
Abstract
The expression and activation of EphA4 in the various cell types in a knee joint was upregulated upon an intraarticular injury. To determine if EphA4 signaling plays a role in osteoarthritis, we determined whether deficient EphA4 expression (in EphA4 knockout mice) or upregulation of the EphA4 signaling (with the EfnA4-fc treatment) would alter cellular functions of synoviocytes and articular chondrocytes. In synoviocytes, deficient EphA4 expression enhanced, whereas activation of the EphA4 signaling reduced, expression and secretion of key inflammatory cytokines and matrix metalloproteases. Conversely, in articular chondrocytes, activation of the EphA4 signaling upregulated, while deficient EphA4 expression reduced, expression levels of chondrogenic genes (e.g., aggrecan, lubricin, type-2 collagen, and Sox9). EfnA4-fc treatment in wildtype, but not EphA4-deficient, articular chondrocytes promoted the formation and activity of acidic proteoglycan-producing colonies. Activation of the EphA4 signaling in articular chondrocytes upregulated Rac1/2 and downregulated RhoA via enhancing Vav1 and reducing Ephexin1 activation, respectively. However, activation of the EphA4 signaling in synoviocytes suppressed the Vav/Rac signaling while upregulated the Ephexin/Rho signaling. In summary, the EphA4 signaling in synoviocytes is largely of anti-catabolic nature through suppression of the expression of inflammatory cytokines and matrix proteases, but in articular chondrocytes the signaling is pro-anabolic in that it promotes the biosynthesis of articular cartilage. The contrasting action of the EphA4 signaling in synoviocytes as opposing to articular chondrocytes may in part be mediated through the opposite differential effects of the EphA4 signaling on the Vav/Rac signaling and Ephexin/Rho signaling in the two skeletal cell types.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Erythropoietin-producing human hepatocellular (Eph) receptors belong to the largest receptor protein-tyrosine kinases family. There are two subfamilies of Eph receptors: EphAs (A1-A8 and A10) and EphBs (B1-B4 and B6). Eph receptors are activated by membrane-bound ligands, ephrins (Efns), located on the cell surface of neighboring cells [1]. There are also two structurally distinct subfamilies of Efn ligands: EfnA (A1-A5) and EfnB (B1-B5). Eph receptors of one particular subfamily demonstrate an ability to bind with high affinity to all Efn ligands of the corresponding subfamily, but in general have little to no cross-binding to Efn ligands of the other subfamily [2]. However, there are a few exceptions. For example, EfnB2 [3] and EfnB3 [4] can bind to and activate EphA4, and EfnA5 can bind to and activate EphB2 [5]. The Efn ligand binding to the Eph receptor triggers autophosphorylation of the juxtamembrane tyrosines [6], which initiates the downstream intracellular signaling [7]. Efn ligands are themselves also capable of receptor-like signaling [8]. Accordingly, the Efn-Eph interaction offers a unique bi-directional signaling mechanism. The Eph receptor-induced signal is known as the forward signaling and the Efn ligand-initiated signal is called the reverse signaling.
The functional importance of Eph-Efn bi-directional signaling has been established in the central nervous system, developing cardiovascular system, insulin secretion, bone homeostasis, immune function, blood clotting, pathological forms of angiogenesis, stem cell differentiation, and cancer [9,10,11,12,13]. With respect to the skeleton, the Efn-Eph bidirectional signaling, particularly that of the EfnB2-EphB4, plays a key role in the maintenance of the size, density, and remodeling of the bone [14], skeletal patterning [15, 16], osteoblast differentiation, and bone formation [17]. EfnB1 ligand and EphA5 receptor have recently been suggested to be mediators of fracture callus cartilage hypertrophy and ossification [18]. In the osteoclast, the reverse signaling of EfnB2 [19] or EfnB1 [20] suppresses osteoclastic differentiation of preosteoclasts via an inhibition of the Fos/Nfatc1 cascade. Conversely, EfnA2 enhances osteoclastic differentiation of preosteoclasts and stimulates bone resorption via its reverse signaling [21]. The forward signaling of EphA4 has also shown to have a potent negative regulatory role in osteoclast activation [22]. With respect to chondrocytes and cartilage metabolism, the EfnB2-EphB4 interaction has been reported to modulate functional activities of articular chondrocytes, albeit some of the findings appear conflicting. On the one hand, the EfnB2-mediated activation of the EphB4 signaling in a subpopulation of subchondral osteoblasts [23] or chondrocytes from patients with osteoarthritis, OA [24] suppressed bone resorption. On the other hand, the EfnB2 treatment in chondrocytes promoted the osteoclast-mediated cartilage destruction [25]. The molecular mechanism(s) attributed to this apparent contrasting effects of EfnB2-mediated activation of EphB4 on bone/cartilage destruction is unclear at this time. Regardless of the mechanistic reasons, the EfnB2-EphB4 bi-directional signaling appears to have an important regulatory role in OA [26], since mice with conditional overexpression of Ephb4 in osteoblasts exhibited less OA-related losses in subchondral bone and articular cartilage than wildtype (WT) littermates [27], and deletion of Efnb2 in chondrocytes led to the development of OA-like phenotype in older mice [28].
