
Abstract
Matrix vesicles (MVs) are extracellular organelles produced by growth plate cartilage cells in a zone-specific manner. MVs are similar in size to exosomes, but they are tethered to the extracellular matrix (ECM) via integrins. Originally associated with matrix calcification, studies now show that they contain matrix processing enzymes and microRNA that are specific to their zone of maturation. MVs produced by costochondral cartilage resting zone (RC) chondrocytes are enriched in microRNA 503 whereas those produced by growth zone (GC) chondrocytes are enriched in microRNA 122. MVs are packaged by chondrocytes under hormonal and factor regulation and release of their contents into the ECM is also under hormonal control, suggesting that their microRNA might have a regulatory role in growth plate proliferation and maturation. To test this, we selected a subset of these enriched microRNAs and transfected synthetic mimics back into RC and GC cells. Transfecting growth plate chondrocytes with select microRNA produced a broad range of phenotypic responses indicating that MV-based microRNAs are involved in the regulation of these cells. Specifically, microRNA 122 drives both RC and GC cells toward a proliferative phenotype, stabilizes the matrix and inhibits differentiation whereas microRNA 22 exerts control over regulatory factor production. This study demonstrates the strong regulatory capability possessed by unique MV enriched microRNAs on growth plate chondrocytes and their potential for use as therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Matrix vesicles (MVs) are small membrane bound organelles present in the extracellular matrix (ECM) of mammalian growth plates, as well as other calcifying tissues including dentine, metastatic cancers, and blood vessels[1, 2]. Like exosomes, they range in size from roughly 50 to 150 nm. Whereas exosomes are found in biological fluids, MVs are tethered to the ECM via integrins [3, 4]. Long associated with matrix calcification [5,6,7], studies show that MVs also play a role in growth plate regulation [6, 8]. Their importance in the avascular growth plate is related to their contents, which vary with the zone of chondrocyte maturation. MVs produced by resting zone cartilage (RC) cells are enriched in neutral metalloproteinases needed for maintenance of a proteoglycan rich matrix [8,9,10]. In contrast, MVs produced by growth zone cartilage (GC) cells contain acid metalloproteinases, which are involved in matrix degradation prior to matrix calcification [8, 10, 11].
Recently, studies have demonstrated that microRNAs are selectively packaged inside these MVs in both the RC and GC regions of the costochondral cartilage growth plate, with 37% of the MV microRNA significantly increased or decreased compared to cell microRNA [12, 13]. Further examination showed that 22% of the microRNA in RC MVs are differentially expressed when compared to GC MVs [12]. This suggests that MV microRNA may also function in the regulation of growth plate chondrocyte proliferation, differentiation and maturation.
The costochondral cartilage growth plate provides an excellent model for assessing the role of individual microRNA in this process. Growth plate chondrocytes express phenotypic changes as they mature with corresponding alterations in the composition of the ECM. Within the costochondral cartilage growth plate the RC encompasses the reservoir of immature chondrocytes and the upper proliferative zone. Cells in the lower proliferating zone, pre and upper hypertrophic zones are referred to as GC chondrocytes. RC and GC chondrocytes can be successfully cultured and retain unique phenotypes through four passages as well as exhibiting zone-specific responses to regulatory factors and hormones [14,15,16,17].
Various factors are involved in growth plate regulation. We selected a handful of the key regulators, described here in greater detail, to investigate if their production is affected by select microRNA transfection. Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP) are two factors that form a feedback loop regulating the proliferation and differentiation of chondrocytes at various stages of the growth plate [18,19,20]. Resting chondrocytes at the periarticular end of the fetal lone bone growth plate express PTHrP that, in turn, maintains chondrocytes in a proliferative state, delaying their differentiation to hypertrophic cells [21, 22]. As chondrocytes move far enough away from the PTHrP production they stop proliferating and begin to express Ihh, marking the transition within the growth plate from proliferating to hypertrophic chondrocytes [19, 22]. This Ihh diffuses through the growth plate and stimulates continued PTHrP production in the resting chondrocytes. This feedback loop functions to regulate the distance between PTHrP and Ihh expressing zones, providing sufficient space for the proliferating chondrocytes [20].
Additional factors are known to play important roles in regulating the growth plate. Transforming growth factor beta 1 (TGF-β1) stimulates bone marrow stromal cell differentiation and regulates chondrocyte maturation [23]. Vascular endothelial growth factor (VEGF) is produced by hypertrophic chondrocytes and is involved in the vascular invasion that occurs in the late hypertrophic region [20, 24, 25]. SRY-Box transcription factor 9 (Sox9) is an early transcription factor active during chondrocyte differentiation and required for the expression of specific matrix proteins [26, 27]. RUNX family transcription factor 2 (RUNX2) is involved in late stage chondrocyte differentiation and needed for the expression of numerous proteins [28].
This study examines the role that MV microRNA may be playing in the regulation of growth plate chondrocytes. We hypothesize that the microRNAs exported with the highest fold change by GC chondrocytes likely play distinct roles in proliferation, differentiation, factor production and ECM composition of cells in the upper growth plate as well as the zone of maturation. Accordingly, we report on the effect that a select subgroup of GC MV microRNAs have on both RC and GC chondrocytes.
Results
Where an effect from treatment was observed it is in relation to the negative control (NC) microRNA group. Unlike the no treatment (NT) group, NC received transfection reagents and a scrambled vector microRNA.
MicroRNA Expression
Comparing the differentially expressed microRNAs in GC MVs vs. cells using a stacked bar chart we were able to identify the microRNAs exhibiting the largest fold change (Fig. 1a). We selected microRNA 22-3p as a positive control due to its abundance in the cell fraction (2.2%) compared to the top three MV microRNA (≤ 0.0005%) and its similar expression profile in RC samples (Fig. 1b). We selected the three microRNA (122-5p, 223-3p and 451-5p) having log2 fold change values below − 10 (Fig. 1c).

