MEK1, MEK2, Akt1 and Akt2 using RNA interference
MC3T3-E1 cells were transfected with small interfering RNAs (siRNA) using Lipofectamine RNAiMAX (Life Technologies Japan) according to the reverse transfection method in the manufacturer’s protocol.
The siRNAs for MEK2, Akt1 and Akt2 and that for MEK1 were purchased from Cell Signaling Technology and Qiagen, respectively, with negative controls for each molecule. MC3T3-E1 cells transfected with siRNA were seeded in 24-well plates at a concentration of 1.0 × 104 cells/cm2 for 48 h. The medium was then replaced with differentiation medium with vehicle or with 20 ng/ml IL-6 and 100 ng/ml sIL-6R and the cells were incubated for 3 days prior to use for further experiments.
Western blotting
Cells cultured in 6-well plates for 2 days were washed twice with PBS and then homogenized with 100 μl of Kaplan buffer (150 mM NaCl, 50 mM Tris–HCL pH 7.4, 1 % NP40, 10 % glycerol, and 1 tablet per 50 ml buffer of protease inhibitor cocktail and phosphatase inhibitor cocktail). The lysates were centrifuged at 13,000 rpm for 20 min at 4 °C, and the supernatants were used for electrophoresis after a protein assay using bovine serum albumin as standard. Western blotting was performed by use of the following antibodies purchased from Cell Signaling Technology, except for phosphate anti-Akt2 antibody from Enogene Biotech (New York, NY, USA): phosphate anti-STAT3 (Tyr705) (1:2000) and anti-STAT3 (1:1000); phosphate anti-Akt (Ser473) (1:2000), phosphate anti-Akt2 (Ser474) (1:1000), anti-Akt1, anti-Akt2, and anti-Akt (1:1000); phosphate anti-ERK (Thr202/Tyr204) (1:2000), anti-MEK1, anti-MEK2 and anti-ERK (1:1000); and phosphate anti-SHP2 (Tyr542) (1:1000). To control for protein loading, blots were additionally stained with anti-β actin antibody (1:1000).
Reverse transcription polymerase chain reaction (RT–PCR)
Total RNA was extracted from cells with an RNeasy Mini Kit (Qiagen), and first-strand cDNA was synthesized using SuperScript II RNase H-reverse transcriptase (Life Technologies Japan). Then PCR was performed using Ex Taq (Takara Bio) and the following primers:
-
Osteocalcin (forward primer 5′-CTCACTCTGCTGGCCCTG-3′; reverse primer 5′-CCGTAGATGCGTTTGTAGGC-3′);
-
Osterix (forward primer 5′-AGGCACAAAGAAGCCATAC-3′; reverse primer 5′-AATGAGTGAGGGAAGGGT-3′);
-
Runx2 (forward primer 5′-GCTTGATGACTCTAAACCTA-3′; reverse primer 5′-AAAAAGGGCCCAGTTCTGAA-3′);
-
GAPDH (forward primer 5′-TGAACGGGAAGCTCACTGG-3′; reverse primer 5′-TCCACCACCCTGTTGCTGTA-3′).
Quantitative real-time PCR analysis
We obtained cDNA by reverse transcription as mentioned above, and proceeded with real-time PCR using a Light Cycler system (Roche Applied Science, Basel, Switzerland). The SYBR Green assay using a Quantitect SYBR Green PCR Kit (Qiagen), in which each cDNA sample was evaluated in triplicate 20-μl reactions, was used for all target transcripts. Expression values were normalized to GAPDH.
Statistical analysis
The results are expressed as the mean ± standard error (SE). Between-group differences were assessed using the ANOVA test. A probability value of <0.05 was considered to indicate statistical significance.
Results
IL-6/sIL-6R does not affect proliferation, but significantly reduces ALP activity and expression of osteoblastic genes in MC3T3-E1 cells
We first measured the proliferation of MC3T3-E1 cells with IL-6. Cell proliferation did not show significant difference in any culture condition (Fig. 1a).

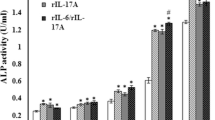
IL-6/sIL-6R significantly reduced ALP activity and expression of osteoblastic genes in MC3T3E1 cells, but did not affect proliferation. a Proliferation of MC3T3-E1 cells with IL-6/sIL-6R was examined. Cells were pre-incubated for 1 day and then the medium was treated with or without IL-6/sIL-6R for 3 days. Cell proliferation assay was performed daily throughout the 4 days of incubation. Cell proliferation did not show significant differences in any culture condition. b ALP staining was performed in MC3T3-E1 cells treated with or without IL-6/sIL-6R for 6 days. Apparently significant reduction of ALP staining was recognized in cells treated with either 10 or 50 ng/ml IL-6. c ALP activity of the lysates of MC3T3-E1 cells treated with or without IL-6/sIL-6R for 6 days was measured using p-nitrophenylphosphate as a substrate. IL-6/sIL-6R significantly reduced ALP activity in a dose-dependent manner. d Total RNA was extracted from MC3T3-E1 cells treated with or without IL-6/sIL-6R for 6 days and subjected to RT-PCR for osteoblastic genes Runx2, osterix, and osteocalcin. Apparently significant reduction of osteoblastic gene expression was recognized in cells treated with either 10 or 50 ng/ml IL-6. e Real-time PCR for Runx2, osterix, and osteocalcin was performed for quantitative analysis. Data were normalized to GAPDH expression and are shown as the ratio of expression compared to control cells treated with vehicle. The expression of osteoblastic genes was significantly downregulated by IL-6/sIL-6R in a dose-dependent manner. Representative data from at least 3 independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; *P < 0.05; **P < 0.001; ***P < 0.001
To investigate the influence of IL-6 treatment on osteoblast differentiation, we examined ALP activity in MC3T3-E1 cells. As shown in Fig. 1b and c, IL-6/sIL-6R significantly reduced ALP activity in a dose-dependent manner. The single addition of sIL-6R did not show a significant difference as compared to the negative control with vehicle. As shown in Fig. 1d and e, gene expression of Runx2, osterix and osteocalcin was significantly downregulated by IL-6/sIL-6R in a dose-dependent manner. Again, the single addition of sIL-6R did not show significant difference as compared to the negative control with vehicle.