We are interested in the potential regulatory role of the EphA4 signaling in OA, because chondrocytes, including articular chondrocytes, expressed high levels of EphA4 and because EphA4 signaling has been suggested to be a positive regulator of endochondral ossification [29]. Our findings that the femur of Epha4 KO mice have shorter growth plate and more disorganized columns of hypertrophic chondrocytes [22], supports the premise that EphA4 may have anabolic actions on longitudinal endochondral bone growth. Our interest in the potential role of EphA4 in OA is piqued further by the observations that: (1) macrophages are one of the major cell types in the inflamed synovium, (2) cells of monocytes–macrophages lineage, including macrophages and osteoclasts, express substantial cellular levels of EphA4, and (3) the EphA4 signaling is a potent negative regulator in osteoclasts and macrophages [22, 30]. Synoviocytes and articular chondroncytes, similar to osteoclasts and macrophages, are key regulatory cell types in the OA pathology. Thus, we sought to investigate whether the EphA4 signaling also has regulatory actions on the functions of synoviocytes and articular chondrocytes. In this study, we present in vitro evidence that the EphA4 signaling has anti-catabolic actions on synoviocytes but conversely and intriguingly, this signaling appears to be pro-anabolic in articular chondrocytes.
Methods
Materials
Culture media were purchased from Life Technologies (Grand Island, NY, USA) and fetal bovine serum (FBS) was obtained from Tissue Culture Biologicals (Los Alamitos, CA, USA) or Atlantic Biologicals (Flowery Branch, GA, USA). Tissue culture supplies were from Falcon (Oxnard, CA, USA). EphA4-fc and EfnA4-fc chimeric proteins were obtained from R&D Systems (Minneapolis, MN, USA). All other reagents were of reagent-grade and were obtained from Millipore-Sigma (St. Louis, MO, USA) or Fisher Scientific (Los Angeles, CA, USA).
Animals
Male C57BL/6J mice were purchased from the Jacksons Laboratory (Bar Harbor, Maine, USA). Homozygous Epha4 KO mice were generated by cross-breeding heterozygous Epha4 KO breeder mice (in 129J genetic background) as described previously [22].
For the investigation of the effects of an intraarticular knee injury on the EphA4 signaling in synovium and articular cartilage, we used a closed intraarticular tibial plateau compression loading-induced injury model of PTOA to create injuries to tibial plateau of the right knee joint of 12-week-old male mice [31]. Briefly, the animals were anesthetized through inhalation of isoflurane (3% isoflurane, 0.5 L/min oxygen delivered through a Bains circuit). The right knee was positioned on an apparatus attached to the load cell of the Instron 8841 servohydraulic mechanical tester (Norwood, MA) in a manner that exposed the tibial plateau. To injure the tibial plateau, the excursion of a blunt indenter blade was set on the top of the tibial plateau. An impact force (controlled by computer using Instron “FastTrack 2” software) was applied under a defined force (55 N) at a speed of 60 N/s to the tibial plateau of the mouse as the leg of the animal is positioned in a cradle. This impact force yielded a compression energy sufficient to create significant injuries to the synovium, the meniscus, and the articular cartilage layer, but not causing obvious fractures, at the tibial plateau. The injured right joint of all mice developed well-defined hallmark characteristics of OA after 5 weeks, including erosion of the articular cartilage layer, deterioration of periarticular/subchondral bone structures, formation of osteophytes, enhanced hypertrophy and calcification of menisci, and loss of trabecular rod and thickening of trabecular plate in subchondral bone [31]. The contralateral uninjured left tibiae were used as internal controls.