MicroRNA expression of resting zone (RC) and growth zone (GC) cells. a Differentially expressed microRNAs (FDR < 0.05) found in MVs as compared to cell isolate. b Expression of select four microRNA in RC and GC samples based on normalized read counts from EdgeR. c Volcano plot of microRNA in GC cells compared to MVs, red denotes differentially expressed microRNA with FDR < 0.05, teal are not differentially expressed. d Nanostring analysis of RC and GC ex-vivo tissue samples, minimum threshold levels determined by Nanostring system. e MicroRNA RT-qPCR cycle threshold values for RC and GC ex-vivo tissue samples. Mean ± SE
Nanostring analysis using a panel of rat microRNA showed that microRNA 22-3p was present in high proportion for both RC and GC sections. MicroRNA 122-5p and 223-3p were not above background levels in either RC or GC samples and microRNA 451-5p was only found above background in the GC samples (Fig. 1d). MicroRNA not above background levels are those that are at or below the threshold levels determined by the system for each sample.
MicroRNA RT-qPCR on RC and GC tissue samples had CT values below 35 for snRNA U6 and microRNA 22-3p. MicroRNA 122-5p and 451-5p groups had three out of four GC samples below 35, all RC samples were between 35 and 40 but one in 451-5p group that was above 40 (Fig. 1e). For microRNA 122-5p the standard curve stopped amplifying at 0.06 ng/μL and for microRNA 451-5p amplification was no longer detected at 0.02 ng/μL with a total cycle count of 60. Examining the melt curves for all eight groups shows tight clustering and a peak appearing within or near to the expected Tm range of 74.5–76 ℃ for these primers (Supplemental Fig. 1).
Response of GC Chondrocytes
Chondrocyte Proliferation and Apoptosis
MicroRNA 122 increased total DNA at 24 and 48 h while microRNA 451 decreased it. MicroRNA 22 and 223 had no effect at 24 h whereas at 48 h microRNA 22 decreased and microRNA 223 increased total DNA (Fig. 2a, b). We normalized microRNA treatments to controls for 12 separate experiments and observed a consistent increase in DNA production as a result of microRNA 122 while being unaffected by microRNA 22, 223 or 451 (Fig. 2c). We examined proliferation rates using 5-ethynyl-2′-deoxyuridine (EdU) incorporation at 24 and 48 h. MicroRNA 122 and 451 increased the proliferation rate at 24 h while all four microRNAs increased proliferation at 48 h (Fig. 2 d, e). We examined whether the acute impact of microRNA 122 on proliferation was present at earlier time points. MicroRNA 122 increased proliferation at 0, 12 and 24 h compared to NC after which it fell below NC for 36 and 48 h (Fig. 2f). Examining metabolic activity, there was increased 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction in GC cells transfected with microRNA 122 at 48 h (Fig. 2g).

Proliferation and apoptosis of GC cells. a, b Total DNA at 24 and 48 h. c Treatment over control of total DNA at 48 h for twelve experiments. d, e Proliferation rate at 24 and 48 h, f time course every 12 h for 48 h. g Metabolism of GC cells at 48 h. h–j Apoptosis indicators (Tunnel, Bax/Bcl2 ratio and p53) were assayed at 48 h. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), NT no treatment, NC negative control microRNA
None of the select microRNA appeared to induce cellular apoptosis. No change was observed between NC and any of the microRNAs with TUNEL (DNA fragmentation) (Fig. 2h), Bcl-2 associated X (Bax)/apoptosis regulator Bcl-2 (Bcl2) levels (Fig. 2i) or tumor suppressor p53 (p53) levels (Fig. 2j).
Alkaline Phosphatase Activity and ECM Composition
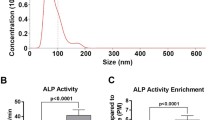
Chondrocytes demonstrated an increase in alkaline phosphatase activity in response to microRNA 223 with 122 and 451 having a significant reduction compared to NC (Fig. 3a). Looking at 5 separate experiments confirmed these results with microRNA 223 increased compared to NC and microRNA 122 and 451 decreased (Fig. 3b).

Alkaline phosphatase activity and ECM composition of GC cells. a, b Alkaline phosphatase activity of GC cells at 48 h & treatment over control of five experiments. c, d sGAG & collagen 2 production by GC cells. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), NT no treatment, NC negative control microRNA
Production of two ECM components—sulfated glycosaminoglycans (sGAGs) and collagen 2—was sensitive to microRNA transfection. MicroRNA 122 increased sGAG levels compared to all other groups while decreasing levels of collagen 2 (Fig. 3c, d). Collagen X was also examined and found to be below the standard for all samples (data not shown).
Growth Plate Factors
Seven different enzyme-linked immunosorbent assays (ELISAs) were used to investigate the production of growth plate proteins (bone morphogenetic protein 2 [BMP-2], Ihh, osteoprotegerin [OPG], PHTrP, receptor activator of NF-κΒ ligand [RANKL], TGF-β1 and VEGF) with protein levels normalized to total DNA per well. BMP-2 was below the standard for all samples (data not shown). VEGF increased following microRNA 22 transfection and decreased in response to microRNAs 122 and 451 (Fig. 4a). OPG production increased in response to microRNA 122 or 451, while RANKL was not detected in any of the samples (Fig. 4b, c). GC cells exhibited an increase in Ihh and PTHrP production following microRNA 22 and a decrease following microRNA 122 (Fig. 4d, e). Transfected chondrocytes increased latent TGF-β1 as a response to microRNA 22 compared to the control cultures, but levels were not different than NT and no change in active TGF-β1 was observed (Fig. 4f, g).