IL-6/sIL-6R significantly inhibits mineralization of extracellular matrix (ECM) in MC3T3-E1 cells
As shown in Fig. 2a, IL-6/sIL-6R significantly inhibited the mineralized area in a dose-dependent manner. The single addition of sIL-6R did not show a significant difference as compared to the negative control with vehicle (Fig. 2a). Quantitative analysis of mineralization by measuring the absorbance of alizarin red revealed a significant decrease by IL-6/sIL-6R in a dose-dependent manner (Fig. 2b).

IL-6/sIL-6R significantly inhibited the mineralization of ECM in MC3T3E1 cells. MC3T3-E1 cells were treated with or without IL-6/sIL-6R and were incubated for 21 days. a After fixation, the cells were stained with alizarin red solution. Apparently significant reduction of alizarin red staining was recognized in the cells treated with either 10, 25, or 50 ng/ml IL-6. b Matrix mineralization was quantified by the measurement of absorbance of alizarin red and normalized by total DNA content. Matrix mineralization was significantly reduced by IL-6/sIL-6R in a dose-dependent manner. Representative data from at least 3 independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; *P < 0.05; **P < 0.001; ***P < 0.001
IL-6/sIL-6R activates ERK, STAT3 and Akt2 signal transduction pathways in MC3T3-E1 cells
When MC3T3-E1 cells were incubated in the presence of IL-6/sIL-6R, phosphorylation of ERK, STAT3 and Akt was clearly observed at 15 min, and their activation became weaker at 30 min. When only sIL-6R was added, there was no apparent activation of ERK, STAT3, or Akt as compared to the negative control (Fig. 3a). As for Akt, the phosphorylation by IL-6/sIL-6R was recognized more strikingly as early as 5 min in a dose-dependent manner, both for whole and for Akt2 only, one of its three isoforms (Fig. 3b).

IL-6/sIL-6R-activated ERK, STAT3, and Akt2 signal transduction pathways in MC3T3-E1 cells. a MC3T3-E1 cells were treated with vehicle or with 10 or 50 ng/ml IL-6 and 100 ng/ml sIL-6R in a time-course experiment (0, 15, and 30 min). Western blot analysis was performed using cell lysates for the detection of ERK, STAT3, and Akt, either phosphorylated or not. IL-6/sIL-6R significantly induced the phosphorylation of ERK, STAT3, and Akt in a dose-dependent manner. b MC3T3-E1 cells were incubated with increasing concentrations of IL-6 and 100 ng/ml sIL-6R for 5 min. Western blotting was performed using cell lysates for the detection of ERK, STAT3, as well as Akt, either non-phosphorylated, phosphorylated, or the phosphorylated isoform Akt2. The phosphorylation of both whole Akt and Akt2 by IL-6/sIL-6R was recognized more strikingly in a dose-dependent manner. Representative data from at least three independent experiments are shown
IL-6-induced activation of ERK is enhanced by blocking the STAT3 signaling pathway, and IL-6-induced ERK and Akt signaling pathways negatively regulate each other reciprocally
The SHP2 inhibitor PHPS1 [35] inhibited IL-6-induced phosphorylation of ERK and Akt to the constitutive level, but did not inhibit STAT3 (Fig. 4a and Supplementary Fig. S1a), suggesting that the downstream pathways of SHP2 are ERK and Akt, not STAT3. The STAT3 inhibitor V Stattic inhibited the phosphorylation of STAT3 but enhanced ERK significantly (Fig. 4a and Supplementary Fig. S1a), suggesting that STAT3 could negatively regulate ERK, which is consistent with previous reports [36]. The MEK/ERK inhibitor U0126 completely inhibited both constitutive and IL-6-induced phosphorylation of ERK but enhanced those of Akt. Moreover, the PI3K/Akt inhibitor LY294002 completely inhibited both constitutive and IL-6-induced phosphorylation of Akt but enhanced those of ERK (Fig. 4b and Supplementary Fig. S1b). From these findings, we concluded that IL-6-induced ERK and Akt signaling pathways, both of which are downstream of SHP2, can negatively regulate each other reciprocally.