The animal protocol was reviewed and approved by the Animal Care and Use Committee of the Pettis Memorial VA Medical Center and also by the Animal Care and Use Review Office (ACURO) of US Army Medical Research and Materiel Command of the Department of Defense. In conducting research using animals, the investigators adhered to the Animal Welfare Act Regulations and other Federal statutes relating to animals and experiments involving animals.
Isolation of Murine Synoviocytes
Murine synoviocytes were isolated from untreated animals according to an established procedure [32]. Briefly, the synovial pad of both knee joints was isolated from the posterior aspect of the knee of C57BL/6J mice at sacrifice and minced. The minced tissues were rinsed once with phosphate-buffered saline (PBS) and treated with trypsin (in PBS) for 15 min at 37 °C with shaking. The trypsin solution was then removed by centrifugation, and the spun down tissue was digested in 0.1% collagenase P, 0.005% DNAse I in F12/DMEM media for 15 min at 37 °C with shaking. The digested medium was collected by centrifugation and discarded, and the tissue was then digested in 1 mL of αMEM containing 0.2% collagenase P and 10% heat-inactivated fetal bovine serum (FBS) for 2–3 h at 37 °C with constant shaking. The digestion was terminated by addition of 25 mL of αMEM containing 10% heat-inactivated FBS, and the digested medium was collected and filtered with a 70-µ filter. The isolated synoviocytes were collected by centrifugation and were cultured at 37 °C under an atmosphere of 5% CO2 in a specialized synoviocyte growth medium purchased from Millipore-Sigma (St. Louis, MO), supplemented with penicillin/streptomycin. Culture medium was replaced with fresh medium after 3 h and on the following day. Medium was changed every 3 to 4 days. Stellar cells-like synoviocyte colonies were visible between 2 days and 2 weeks in culture. After confluent, cells were frozen for preservation or immediately expanded for in vitro studies.
The overall yield of murine synoviocytes from a single mouse was low in part because of the very limited amounts of synovial tissues that could be isolated from each mouse. Thus, synoviocytes from both knee joints of two to three mice were pooled together for use as a single sample in the subsequent in vitro studies. The isolated synoviocytes expressed high mRNA levels of S100A4 (a member of the S100 calcium-binding proteins—a marker of activated synoviocytes at sites of inflamed joints [33]), type II collagen (an extracellular protein synthesized by synoviocytes), and α-SMA (a smooth muscle gene that is highly expressed in inflamed synoviocytes and has been used as one of the marker genes of inflamed synoviocytes in rheumatoid arthritis [34, 35]) (Fig. 1a), but with very low levels of type I collagen mRNA (a marker gene of fibroblasts) (not shown). The foregoing characteristics support our premise that they were bona fide synoviocytes. The expression level of S100a4 and Col2α1 mRNA in synoviocytes of the injured inflamed synovium was increased 190- and sevenfold, respectively, compared to control synoviocytes from uninjured synovium (Fig. 1b), indicating that synoviocytes in the injured synovia were highly activated. However, although the expression of α-SMA in synoviocytes is upregulated in rheumatoid arthritis [35], its expression in synoviocytes isolated from injured knee joints was not only not increased but drastically reduced (Fig. 1b), raising the possibility that α-SMA might not be a marker for inflamed synoviocytes in OA.