Growth plate factor production of GC cells at 48 h a VEGF, b OPG, c RANKL, d Ihh, e PTHrP, f Active TGF-β1 and g Latent TGF-β1. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), NT no treatment, NC negative control microRNA
Response of RC Chondrocytes
Proliferation and Apoptosis
Total DNA, TUNEL and p53 of RC chondrocytes were assessed at 48 h. Total DNA increased in cells following microRNA 122 transfection whereas it was decreased by microRNAs 22, 223 and 451 (Fig. 5a). Apoptosis as measured by TUNEL and p53 was unaffected by the four microRNAs (Fig. 5b, c).

Response of RC cells at 48 h. a Total DNA quantity, b, c apoptosis indicators TUNEL & p53. d Alkaline phosphatase activity of RC cells and e treatment over control for three experiments. f sGAG production of RC cells. g, h Ihh & PTHrP factor production of RC cells. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), NT no treatment, NC negative control microRNA
Alkaline Phosphatase Activity and ECM Composition
RC chondrocytes exhibited a reduction in alkaline phosphatase activity for all four microRNA when compared to control with microRNA 122 resulting in a significantly greater decrease (Fig. 5d). We normalized the five microRNA treatments over control (NC) for three separate experiments at 48 h and observed a similar response (Fig. 5e). ECM composition was assessed by sGAG levels with microRNA 22 resulting in a decrease in sGAG levels and microRNA 122 an increase (Fig. 5f).
Growth Plate Factors
Ihh and PTHrP were found to be increased as a result of microRNA 22 transfection and decreased following transfection with microRNA 122 (Fig. 5g, h).
Selective MicroRNA Inhibitors
In order to examine the sensitivity of the mimics used in the transfection experiments, RC and GC chondrocytes were transfected with a select microRNA inhibitor (22-3p or 122-5p) at three different concentrations, with and without the corresponding mimic along with a NT group and a mimic only group. Alkaline phosphatase activity and total DNA production were assayed.
Response of GC Chondrocytes
MicroRNA 22 decreased DNA production and this effect was reversed by transfection alongside the inhibitor with inhibitor alone having a slight reduction in DNA production compared to NT (Fig. 6a). Alkaline phosphatase activity was not significantly different for all but the mimic plus inhibitor (30 or 50 nM) groups where slight decreases were observed (Fig. 6b).

Selective microRNA inhibitors effect on GC and RC cells at 48 h. a–d Response of GC cells, a, b total DNA & alkaline phosphatase activity following transfection with 22-3p, c, d following transfection with 122-5p. e, f Response of RC cells, e, f total DNA and alkaline phosphatase activity following transfection with 22-3p, g, h following transfection with 122-5p. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), NT no treatment
MicroRNA 122 inhibitor alone had no effect on total DNA production, 122 mimic alone increased DNA production and mimic + inhibitor was able to reduce DNA production but not back to NT levels (Fig. 6c). Alkaline phosphatase activity underwent no change with inhibitor alone but was significantly reduced when transfected with 122 mimic on its own. The addition of 122 inhibitor to the mimic increased the alkaline phosphatase activity but was unable to return it to the NT level (Fig. 6d).
Response of RC Chondrocytes
MicroRNA 22 decreased total DNA production when compared to all other groups. The addition of inhibitor to the mimic was able to bring DNA production nearly back to the NT group. Transfection with inhibitor alone resulted in a slightly greater decrease of total DNA production compared to mimic combined with inhibitor (Fig. 6e). Alkaline phosphatase activity was largely unaffected by the 22 inhibitor alone with the highest concentration (50 nM) resulting in a slight decrease. Mimic alone resulted in a significant decrease that was largely reversed by addition of inhibitor (Fig. 6f).
MicroRNA 122 inhibitor alone had no significant effect on total DNA production, mimic alone produced a significant increase in DNA production that was partially reversed by addition of inhibitor (Fig. 6g). Similarly, the addition of inhibitor on its own resulted in no significant difference in alkaline phosphatase activity, whereas the 122 mimic on its own significantly reduced alkaline phosphatase activity, which was partially reversed by addition of inhibitor (Fig. 6h).
Angiogenesis
MicroRNA 122 had a clear visual effect on human umbilical vein endothelial cells (HUVECs) when compared to scrambled vector transfection (Fig. 7a) and also resulted in a decrease in total DNA production when compared to NC (Fig. 7b). MicroRNA 22, 122, 223 and 451 all resulted in a decrease in the connection length at 20 h with only microRNA 122 and 451 driving a reduction in connection number (Fig. 7c).

Angiogenesis of HUVEC cells following transfection. a Representative images of NC and 122 transfection at 20 h. b Total DNA production by HUVEC cells at 48 h. c, d Connection length and connection number counted every 4 h over a 20 h time period. Mean ± SE, groups not sharing a letter are statistically different (α = 0.05), * and x are statistically different from NC (α = 0.05), NC negative control microRNA
Discussion
This study using rat costochondral cartilage cell cultures as a model system, demonstrates the potential of growth plate chondrocyte microRNA, packaged in MVs and exported into the ECM, to exert a regulatory influence back on the chondrocytes in a paracrine and potentially autocrine fashion. These selectively packaged microRNA produce clear and maturation zone-dependent effects on growth plate chondrocytes [12, 13]. Establishing regulatory capability for MV microRNAs lays out additional roles for MVs, providing protection against degradation for microRNA and directly modulating chondrocyte gene expression [11, 29]. MicroRNA have robust potential as a regulatory molecule as a result of each microRNA’s ability to impact the production of multiple proteins within a cell.
Our study examined the effects that MV microRNAs can exert on growth plate chondrocytes. Previous work examined the importance of microRNA for a functioning growth plate and the role that microRNAs have in regulating factor production [30, 31]. This study found two microRNA that exhibited significant maturation-dependent regulatory effects on chondrocytes. MicroRNA 22 demonstrated an ability to modulate ECM and factor production by both RC and GC chondrocytes. MicroRNA 122 demonstrated a maintenance type of effect increasing production of components associated with healthy cartilage while delaying terminal differentiation and increasing proliferation.