IL-6-induced activation of ERK was enhanced by blocking the STAT3 signaling pathway, and IL-6-induced ERK and Akt signaling pathways negatively regulated each other reciprocally. a MC3T3-E1 cells were stimulated with 10 ng/ml IL-6 and 100 ng/ml sIL-6R (15 min) after pretreatment either with PHPS1 (5, 20, 40 μM; 1 h), with U0126 (5 μM; 1 h), or with V Stattic (5 μM; 1 h), and the cell lysates were subjected to Western blotting. PHPS1 inhibited IL-6-induced phosphorylation of ERK and Akt to the constitutive level, but not of STAT3. IL-6-induced activation of ERK was enhanced by V Stattic. b MC3T3-E1 cells were treated with vehicle or with 10 ng/ml IL-6 and 100 ng/ml sIL-6R (15 min) after pretreatment either with U0126 (5 μM; 1 h) or with LY294002 (10 μM; 1 h), and the cell lysates were subjected to Western blotting. Both constitutive and IL-6-induced phosphorylation of Akt and ERK were enhanced by treatment with U0126 and LY294002, respectively. Representative data from at least three independent experiments are shown
The negative effects of IL-6 on osteoblast differentiation are restored by inhibition of MEK, PI3K and SHP2, while they are enhanced by inhibition of STAT3
To identify the intracellular signaling pathways associated with the downregulation of osteoblast differentiation, the effects of various signal transduction inhibitors, consisting of a MEK inhibitor (U0126), PI3K inhibitor (LY294002), SHP2 inhibitor (PHPS1), and STAT3 inhibitor (V Stattic), were assessed for ALP activity, the expression of osteoblastic genes (Runx2, osterix and osteocalcin), and the mineralization of ECM.
The negative effect of IL-6/sIL-6R on ALP activity was restored by treatment with either U0126, LY294002, or PHPS1 in a dose-dependent manner. On the other hand, the negative effect of IL-6/sIL-6R on ALP activity was enhanced by treatment with V Stattic (Fig. 5a). These results indicate that the SHP2-associated signal transduction molecules MEK/ERK and PI3K/Akt have a negative effect on osteoblast differentiation, whereas the JAK-associated molecule STAT3 has a positive effect.

The negative effects of IL-6 on ALP activity and the expression of osteoblastic genes were restored by inhibition of MEK, PI3K, and SHP2, while they were enhanced by inhibition of STAT3. MC3T3-E1 cells were pretreated either with U0126 (1, 2.5, 5 μM; 1 h), LY294002 (1, 2.5, 5 μM; 1 h), PHPS1 (5, 20 μM; 1 h), or V Stattic (5 μM; 1 h), then stimulated either with 10 ng/ml IL-6 and 100 ng/ml sIL-6R or with vehicle and incubated for 6 days. (a) ALP activity of the cell lysates was measured using p-nitrophenylphosphate as a substrate. The negative effect of IL-6 on ALP activity was restored by treatment with either U0126, LY294002, or PHPS1 in a dose-dependent manner, while it was enhanced by treatment with V Stattic. b Total RNA was extracted and real-time PCR for Runx2, osterix, and osteocalcin was performed. Data were normalized to GAPDH expression and are shown as the ratio of gene expression compared to control cells treated with vehicle. The negative effect of IL-6 on expression of osteoblastic genes was restored by treatment either with U0126, LY294002, or PHPS1 in a dose-dependent manner, while it was enhanced by treatment with V Stattic. Representative data from at least three independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; # P < 0.05; ## P < 0.001; ### P < 0.001, compared to the group treated with vehicle. *P < 0.05; **P < 0.001; ***P < 0.001, compared to group treated with IL-6/sIL-6R
The negative effect of IL-6/sIL-6R on the expression of osteoblastic genes (Runx2, osterix and osteocalcin) was also restored by treatment with either U0126, LY294002, or PHPS1 in a dose-dependent manner, while it was enhanced by treatment with V Stattic (Fig. 5b). Moreover, a high dose of PHPS1, 20 μM, caused significantly upregulated expression of osteocalcin.
For mineralization of ECM, the negative effect of IL-6/sIL-6R was restored by treatment with either U0126, LY294002, or PHPS1. As with ALP activity and osteoblastic gene expression, the negative effect of IL-6/sIL-6R on mineralization was enhanced by treatment with V Stattic (Fig. 6a, b). ALP activity, osteoblastic gene expression, and mineralization of ECM in cells treated only with each inhibitor demonstrated the same behavior (Figs. 5, 6).