Primary mouse synovial fibroblasts (synoviocytes) expressed high levels of three known marker gene of synoviocytes, i.e., S100a4 Col2α1, and αSMA (a marker gene of smooth muscle cells but highly expressed in inflamed synoviocytes in rheumatoid arthritis (a), and an intraarticular tibial plateau compression loading-induced injury drastically upregulated S100A4 and Col2α1, but not αSMA, in synoviocytes (b). Conversely, primary mouse articular chondrocytes expressed high levels of Col2 but very low levels of Col1 and Col10, as well as substantial levels of Sox9 and ROCK (c). In a and b, the assays were performed on a single pooled sample of synoviocytes generated from three adult uninjured, untreated C57BL/6J mice. In c, the assays were performed in four separate preparations of articular chondrocytes each isolated from a single adult untreated, uninjured C57BL/6J mouse. Results are shown as mean ± SEM (n = 4)
Isolation of Mouse Articular Chondrocytes
Articular chondrocytes were isolated according to a published procedure [36]. Briefly, untreated C57BL/6J mice were sacrificed and articular cartilages from both knee joints were collected, rinsed in PBS, and incubated with 2.5% trypsin (Atlanta Biologicals, Flowery Branch, GA) in 3 mL of PBS at 37 °C for an hour. The trypsin was removed and the tissue was digested with Pronase (2 mg/mL) in minimal essential medium α medium (α-MEM) containing penicillin and streptomycin at 37 °C for 1 h. The Pronase was removed and the exposed articular cartilage layer was then digested overnight with 0.25 mg/mL of collagenase IV in α-MEM containing 5% heat-inactivated FBS and antibiotics. The digestion was terminated by the addition of α-MEM containing 10% FBS. The released articular chondrocytes were filtered through a 70-μ sterile filter, collected by centrifugation, and expanded in α-MEM supplemented with 10% FBS. The isolated cells expressed predominantly type II collagen (a marker for proliferative chondrocytes) with 400- to 500-fold lower levels of type X collagen (a marker for hypertrophic chondrocytes) or type I collagen (a marker for fibroblasts and osteoblasts) (Fig. 1c), confirming that these are bona fide chondrocytes. These cells also expressed substantial amounts of Sox9 and Rho-associated protein kinase (ROCK), two of the genes that are essential for cartilage matrix production [37].
Acidic Proteoglycan-Expressing Articular Chondrocyte Colony Nodule Assay
Briefly, chondrocytes were plated at a density of 10,500 cells/cm2 in a 24-well plate in α-MEM supplemented with 10% FBS and penicillin/streptomycin. At ~ 80% confluency, the test agent (EfnA4-fc or EphA4-fc, each at 10 ng/mL) or PBS (vehicle control) was added. The cells were cultured in α-MEM supplemented with 10% FBS for about 4 weeks. To identify acidic proteoglycans, cells were washed once with Tris-buffered saline containing calcium and were fixed for 30 min at room temperature in the modified Kahle’s fixative (15% ethanol, 6% formaldehyde, and 1% acetic acid). Acidic proteoglycan-expressing colonies were stained with 1% Alcian Blue 8GX in 3% acetic acid overnight at room temperature. The bluish-stained cells were washed 3 times in distilled deionized water for 2 min each. A light microscope was used to capture images of the colonies for analysis with ImagePro Plus 4.5 to determine the number and area of the colonies.
Quantitative Polymer Chain Reaction (qRT-PCR) Assays
qPCR reactions were carried out with the SYBR Green method on the MJ Research DNA Engine Opticon 2 system (Waltham, MA, USA). Total RNA was extracted from cells or tissues with the Qiagen RNeasy kit. The purified RNA was used as the template for synthesizing cDNA by reverse transcription using oligo dT(20) primers and ThermoScript RT-PCR kit (Invitrogen, Carlsbad, CA, USA). An aliquot of the cDNA was subjected to qPCR amplification using gene-specific primer sets listed in Table 1. The reaction mixture (25 µL) in each assay consisted of 12.5 µL of 2 × GoTag qPCR master mix (Promega, Madison, WI, USA), which included the Hot Start Taq polymerase, and the inclusion of 3 µM of primers with 3 µL of cDNA template. The PCR amplification condition consisted of an initial 10-min hot start at 95 °C, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing and extension for 30 s at an appropriate temperature for a given gene, and a final step of melting curve analysis from 60 to 72 °C. The data [normalized against cyclophilin A (Ppia) mRNA] were analyzed with Opticon Monitor Software 2.0. Data and the relative folds change were calculated by the threshold cycle (\(\Delta \Delta C_{{\text{t}}}\)) method.