MicroRNA 122 transfection resulted in consistent responses from both RC and GC cells that are characteristic of proliferating chondrocytes. Cell proliferation increased, alkaline phosphatase activity decreased and sGAG production increased (with GC cells approaching levels found in RC cells). To further study the maintenance aspect of microRNA 122 we used HUVEC cells to examine the potential angiogenic effect of microRNA 122 in the growth plate (an avascular tissue). When transfected into HUVEC cells, microRNA 122 drove a reduction in both connection number and length between the cells indicating a potential inhibitory effect on blood vessel formation useful for avascular tissue maintenance.
A stark response demonstrated by both RC and GC chondrocytes to microRNA 122 transfection was an increase in proliferation that may be connected to the reduction of both Ihh and PTHrP production that was also observed. Proliferating cells of the growth plate are located within the PTHrP and Ihh feedback loop sandwiched by resting cells producing PTHrP on one end and cells differentiating to a hypertrophic phenotype and producing Ihh on the other. Between these two regions chondrocytes are proliferating with expression of Ihh and PTHrP largely absent [20]. Whether the reduction in PTHrP and Ihh is driving the increase in proliferation or if other effectors are involved is unknown. What was made very clear is that microRNA 122 has numerous impactful effects on growth plate chondrocytes (increasing proliferation, stopping vascularization and maintaining production of ‘healthy’ ECM).
The response of both RC and GC cells to microRNA 22 was unexpected. Transfection with microRNA 22 was able to impact the ECM composition of both chondrocyte populations, as well as proliferation and differentiation markers of RC cells. Its strongest impact was on factors produced by cells from both growth plate zones. Although microRNA 22 was active in both RC and GC chondrocytes, it did not appear to drive either population of cells toward a more proliferative or hypertrophic phenotype. Instead, it increased the production of proteins that are characteristic of both regions. MicroRNA 22’s relatively abundant presence in RC and GC cells and their corresponding MVs, the observed response of chondrocytes to microRNA 22 transfection not being characteristic of either zone indicate that it is potentially functioning as part of a larger microRNA regulatory network within growth plate chondrocytes [32].
As a result of microRNA 22 and 122 exhibiting the majority of effects we decided to assess the specificity of these mimics by transfecting them along with their specific inhibitor. Both mimics and inhibitor are transfected as double stranded RNA molecules that require cellular processing in order to be activated. We observed partial or complete reversal of the effects of the mimic when transfected alongside specific inhibitors. It is likely that a portion of the mimics are loaded into the RNA-induced silencing complex and able to regulate available mRNA before being sequestered by the co-transfected inhibitors even when the inhibitor is administered at three times the concentration of the mimic.
In addition to examining the specificity of the mimics, we wanted to verify the presence of these microRNA in tissue samples to ensure they are not an artifact of cell culture. Our first approach was using a NanoString microRNA panel to examine RNA isolated from GC and RC tissue samples. At first glance, the absence of microRNA 122 and 223 in any of the tissue samples and microRNA 451 in the RC tissue samples are concerning. However, in the RNAseq data these microRNA range from 0 to 4.9 × 10−4% in the cell samples and 4.3 × 10−3 to 1.1% in the MV samples. The quantity of MV RNA that is isolated from cell culture preparations is very low compared to the corresponding cell pellet (average of 300 times less according to bioanalyzer analysis, data not shown). These three microRNA (122, 223 and 451) were found primarily if not exclusively in the MV isolation and so are likely to be present in extremely low numbers in actual tissue. Results from the NanoString are not conclusive that the microRNA are not present in native tissue given the low read numbers in the RNAseq data from tissue culture. Colleagues in our lab have found microRNA 122 and 451 in articular cartilage using qPCR from ex vivo samples [33]. Additionally, a recent publication comparing four different microRNA quantification methods (small RNAseq, FirePlex, EdgeSeq and nCounter) found the nCounter system to be the least sensitive of the four with fewer microRNAs determined to be above background levels [34]. MicroRNA 22 was found by the NanoString panel in both RC and GC tissue samples (average of 11.19 and 5.15%, respectively). These values are similar to the results from the small RNAseq data (cells: 2.2% RC and GC, MV: 20% RC and 17% GC). These data do confirm the presence of both microRNA 22 and 451 in ex-vivo GC tissue samples.
In order to further validate the presence of the microRNA in tissue we performed RT-qPCR on RNA isolated from RC and GC tissue. The standard curve generated produced an upward trend in CT values as the dilution increased for snRNA U6 (positive control) and microRNA 22. There is no clearly discernable trend for microRNAs 122 and 451 however the ability to amplify a detectable product was lost as dilutions increased. In a similar fashion all snRNA U6 and microRNA 22 samples fell below 35 CT (a commonly used cut off point for mRNA RT-qPCR) along with all but one of the GC microRNA 122 and 451 groups. Both the increase in CT values from microRNA 22 to microRNAs 122 and 451 and the standard curve becoming undetectable for microRNAs 122 and 451 follows the trend that was observed in the small RNAseq data with microRNA 22 being more abundant than either microRNAs 122 or 451. These RT-qPCR results provide compelling evidence of their presence at a low level in tissue (Supplemental Fig. 1).
Investigating the in vivo chondrocyte response to microRNA 22 and 122 is an intriguing next step. MicroRNA 122 transfection was able to generate a response from both RC and GC cells that was analogous to proliferating chondrocytes while the strong factor response to microRNA 22 makes both microRNAs interesting candidates for future animal studies. Delivery of targeted cocktails of microRNA packaged into vesicles look to be potential methods for treating cartilage disorders. A more detailed understanding of MV packaging and release would be beneficial in designing treatment delivery mechanisms that closely mimic existing biological systems.