The negative effect of IL-6 on mineralization of ECM was restored by inhibition of MEK, PI3K, and SHP2, while it was enhanced by inhibition of STAT3. MC3T3-E1 cell were pretreated either with U0126 (1 μM; 1 h), LY294002 (1 μM; 1 h), PHPS1 (20 μM; 1 h), or V Stattic (2.5 μM; 1 h), then stimulated with either 10 ng/ml IL-6 and 100 ng/ml sIL-6R or with vehicle and incubated for 21 days. a After fixation, the cells were stained with alizarin red solution. The reduction of alizarin red staining by IL-6/sIL-6R was restored in cells treated with either U0126, LY294002, or PHPS1, while it was enhanced in those treated with V Stattic. b Quantification of matrix mineralization was by measurement of absorbance for alizarin red normalized by total DNA content. The reduction of matrix mineralization by IL-6/sIL-6R was restored in cells treated with either U0126, LY294002, or PHPS1, while it was enhanced in those treated with V Stattic. Representative data from at least three independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; # P < 0.05; ## P < 0.001; ### P < 0.001, compared to the group treated with vehicle. *P < 0.05; **P < 0.001; ***P < 0.001, compared to group treated with IL-6/sIL-6R
Furthermore, the negative effects of ALP activity, osteoblastic gene expression and mineralization of ECM by stimulation with IL-6/sIL-6R were compared for levels in the presence and in the absence of each inhibitor. The negative effects on osteoblast differentiation by IL-6/sIL-6R showed a tendency to decrease in the presence of each inhibitor, as compared to the absence of inhibitors (Figs. 5, 6). The negative effects were decreased by 15–44, 20–61, 7–140, and 21–80 % in the presence of U0126, LY294002, PHPS1 and V Stattic, respectively, as compared to the absence of inhibitors. These results indicate that the effects of IL-6/sIL-6R on osteoblast differentiation might be mediated either by MEK/ERK, PI3K/Akt, or JAK/STAT3 pathways.
Knockdown of MEK2 and Akt2 via siRNA transfection restores ALP activity and Runx2 gene expression
To further confirm the effects of MEK and Akt inhibition on osteoblast differentiation in MC3T3-E1 cells, we studied cell differentiation after knockdown of MEK and Akt. For each protein, RNAs of two isoforms were separately blocked: MEK1 and MEK2 for MEK, and Akt1 and Akt2 for Akt.
The protein expression level of each molecule was found to be diminished selectively at 48 h after transfection of the respective siRNAs (Fig. 7a). The ALP activity in MC3T3-E1 cells treated with IL-6/sIL-6R was restored by knockdown of MEK2 and Akt2 as compared to that in cells transfected with negative control siRNA. On the other hand, knockdown of MEK1 and Akt1 enhanced the negative effects of IL-6/sIL-6R on ALP activity (Fig. 7b) (ALP activity after transfection with each siRNA without IL-6/sIL-6R demonstrated the same behavior; Fig. 7b) Quantitative real-time PCR analysis revealed that the gene expressions of Runx2, osterix, and osteocalcin were restored by knockdown of MEK2. On the other hand, knockdown of Akt2 also restored Runx2, but decreased osteocalcin expression (Fig. 7c), while knockdown of Akt2 without IL-6/sIL-6R caused no significant difference in Runx2 expression (Fig. 7b). As was recognized for ALP activity, knockdown of MEK1 and Akt1 enhanced the downregulation of osteocalcin expression (Fig. 7b, c). Also, the negative effects of IL-6/sIL-6R on osteoblast differentiation showed some tendency to decrease with each knockdown compared to those without knockdown. The negative effects were decreased by 2–24, 4–27, 7–43, and 21–26 % with knockdown of MEK1, MEK2, Akt1, and Akt2, respectively, as compared to those without knockdown. These results indicate that IL-6 may suppress osteoblast differentiation through MEK2 and Akt2.

Knockdown of MEK2 and Akt2 in cells transfected with siRNA restored ALP activity and Runx2 gene expression. a MC3T3-E1 cells transfected with respective siRNAs were cultured for 48 h. Western blotting was performed using cell lysates stimulated with vehicle or with 20 ng/ml IL-6 and 100 ng/ml sIL-6R (15 min). Expression levels of each protein, MEK1, MEK2, Akt1, and Akt2, were selectively diminished at 48 h after transfection with respective siRNAs. b MC3T3-E1 cells transfected with respective siRNAs were incubated for 48 h after which the medium was changed to differentiation medium with vehicle or with 20 ng/ml IL-6 and 100 ng/ml sIL-6R. The cells were then incubated for 3 days to evaluate osteoblast differentiation. ALP activity in MC3T3-E1 cells treated with IL-6/sIL-6R was restored by knockdown of MEK2 and Akt2 as compared to that in cells transfected with negative control siRNA. c Expression of osteoblastic genes in MC3T3-E1 cells transfected with respective siRNAs was assessed by real-time PCR. The expression of each gene was normalized against GAPDH expression. The gene expressions of Runx2, osterix, and osteocalcin were restored by knockdown of MEK2. Knockdown of Akt2 also restored Runx2, but decreased osteocalcin. Representative data from at least three independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; # P < 0.05; ## P < 0.001; ### P < 0.001, compared to negative control group treated with vehicle. *P < 0.05; **P < 0.001; ***P < 0.001, compared to negative control group treated with IL-6/sIL-6R
IL-6/sIL-6R inhibits the differentiation of primary murine calvarial osteoblasts by activating phosphorylation of ERK, Akt2, and STAT3
Experiments were repeated with murine calvarial osteoblasts isolated from the calvariae of 3-day-old C57BL/6 mice. As was recognized in MC3T3-E1 cells, IL-6 inhibited ALP activity (Fig. 8a), the expression of osteoblastic genes (Fig. 8b), and mineralization (Fig. 8c, d) in a dose-dependent manner. Furthermore, IL-6 induced phosphorylation of ERK, Akt2, and STAT3 (Fig. 8e), which was exactly the same as with MC3T3-E1 cells.