This study demonstrates that microRNA, first discovered in cell culture isolations, are likely involved in the complex regulation of growth plate chondrocytes as they proliferate and mature. These microRNA that are exported into the ECM within MVs are able to produce phenotypic effects on growth plate chondrocytes that fall within understood zonal behaviors for these cells. Gaining a better understanding of the regulation that microRNA production and export falls under as well as a more precise view into the regulatory effects that individual microRNA are able to elicit will be fundamental in selecting microRNA for potential treatment avenues.
Experimental Procedures
Chondrocyte Cultures
Costochondral cartilage was removed from 100 to 125 g male Sprague Dawley rats, isolated by enzymatic digestion and cultured as previously described by Boyan et al. [14, 17]. All animal procedures followed a protocol approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University. Animals were killed by CO2 asphyxiation followed by cervical dislocation. Rib cages were removed by sharp dissection and placed in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Carlsbad, CA) containing 1 g/L glucose with 150 U penicillin/mL and 150 μg streptomycin/mL (Lonza, Basel, Switzerland). Tissue was removed from around the ribs and RC and GC cartilage sections were dissected out. A small region of GC tissue was between the bone and RC tissue and the three different regions were clearly visible under a dissection scope. One or two transition slices between the RC and GC regions were discarded and the dissected tissue incubated overnight in DMEM. RC and GC slices were incubated in 0.25% trypsin–EDTA (Gibco, Gaithersburg, MD) for 1 h, washed and incubated in 0.2% collagenase type II (Worthington Biochemical, Lakewood, NJ) for 3 h on a shaker. Cells were filtered through a 40 µm nylon mesh strainer, collected by centrifugation (751 g for 10 min) and re-suspended in DMEM FM (1 g/L glucose DMEM with 10% FBS, 50 U/mL penicillin, 50 μg/mL streptomycin and 50 µg/mL ascorbic acid). Cells were plated at a density of 20,000 GC cells/cm2 or 10,000 RC cells/cm2 and incubated at 37 ℃ and 5% CO2. The culture media were changed 24 h after plating and then every 48 h thereafter. Confluent cells were passaged using 0.25% trypsin and plated as above. Fourth passage cells were used for all experiments.
Bioinformatic Analysis
Small RNA-seq data from chondrocytes and MVs that were isolated in a previous study (Lin et al. 2016) were used for this study [13]. Reads were aligned against the rat genome and analyzed using the miARma-Seq tool [35]. In brief the read quality was assessed using FASTQC; reads were aligned to the NCBI Rattus norvegicus annotation release 105 (Rnor_6.0) using Bowtie2. Resulting bam files were processed by miARma-Seq to quantify microRNAs and differential expression calculated using edgeR [36]. Normalized read counts from edgeR were used to determine the percent of each microRNA in the sample \(percent \times microRNA = \left( {\frac{specific microRNA}{{total microRNA}} \times 100} \right)\). Log twofold change of GC microRNA in cell vs. MV was visualized against − log 10 false discovery rate in a volcano plot and the three microRNA with a log 2 fold change less than − 10 highlighted. Differentially expressed microRNA with a false discovery rate (FDR) less than 0.05 were loaded into a stacked bar chart with \(\% {\text{ }}GC{\text{ }}MV = \frac{{percent{\text{ }}MV{\text{ }}microRNA}}{{percent{\text{ }}total\left( {MV + Cell} \right)microRNA}}{\text{over }}\% {\text{ }}GC{\text{ }}Cell = \frac{{percent{\text{ }}Cell{\text{ }}microRNA}}{{percent{\text{ }}total\left( {MV + Cell} \right)microRNA}}\) for each of the 111 DE microRNA.
Nanostring
We used NanoString’s nCounter system to quantify the presence of microRNA in costochondral tissue. Tissue slices were harvested, cleaned and directly transferred to Qiazol lysis reagent (Qiagen, Hilden, Germany) for homogenization with no intermediate. In brief, the RC and GC zones were carefully cut into thin slices taking extra care to remove surrounding tissue from the ribs and discarding one or two slices separating the zones. The tissue was not exposed to media or FBS. Cartilage slices (avg ~ 15 mg per sample) were transferred to BeadBug homogenizer tubes (Benchmark Scientific, Sayreville, NJ) with 6.0 mm zirconium bead (previously treated to degrade any contaminating RNA: 2 h under UV light and 2 h in 75 ℃). 700 μL Qiazol was added to the tube and slices were homogenized on a BeadBug (cycle settings: speed = 400, time = 40 s) with GC slices requiring 19 cycles for homogenization and 8 cycles for RC samples. RNA was extracted using Qiagen’s miRNeasy micro kit and resuspended in nuclease-free water. Samples were submitted for analysis at the Biobehavioral Research Lab at VCU using Nanostring’s Rat v1.5 miRNA panel.
Ex-Vivo RT-qPCR
Tissue was removed in identical manner to ‘chondrocyte cultures’ above. Following trypsin digestion, tissue slices were washed with Hank’s Balanced Salt Solution (HBSS) and deposited into a BeadBug homogenizer tube (same tube and treatment as ‘Nanostring’). 700 μL of Qiazol was added to the tube and allowed to sit at room temperature for 5 min. GC and RC tubes were then run on the BeadBug homogenizer (cycle settings: speed = 400, time = 60 s) for four cycles. RNA was purified using Qiagen’s miRNeasy micro kit and resuspended in nuclease-free water. RT-PCR was performed on four GC and RC samples normalized to 1,250 ng of RNA per well using Qiagen’s HiSpec Buffer and incubating in thermocycler at 37 °C for 60 min followed by 95 °C for 5 min. qPCR was run on the resulting cDNA diluted to a final working concentration of 3.125 ng/μL using Qiagen’s miScript II RT kit and primers for snRNA U6, microRNA 22-3p (Qiagen), 122-5p and 451-5p (Eurofins Scientific, Luxembourg). An eight-point standard curve was generated by mixing 4 μL from each of the eight samples together and performing a 1:3 serial dilution starting at 5 ng/μL and run in duplicate. 10 μL reactions were run in a 96-well plate using a QuantStudio 3 RT-PCR system (ThermoFisher): activation: 95 ℃ for 15 min, 60 cycles of (denaturation: 94 ℃ for 15 s, annealing: 55 ℃ for 30 s, extension: 70 ℃ for 30 s) followed by melt curve. Data were collected on the extension and melt curve steps.
MicroRNA Transfection
Once fourth passage chondrocytes were 60–70% confluent they were transfected with microRNA mimics (mirVana; Invitrogen, Carlsbad, CA) at final concentration of 14.5 nM using Lipofectamine RNAiMAX transfection reagent (Invitrogen) at a final dilution of 1:500. Four microRNA mimics were used for these experiments: (microRNA 22-3p, 122-5p, 223-3p and 451-5p). Transfection was carried out in DMEM 1 g/L glucose with 10% FBS and incubated at 37 ℃ and 5% CO2 for 24 h. At transfection completion the media were changed to DMEM FM until harvest. Harvest was carried out according to the requirements of each specific assay with reported hours being hours post transfection completion. Two control groups were included in the experiments: a NT group that received neither lipofectamine nor microRNA mimics and a NC group that included lipofectamine along with a mimic designed not to interact with known mRNA.
Specific microRNA inhibitors (22-3p and 122-5p miRCURY LNA miRNA Inhibitors, Qiagen) were transfected following the same procedure as for the microRNA mimics. Final concentration of microRNA inhibitors was 10, 30 or 50 nM. When transfected along with microRNA mimics the inhibitors were prepared and administered together with the mimics in one treatment solution. Lipofectamine concentration and media formulations unchanged. MicroRNA inhibitors for 223-3p and 451-5p were not investigated.
Cell Response
DNA Content
Double stranded DNA was quantified after sonicating the cell monolayer (40 amps, 10 s per well) and assaying bound fluorescent dye (QuantiFluor® dsDNA system; Promega, Madison, WI) using a plate reader (Synergy H1 Hybrid Reader; BioTek, Winooski VT) with excitation of 485 nm and emission of 538 nm.
Proliferation Rate
Four hours before indicated timepoint cells were pulsed with EdU at 10 μM concentration and incubated at 37 ℃ for four hours. Cells were then fixed, labeled and read (excitation 568 nm emission 585 nm) according to manufacturer’s protocol (Click-iT® EdU Microplate Assay; Invitrogen) to quantify all EdU that was incorporated by the actively proliferating cells during the four hour pulse.
Metabolic Activity
100 μL of MTT solution (5 mg/mL in PBS) was added to the 500 μL of serum-free media per well and incubated at 37 ℃ for four hours. Media was aspirated and replaced with 500 μL of DMSO per well and placed on a shaker for 5 min. 200 μL of the DMSO per well was transferred to a 96-well plate and absorbance read at 570 nm.
Production of Proteins
Proteins were quantified by ELISA normalized to DNA content. Assays were carried out following accompanying protocols for: BMP-2 (PeproTech, Rocky Hill, NJ), type II collagen, type X collagen, Ihh, p53 and PTHrP (LS Bio, Seattle, WA), OPG, TGF-β1 and VEGF (R&D Systems, Minneapolis, MN). Media and cell monolayer were isolated at 48 h.
In cell western analysis was used to quantify specific protein levels with results normalized to DNA content. At 48 h cells were fixed using 3.7% formaldehyde, permeabilized with 0.1% Triton X-100, blocked for 1.5 h (Odyssey Blocking Buffer) and incubated overnight at 4 ℃ with primary antibody specific for Bax or Bcl at 1:100 and 1:20 dilution, respectively. Wells were washed and incubated for 1 h at room temperature with DRAQ5 (1:10,000 dilution) and corresponding secondary antibody (1:1,000 dilution). Wells were washed and imaged on the Li-Cor Odyssey CLx (LI-COR, Lincoln, NE).
TUNEL
Cells were fixed with 3.7% buffered formaldehyde solution, permeabilized with proteinase K and labeled with TdT labeling buffer. Absorbance was measured at 450 nm on a plate reader.
Alkaline Phosphatase Activity
At 48 h media were aspirated and cell monolayer washed twice with 1 X PBS. Monolayer was removed with 0.05% Triton X 100 and lysed by three cycles of freeze thawing. Alkaline phosphatase activity was quantified using a colorimetric assay that determines the amount of enzyme needed to hydrolyze 1 µmole of p-nitrophenyl phosphate to 1 µmole of p-nitrophenol at 37 ℃. Absorbance was read in plate reader at 405 nm. Activity was normalized to protein content as determined by Pierce BCA assay (ThermoFisher, Waltham, MA).
Sulfated Glycosaminoglycan Production
At 48 h media were aspirated and cell monolayer washed twice with 1 X PBS. Cell monolayer was removed from wells with papain digest solution (3.8 U/mL papain in 55 mM sodium citrate, 10 mM cysteine) and digested overnight at 60 ℃. sGAG content was assessed using 1,9-dimethylmethylene blue dye with pH of 1.5 and absorbance measured at 525 nm on a plate reader.
Regulation of Vasculogenesis
HUVECs were grown on Geltrex and cultured using endothelial cell growth media from the Angiogenesis Starter Kit (ThermoFisher). The four select microRNA were transfected into HUVECs, as detailed above, and the connection length (measured in pixels) and the connection count taken every four hours over a 20 h period.
Statistical Analysis
DNA content, proliferation rate, MTT, ELISA, in cell western, TUNEL, alkaline phosphatase activity and sGAG are presented as mean ± standard error for six independent cultures per variable. Differences between groups were examined by ANOVA with post-hoc Tukey HSD. A p-value less than 0.05 was considered significant. Experimental observations validated with at least one repeat experiment. Connection length and number examined by ANOVA with post-hoc Tukey HSD. Significance discussed is between treatment and NC group based on a p-value less than 0.05. Statistical analysis and presentation for all examinations were carried out using R 3.6.0 [37]. Following R packages were used: egg, grid, gridExtra, ggplot2, ggsignif, magrittr, multcompview, outliers, plyr and sfsmisc [37,38,39,40,41,42,43,44,45,46].