IL-6/sIL-6R inhibited the differentiation of primary murine calvarial osteoblasts with the activated phosphorylation of ERK, Akt2, and STAT3. a ALP activity of lysates of murine calvarial osteoblasts treated with or without IL-6/sIL-6R for 6 days was measured using p-nitrophenylphosphate as a substrate. IL-6/sIL-6R significantly reduced ALP activity in a dose-dependent manner. b Total RNA was extracted from murine calvarial osteoblasts treated with or without IL-6/sIL-6R for 6 days, and real-time PCR for Runx2, osterix, and osteocalcin was performed. Data were normalized to GAPDH expression and are shown as the ratio of gene expression as compared to control cells treated with vehicle. The expression of osteoblastic genes was significantly downregulated by IL-6/sIL-6R in a dose-dependent manner. c Murine calvarial osteoblasts were treated with or without IL-6/sIL-6R and were cultured for 21 days. After fixation, the cells were stained with alizarin red solution. Apparently significant reduction of alizarin red staining was recognized in cells treated with either 10 or 50 ng/ml IL-6. d Matrix mineralization was quantified by measurement of absorbance for alizarin red normalized by total DNA content. IL-6/sIL-6R significantly inhibited mineralization of ECM in a dose-dependent manner. e Primary murine calvarial osteoblasts were treated with vehicle or 10 or 50 ng/ml IL-6 and 100 ng/ml sIL-6R in a time-course experiment (5, 15, and 30 min). Western blotting was performed using cell lysates. IL-6 significantly induced the phosphorylation of ERK, Akt2, and STAT3 in a dose-dependent manner. Representative data from at least three independent experiments are shown. Data are shown as mean ± SE. n.s. not significant; *P < 0.05; **P < 0.001; ***P < 0.001
Discussion
We examined the effects of IL-6 and its soluble receptor on the proliferation and differentiation of murine MC3T3-E1 osteoblastic cells and primary murine calvarial osteoblasts. Our results showed that they significantly reduced ALP activity, bone mineralization, and expression of the osteoblastic genes Runx2 osterix and osteocalcin, in a dose-dependent manner. From these experiments, we clearly demonstrated that IL-6 inhibited osteoblast differentiation of MC3T3-E1 cells and primary murine calvarial osteoblasts.
It has been demonstrated that the JAK/STAT3 signaling pathway has important roles both, in vivo and in vitro, in the differentiation of osteoblasts [37, 38]. Our results are consistent with previous reports and imply that the activation of STAT3 induced by IL-6 may induce osteoblast differentiation.
IL-6 activates another major intracellular signaling pathway, SHP2/ERK, and can also lead to the activation of an additional signaling cascade involving SHP2/PI3K/Akt. IL-6-induced activation of PI3K and downstream protein kinase Akt/PKB has been reported to play important roles in the proliferation of prostate cancer cells [30, 31], hepatoma cells [32], and multiple myeloma cells [29]. They were also reported to associate with neuroendocrine differentiation of prostate cancer cells induced by IL-6 [32]. In this study, we focused on the PI3K/Akt pathway triggered by IL-6, because no reports have demonstrated the role of IL-6 in the activation of PI3K/Akt signaling pathway in osteoblasts. We have demonstrated for the first time that IL-6-induced activation of Akt2, one of the downstream pathways of SHP2, may be a key player in the negative regulation of osteoblast differentiation induced by IL-6. Among the three isoforms of Akt, Akt1 and Akt2 are highly expressed in osteoblasts [39]. Mice lacking Akt1, the major isoform in bone tissue, exhibit osteopenia [40, 41], and the impact of Akt1 deficiency in osteoblast differentiation and bone development have also been published [39, 42–44], all of which are consistent with our results showing that knockdown of Akt1 signaling by siRNA inhibited osteoblast differentiation. In contrast, Mukherjee et al. [44] reported enhanced osteogenic differentiation in the absence of Akt1 in cell lines. Moreover, they reported that Akt2 was required for BMP2-initiated osteoblast differentiation of cultured murine mesenchymal stem cells, but that Akt1 was dispensable in this assay [45], which is inconsistent with our results showing that knockdown of Akt2 signaling by siRNA promoted osteoblast differentiation. These discrepancies might be due to the difference between cell types, i.e. intramembranous (calvariae) cells and endochondral (long bones) cells.
In this study, gene expression of osteocalcin, a late osteoblastic differentiation marker, was upregulated by treatment with a PI3K/Akt inhibitor, but was downregulated by knockdown of both Akt1 and Akt2. Moreover, a complete blockade with a high dose (more than 10 μM) of the PI3K/Akt inhibitor conversely downregulated the expression of osteocalcin (data not shown). This discrepancy may be due to the difference between the temporary or partial blockade by the inhibitor and constitutive knockdown by siRNA. Since bone formation has been reported to increase without impairment of mineralization and resorption even in osteocalcin-deficient mice [46], the expression of osteocalcin may not directly affect bone formation.