References
Anderson HC, Garimella R, Tague SE (2005) The role of matrix vesicles in growth plate development and biomineralization. Front Biosci 10:822–837. https://doi.org/10.2741/1576
Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D (2016) Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30:836–848. https://doi.org/10.1016/j.ccell.2016.10.009
Wuthier RE, Lipscomb GF (2011) Matrix vesicles: structure, composition, formation and function in calcification. Front. Biosci. 16:2812–2902. https://doi.org/10.1095/biolreprod.107.067082
Rilla K, Mustonen A-M, Arasu UT, Härkönen K, Matilainen J, Nieminen P (2017) Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix Biol. 75:201–219. https://doi.org/10.1016/j.matbio.2017.10.003
Anderson HC (1967) Electron microscopic studies of induced cartilage development and calcification. J Cell Biol 35:81–101. https://doi.org/10.1083/jcb.35.1.81
Golub EE (2009) Role of matrix vesicles in biomineralization. Biochim Biophys Acta Gen Subj 1790:1592–1598. https://doi.org/10.1016/j.bbagen.2009.09.006
Kirsch T, Wuthier RE (1994) Stimulation of calcification of growth plate cartilage matrix vesicles by binding to type II and X collagens. J Biol Chem 269: 11462–11469. http://www.ncbi.nlm.nih.gov/pubmed/8157677.
Dean DD, Schwartz ZV, Muniz OE, Gomez R, Swain LD, Howell DS, Boyan BD (1992) Matrix vesicles contain metalloproteinases that degrade proteoglycans. Bone Miner 17:172–176. https://doi.org/10.1007/BF00301632
Krane SM, Inada M (2008) Matrix metalloproteinases and bone. Bone 43:7–18. https://doi.org/10.1016/j.bone.2008.03.020
Nishimura R, Wakabayashi M, Hata K, Matsubara T, Honma S, Wakisaka S, Kiyonari H, Shioi G, Yamaguchi A, Tsumaki N, Akiyama H, Yoneda T (2012) Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J Biol Chem 287:33179–33190. https://doi.org/10.1074/jbc.M111.337063
Dean DD, Schwartz Z, Muniz OE, Gomez R, Swain LD, Howell DS, Boyan BD (1992) Matrix vesicles are enriched in metalloproteinases that degrade proteoglycans. Calcif Tissue Int 50:342–349. https://doi.org/10.1007/BF00301632
Lin Z, McClure MJ, Zhao J, Ramey AN, Asmussen N, Hyzy SL, Schwartz Z, Boyan BD (2018) MicroRNA contents in matrix vesicles produced by growth plate chondrocytes are cell maturation dependent. Sci Rep 8:3609. https://doi.org/10.1038/s41598-018-21517-4
Lin Z, Rodriguez NE, Zhao J, Ramey AN, Hyzy SL, Boyan BD, Schwartz Z (2016) Selective enrichment of microRNAs in extracellular matrix vesicles produced by growth plate chondrocytes. Bone 88:47–55. https://doi.org/10.1016/j.bone.2016.03.018
Boyan BD, Schwartz Z, Swain LD, Carnes DL, Zislis T (1988) Differential expression of phenotype by resting zone and growth region costochondral chondrocytes in vitro. Bone 9:185–194. https://doi.org/10.1016/8756-3282(88)90008-7
Boyan BD, Schwartz Z, Carnes DL, Ramirez V (1988) The effects of vitamin D metabolites on the plasma and matrix vesicle membranes of growth and resting cartilage cells in vitro. Endocrinology 122:2851–2860. https://doi.org/10.1210/endo-122-6-2851
Schwartz Z, Swain LD, Ramirez V, Boyan BD (1990) Regulation of arachidonic acid turnover by 1,25-(OH)2D3 and 24,25-(OH)2D3 in growth zone and resting zone chondrocyte cultures. Biochim Biophys Acta 1027:278–286. https://doi.org/10.1016/0005-2736(90)90319-j
Boyan BD, Schwartz Z, Park-Snyder S, Dean DD, Yang F, Twardzik D, Bonewald LF (1994) Latent transforming growth factor-beta is produced by chondrocytes and activated by extracellular matrix vesicles upon exposure to 1,25-(OH)2D3. J. Biol. Chem. 269: 28374–28381. http://www.ncbi.nlm.nih.gov/pubmed/7961777
Kobayashi T, Chung U, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM (2002) PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 129: 2977–2986. http://dev.biologists.org/content/129/12/2977.short.
Mak KK, Kronenberg HM, Chuang P-T, Mackem S, Yang Y (2008) Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135:1947–1956. https://doi.org/10.1242/dev.018044
van der Eerden BCJ, Karperien M, Wit JM (2003) Systemic and local regulation of the growth plate. Endocr Rev 24:782–801. https://doi.org/10.1210/er.2002-0033
Burdan F, Szumiło J, Korobowicz A, Farooquee R, Patel S, Patel A, Dave A, Szumiło M, Solecki M, Klepacz R, Dudka J (2009) Morphology and physiology of the epiphyseal growth plate. Folia Histochem Cytobiol 47:5–16. https://doi.org/10.2478/v10042-009-0007-1
Kronenberg HM (2006) PTHrP and skeletal development. Ann N Y Acad Sci 1068:1–13. https://doi.org/10.1196/annals.1346.002
Schwartz Z, Sylvia VL, Dean DD, Boyan BD (1998) The synergistic effects of vitamin D metabolites and transforming growth factor-beta on costochondral chondrocytes are mediated by increases in protein kinase C activity involving two separate pathways. Endocrinology 139:534–545. https://doi.org/10.1210/endo.139.2.5753
Ortega N, Wang K, Ferrara N, Werb Z, Vu TH (2010) Complementary interplay between matrix metalloproteinase-9, vascular endothelial growth factor and osteoclast function drives endochondral bone formation. Dis Model Mech 3:224–235. https://doi.org/10.1242/dmm.004226
Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S, Olsen BR (2001) Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev 106:97–106. https://doi.org/10.1016/S0925-4773(01)00428-2
Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B (1999) Sox9 is required for cartilage formation. Nat Genet 22:85–89. https://doi.org/10.1038/8792
Ballock RT, O’Keefe RJ (2003) The biology of the growth plate. J. Bone Joint Surg. Am. 85: 715–726. http://www.ncbi.nlm.nih.gov/pubmed/12672851.