We have previously reported that osteoblast differentiation was significantly promoted by MEK inhibitor in BMP-2-treated C2C12 cells and MC3T3-E1 cells [47]. Our findings in the present study are consistent with our previous report and others [47–49] at the point that IL-6-induced activation of ERK significantly downregulated osteoblast differentiation. In addition, our results suggest that there might be different roles in osteoblast differentiation between MEK1 and MEK2. Constitutively active expression of MEK1 has been reported to accelerate bone development both in vitro [50] and in vivo [51], which is consistent with the results showing that knockdown of MEK1 inhibited osteoblast differentiation in the present study. As for MEK2, there are no reports concerning its roles in osteoblast differentiation, and we are the first to demonstrate that MEK2 may also be a key player in the negative regulation of osteoblast differentiation induced by IL-6. The effects of an MEK inhibitor that inhibits both MEK1 and MEK2 on bone formation are still controversial [52]. These controversies might be due to different roles played between MEK1 and MEK2 in osteoblast differentiation, and the effects of MEK inhibitors could depend on which pathway is predominantly inhibited in each study.
With respect to intracellular signaling pathways, our results showed that IL-6 triggers three signaling pathways, one of which has a conflicting function with the others. SHP2/ERK and SHP2/Akt2 negatively affects osteoblast differentiation, whereas JAK/STAT3 positively affects it (Fig. 9). In other cells, it is often that simultaneous activation of the SHP2/ERK and JAK/STAT3 cascades generate opposing, or at least different signals. In osteoclasts, for example, SHP2/ERK activation inhibits osteoclastogenesis [53], whereas STAT3 is a pro-osteoclastic molecule after phosphorylation on serine727 [54]. In myeloid leukemic M1 cells, STAT3 induces differentiation in vitro [55], whereas the SHP2/ERK pathway promotes their proliferation [56]. These examples suggest that the integration of opposing activities transduced by more than one pathway could provide a biologically balanced state in the end, leaving availability to respond to another physiological situation. Indeed, Hirano and colleagues [57] have proposed a “signaling orchestration” model in a single cell, where the balance or interplay of simultaneously generated contradictory signals eventually determines the biological outcome. Thus, the inconsistent results regarding the effects of IL-6 on osteoblast differentiation in previous reports could be explained by which intracellular signaling pathway was predominantly activated in each study. The balance of three signaling pathways could be influenced by such conditions as the variety of cultured cells, the stage of cell differentiation, and the employed culture conditions.

Schematic presentation of signaling pathways involved in osteoblast differentiation induced by IL-6. IL-6-induced novel SHP2/MEK2/ERK and SHP2/PI3K/Akt2 signal crosstalk in osteoblastic cells; ERK and Akt signaling pathways, both of which are downstream of SHP2, negatively regulate each other reciprocally. On the other hand, the STAT3 signaling pathway negatively regulates the ERK signaling pathway. MEK2/ERK and PI3K/Akt2 have negative effects on osteoblast differentiation, whereas STAT3 has a positive effect. Overall, IL-6 inhibits osteoblast differentiation through MEK2 and Akt2 signaling pathways
To the best of our knowledge, this is the first report of signal crosstalk in which IL-6-induced ERK and Akt signaling pathways negatively regulated each other in cultured osteoblastic cells. In this study, however, cancellation of the negative effects of IL-6/sIL-6R on osteoblast differentiation by inhibitors was incomplete as compared to the absence of inhibitor (Figs. 5, 6). This might be because ERK, Akt and STAT3 are all critical pathways in osteoblast differentiation even in the absence of IL-6/sIL-6R, and even though one pathway is blocked, another pathway is enhanced by reciprocal regulation in the crosstalk between IL-6-activated signaling pathways (Fig. 9). Our results demonstrated that a STAT3 inhibitor significantly enhanced IL-6-induced activation of ERK and SHP2, but not of Akt (Fig. 4a). SHP2 could predominantly lead to the activation of the ERK signaling pathway as compared to Akt, and the enhanced signaling of ERK may restrain the enhancement of the Akt signaling pathway in a negative feedback manner.
The results obtained from the present study show that SHP2, MEK and PI3K inhibitors would be of potential use for the treatment of osteoporotic changes in RA patients. In particular, SHP2 inhibitors not only could inhibit the negative effect of IL-6-induced MEK/ERK and PI3K/Akt2 signaling, but also enhance the positive effect of IL-6-induced STAT3 signaling on osteoblast differentiation [37]. However, since a pro-inflammatory effect of STAT3 on synovitis has been reported [36, 58], selective inhibition of MEK2 and Akt2 signaling in osteoblasts may be more promising in order to avoid the enhancement of synovitis and consequent joint destruction.
In conclusion, our study provides new insights in the pathophysiology as well as potential treatment options for bone loss in RA, focusing on osteoblast differentiation in vitro. Our results demonstrated that IL-6 could inhibit osteoblast differentiation through MEK2/ERK and PI3K/Akt2 signaling pathways, both of which are SHP2-dependent downstream signaling pathways.
References
Hashizume M, Mihara M (2011) The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis 2011:765624
Ito A, Itoh Y, Sasaguri Y, Morimatsu M, Mori Y (1992) Effects of interleukin-6 on the metabolism of connective tissue components in rheumatoid synovial fibroblasts. Arthritis Rheum 35:1197–1201
Nishimoto N, Kishimoto T (2004) Inhibition of IL-6 for the treatment of inflammatory diseases. Curr Opin Pharmacol 4:386–391
De Benedetti F, Robbioni P, Massa M, Viola S, Albani S, Martini A (1992) Serum interleukin-6 levels and joint involvement in polyarticular and pauciarticular juvenile chronic arthritis. Clin Exp Rheumatol 10:493–498
De Benedetti F, Massa M, Pignatti P, Albani S, Novick D, Martini A (1994) Serum soluble interleukin 6 (IL-6) receptor and IL-6/soluble IL-6 receptor complex in systemic juvenile rheumatoid arthritis. J Clin Invest 93:2114–2119
Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I, Yamaguchi A, Kishimoto T, Suda T, Kashiwazaki S (1996) Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res 11:88–95
Kwan Tat S, Padrines M, Theoleyre S, Heymann D, Fortun Y (2004) IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev 15:49–60
Palmqvist P, Persson E, Conaway HH, Lerner UH (2002) IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol 169:3353–3362
Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y (2010) Role for interleukin-6 in structural joint damage and systemic bone loss in rheumatoid arthritis. Joint Bone Spine 77:201–205
Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M et al (1988) Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 18:1797–1801
Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M, Kishimoto T (1998) Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA 95:8222–8226
Dasgupta B, Corkill M, Kirkham B, Gibson T, Panayi G (1992) Serial estimation of interleukin 6 as a measure of systemic disease in rheumatoid arthritis. J Rheumatol 19:22–25
Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan GA, Costantini F (1994) Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J 13:1189–1196
Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP (2007) Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone 41:928–936
Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368:339–342
De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, Vivarelli M, Muratori F, Berni S, Ballanti P, Ferrari S, Teti A (2006) Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 54:3551–3563
Naka T, Nishimoto N, Kishimoto T (2002) The paradigm of IL-6: from basic science to medicine. Arthritis Res 4(Suppl 3):S233–S242
Wong PK, Campbell IK, Egan PJ, Ernst M, Wicks IP (2003) The role of the interleukin-6 family of cytokines in inflammatory arthritis and bone turnover. Arthritis Rheum 48:1177–1189
Garnero P, Thompson E, Woodworth T, Smolen JS (2010) Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum 62:33–43
Franchimont N, Wertz S, Malaise M (2005) Interleukin-6: an osteotropic factor influencing bone formation? Bone 37:601–606
Li YP, Stashenko P (1992) Proinflammatory cytokines tumor necrosis factor-alpha and IL-6, but not IL-1, down-regulate the osteocalcin gene promoter. J Immunol 148:788–794
Peruzzi B, Cappariello A, Del Fattore A, Rucci N, De Benedetti F, Teti A (2012) c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat Commun 3:630
Hughes FJ, Howells GL (1993) Interleukin-6 inhibits bone formation in vitro. Bone Miner 21:21–28
Nishimura R, Moriyama K, Yasukawa K, Mundy GR, Yoneda T (1998) Combination of interleukin-6 and soluble interleukin-6 receptors induces differentiation and activation of JAK-STAT and MAP kinase pathways in MG-63 human osteoblastic cells. J Bone Miner Res 13:777–785
Taguchi Y, Yamamoto M, Yamate T, Lin SC, Mocharla H, DeTogni P, Nakayama N, Boyce BF, Abe E, Manolagas SC (1998) Interleukin-6-type cytokines stimulate mesenchymal progenitor differentiation toward the osteoblastic lineage. Proc Assoc Am Physicians 110:559–574
Ishihara K, Hirano T (2002) Molecular basis of the cell specificity of cytokine action. Biochim Biophys Acta 1592:281–296
Takahashi-Tezuka M, Yoshida Y, Fukada T, Ohtani T, Yamanaka Y, Nishida K, Nakajima K, Hibi M, Hirano T (1998) Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase. Mol Cell Biol 18:4109–4117
Hideshima T, Nakamura N, Chauhan D, Anderson KC (2001) Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene 20:5991–6000
Tu Y, Gardner A, Lichtenstein A (2000) The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res 60:6763–6770
Chung TD, Yu JJ, Kong TA, Spiotto MT, Lin JM (2000) Interleukin-6 activates phosphatidylinositol-3 kinase, which inhibits apoptosis in human prostate cancer cell lines. Prostate 42:1–7
Qiu Y, Robinson D, Pretlow TG, Kung HJ (1998) Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci USA 95:3644–3649
Chen RH, Chang MC, Su YH, Tsai YT, Kuo ML (1999) Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J Biol Chem 274:23013–23019
Schmidt K, Schinke T, Haberland M, Priemel M, Schilling AF, Mueldner C, Rueger JM, Sock E, Wegner M, Amling M (2005) The high mobility group transcription factor Sox8 is a negative regulator of osteoblast differentiation. J Cell Biol 168:899–910
Ratisoontorn C, Seto ML, Broughton KM, Cunningham ML (2005) In vitro differentiation profile of osteoblasts derived from patients with Saethre-Chotzen syndrome. Bone 36:627–634
Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W (2008) Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci USA 105:7275–7280
Ernst M, Inglese M, Waring P, Campbell IK, Bao S, Clay FJ, Alexander WS, Wicks IP, Tarlinton DM, Novak U, Heath JK, Dunn AR (2001) Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J Exp Med 194:189–203
Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, Ishihara K (2006) A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 39:505–512
Sims NA (2004) Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Investig 113:379–389
Kawamura N, Kugimiya F, Oshima Y, Ohba S, Ikeda T et al (2007) Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS One 2:e1058
Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N (2001) Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 15:2203–2208
Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA (2003) Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem 278:32124–32131
Vandoorne K, Magland J, Plaks V, Sharir A, Zelzer E, Wehrli F, Hemmings BA, Harmelin A, Neeman M (2010) Bone vascularization and trabecular bone formation are mediated by PKB alpha/Akt1 in a gene-dosage-dependent manner: in vivo and ex vivo MRI. Magn Reson Med 64:54–64
Choi YH, Choi HJ, Lee KY, Oh JW (2012) Akt1 regulates phosphorylation and osteogenic activity of Dlx3. Biochem Biophys Res Commun 425:800–805
Mukherjee A, Rotwein P (2012) Selective signaling by Akt1 controls osteoblast differentiation and osteoblast-mediated osteoclast development. Mol Cell Biol 32:490–500
Mukherjee A, Wilson EM, Rotwein P (2010) Selective signaling by Akt2 promotes bone morphogenetic protein 2-mediated osteoblast differentiation. Mol Cell Biol 30:1018–1027
Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, Bradley A, Karsenty G (1996) Increased bone formation in osteocalcin-deficient mice. Nature 382:448–452
Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, Itoh K (2002) Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res 17:1785–1794
Chaudhary LR, Avioli LV (2000) Extracellular-signal regulated kinase signaling pathway mediates downregulation of type I procollagen gene expression by FGF-2, PDGF-BB, and okadaic acid in osteoblastic cells. J Cell Biochem 76:354–359
Lin FH, Chang JB, Brigman BE (2011) Role of mitogen-activated protein kinase in osteoblast differentiation. J Orthop Res 29:204–210
Ge C, Xiao G, Jiang D, Franceschi RT (2007) Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol 176:709–718
Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S (2009) Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol 29:5843–5857
Schindeler A, Little DG (2006) Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res 21:1331–1338
Sims NA, Jenkins BJ, Quinn JM, Nakamura A, Glatt M, Gillespie MT, Ernst M, Martin TJ (2004) Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest 113:379–389
Duplomb L, Baud’huin M, Charrier C, Berreur M, Trichet V, Blanchard F, Heymann D (2008) Interleukin-6 inhibits receptor activator of nuclear factor kappaB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of Serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology 149:3688–3697
Yamanaka Y, Nakajima K, Fukada T, Hibi M, Hirano T (1996) Differentiation and growth arrest signals are generated through the cytoplasmic region of gp130 that is essential for Stat3 activation. EMBO J 15:1557–1565
Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T (1996) A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J 15:3651–3658
Hirano T, Matsuda T, Nakajima K (1994) Signal transduction through gp130 that is shared among the receptors for the interleukin 6 related cytokine subfamily. Stem Cells 12:262–277
Krause A, Scaletta N, Ji JD, Ivashkiv LB (2002) Rheumatoid arthritis synoviocyte survival is dependent on Stat3. J Immunol 169:6610–6616
Conflict of interest
All authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
774_2013_514_MOESM1_ESM.tif
Supplementary Fig. S1 IL-6-induced activation of ERK was enhanced by blocking the STAT3 signaling pathway, and IL-6-induced ERK and Akt signaling pathways negatively regulated each other reciprocally. (a) MC3T3-E1 cells were stimulated with 10 ng/ml IL-6 and 100 ng/ml sIL-6R (30 min) after pretreatment either with PHPS1 (5, 20, 40 μM; 1 h), U0126 (5 μM; 1 h), or V Stattic (5 μM; 1 h), and the cell lysates were subjected to Western blotting. PHPS1 inhibited IL-6-induced phosphorylation of ERK and Akt to the constitutive level, but not phosphorylation of STAT3. IL-6-induced activation of ERK was enhanced by V Stattic. (b) MC3T3-E1 cells were treated with vehicle or with 10 ng/ml IL-6 and 100 ng/ml sIL-6R (30 min) after pretreatment with either U0126 (5 μM; 1 h) or LY294002 (10 μM; 1 h), and and the cell lysates were subjected to Western blotting. Both constitutive and IL-6-induced phosphorylation of Akt and ERK were enhanced by treatment with U0126 and LY294002, respectively. Representative data from at least three independent experiments are shown. (TIFF 536 kb)
About this article
Cite this article
Kaneshiro, S., Ebina, K., Shi, K. et al. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Metab 32, 378–392 (2014). https://doi.org/10.1007/s00774-013-0514-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-013-0514-1