Komori T (2010) Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 339:189–195. https://doi.org/10.1007/s00441-009-0832-8
Maeda S, Dean DD, Gay I, Schwartz Z, Boyan BD (2001) Activation of latent transforming growth factor beta1 by stromelysin 1 in extracts of growth plate chondrocyte-derived matrix vesicles. J Bone Miner Res 16:1281–1290. https://doi.org/10.1359/jbmr.2001.16.7.1281
Lui JC (2017) Regulation of body growth by microRNAs. Mol Cell Endocrinol 456:2–8. https://doi.org/10.1016/j.mce.2016.10.024
Jee YH, Wang J, Yue S, Jennings M, Clokie SJ, Nilsson O, Lui JC, Baron J (2018) mir-374-5p, mir-379-5p, and mir-503-5p regulate proliferation and hypertrophic differentiation of growth plate chondrocytes in male rats. Endocrinology 159:1469–1478. https://doi.org/10.1210/en.2017-00780
Nalluri JJ, Kamapantula BK, Barh D, Jain N, Bhattacharya A, De Almeida SS, Ramos RTJ, Silva A, Azevedo V, Ghosh P (2015) DISMIRA: Prioritization of disease candidates in miRNA-disease associations based on maximum weighted matching inference model and motif-based analysis. BMC Genomics 16(Suppl 5):S12. https://doi.org/10.1186/1471-2164-16-S5-S12
Scott KM, Cohen DJ, Hays M, Nielson DW, Grinstaff MW, Lawson TB, Snyder BD, Boyan BD, Schwartz Z (2021) Regulation of inflammatory and catabolic responses to IL-1β in rat articular chondrocytes by microRNAs miR-122 and miR-451. Osteoarthr Cartil 29:113–123. https://doi.org/10.1016/j.joca.2020.09.004
Godoy PM, Barczak AJ, DeHoff P, Srinivasan S, Etheridge A, Galas D, Das S, Erle DJ, Laurent LC (2019) Comparison of reproducibility, accuracy, sensitivity, and specificity of miRNA quantification platforms. Cell Rep 29:4212-4222.e5. https://doi.org/10.1016/j.celrep.2019.11.078
Andrés-León E, Núñez-Torres R, Rojas AM (2016) miARma-Seq: a comprehensive tool for miRNA, mRNA and circRNA analysis. Sci Rep 6:25749. https://doi.org/10.1038/srep25749
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a 7Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
R Core Team (2019) R: a language and environment for statistical computing. https://www.r-project.org/
Auguie B (2019) Egg: extensions for “ggplot2”: custom geom, custom themes, plot alignment, labelled panels, symmetric scales, and fixed panel size. https://cran.r-project.org/package=egg
Auguie B (2017) gridExtra: miscellaneous functions for “grid” graphics. https://cran.r-project.org/package=gridExtra
Wickham H (2016) ggplot2: elegant graphics for data analysis. https://ggplot2.tidyverse.org
Ahlmann-Eltze C (2019) ggsignif: Significance brackets for “ggplot2”. https://cran.r-project.org/package=ggsignif
Bache SM, Wickham H (2014) magrittr: a forward-pipe operator for R. https://cran.r-project.org/package=magrittr
Graves S, Piepho HP, Selzer L, Dorai-Raj S (2015) multcompView: visualizations of paired comparisons. https://cran.r-project.org/package=multcompView
Komsta L (2011) Outliers: tests for outliers. https://cran.r-project.org/package=outliers.
Wickham H (2011) The split-apply-combine strategy for data analysis. J Stat Softw 40:1–29. https://doi.org/10.18637/jss.v040.i01
Maechler M (2019) sfsmisc: utilities from “seminar fuer statistik” ETH Zurich. https://cran.r-project.org/package=sfsmisc.
Acknowledgement
The authors acknowledge resources provided by the High Performance Research Computing (HPRC) Core Facility at Virginia Commonwealth University (https://chipc.vcu.edu) for conducting analyses reported in this paper.
Funding
This research was funded in part by a grant from the National Institutes of Health (NIH/NIDCR R03DE027146 to ZL).
Author information
Authors and Affiliations
Contributions
Niels C. Asmussen: Conceptualization, Investigation, Formal analysis, Writing- Original Draft. David J. Cohen: Conceptualization, Resources. Zhao Lin: Conceptualization, Investigation. Michael J. McClure: Conceptualization, Investigation. Barbara D. Boyan: Supervision, Data analysis, Writing- Review & Editing, Funding acquisition. Zvi Schwartz: Conceptualization, Data analysis, Project administration.
Corresponding author
Ethics declarations
Disclosure
The authors report no conflict of interest.
Human and Animal Rights and Informed Consent
All animal procedures followed a protocol approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Asmussen, N.C., Cohen, D.J., Lin, Z. et al. Specific MicroRNAs Found in Extracellular Matrix Vesicles Regulate Proliferation and Differentiation in Growth Plate Chondrocytes. Calcif Tissue Int 109, 455–468 (2021). https://doi.org/10.1007/s00223-021-00855-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-021-00855-y