Immunohistochemical (IHC) Staining for EphA4 Receptor
Briefly, 5-µm sections from the formalin-fixed, decalcified tibial joint were cut from a paraffin block medially and sagittally. The sections were baked for 1 h at 60 °C for enhanced attachment and deparaffinized with xylene/graded ethanol. Following washing three-times with PBS, the endogenous peroxidase activities and non-specific binding sites were then blocked with BLOXALL (endogenous peroxidase and alkaline phosphatase blocking solution, Vector Labs, Burlingame, CA) at room-temperature for 20 min followed by rinsing for three times with PBS. The non-specific binding sites were further blocked with normal horse serum for 30 min at 37 °C. The slides were then incubated with an appropriately diluted control rabbit IgG or rabbit antibody against EphA4 or pY-596-EphA4 (ThermoFisher Scientific, Los Angeles, CA, USA) in PBS containing 1% bovine serum albumin (in 0.05% Tween 20) for 1 h at 37 °C. The slides were rinsed three times with PBST for 3 min each. The appropriately diluted horseradish-conjugated anti-rabbit IgG (in PBS) was then added to each slide and were incubated at 37 °C for 30 min. After washing three times with PBS, the substrate of horseradish peroxidase (DAB) was added to the surface of the slides, incubated in dark for 2 to 10 min. The slides were washed twice with water for 1 min each time and counter-stained with hematoxylin for 18 s. They were then washed gently in running water followed by 95% ethanol, 100% ethanol, and xylene, air-dried and sealed with cytoseal.
Assay of Activation of Rac and Rho GTPases
The activation status of Rac or Rho was determined by measuring the relative GTP-bound Rac or Rho levels in 1 mg of each cellular extract proteins of pooled articular chondrocytes or synoviocytes from 3 mice of each EphA4 KO mice and WT littermates using the immune-pulldown-based Rac Activation Assay Kit or Rho Activation Assay Kit (ThermoFisher Scientific), respectively, as instructed by the supplier.
Western Immunoblot Assays
Relative levels of Rac1, RhoA, pY174-Vav1, Vav1, pY87-Ephexin, and Ephexin were determined by Western immunoblots using respective specific antibody (Santa Cruz Biotech., Santa Cruz, CA) as described previously [30]. Briefly, confluent synoviocytes or chondrocytes from EphA4 KO mice or WT littermates were treated with or without the 10 ng/mL EfnA4-fc chimera for 24 h. The protein was extracted with RIPA supplemented with the protease inhibitors mix (Millipore-Sigma), 1 mM PMSF, and 1 mM vanadate. The lysate was boiled in 1 × loading buffer containing β-mercaptoethanol. 30 (for Vav1) or 40 µg (for Ephexin) of cellular protein of each sample was separated on 10% SDS-PAGE gels and transblotted onto nitrocellulose membranes. Nonspecific sites were blocked with 5% fetal bovine serum or 10% non-fat milk in Tris-buffered saline Triton (TBST) overnight. The corresponding appropriately diluted antibody was then added to the blots and incubated for 4 h. After extensively washing, the blots were incubated with an appropriate horseradish peroxidase-conjugated secondary antibody for 1 h and chemiluminescent substrates were added. Quantification of chemiluminescence was done with the FluorChem Q imaging system (Proteinsimple, Inc., Santa Clara, CA).
Cell Proliferation/Viability and Apoptosis Assays
Cell proliferation/viability was determined with the MTT[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]-based Roche Cell Proliferation Kit I (obtained through Millipore-Sigma) according to the supplier’s instruction. Cell apoptosis was measured with the Roche Cell Death Detection ELISAPLUS kit, which was obtained through Millipore-Sigma.
Statistical Analysis
Results are shown as mean ± SEM. Statistical significance was analyzed with two-tailed Student’s t test between two test groups. Statistical significance of more than two groups was determined by one-way ANOVA followed by the Tukey Post Hoc test using Systat11 (Systat, Inc., Chicago, IL, USA). The difference was considered significant when P < 0.05.
Results
Intraarticular Tibial Plateau Injury Upregulated the Expression and Phosphorylation of EphA4 in Relevant Cells of the Knee Joint
We performed IHC experiments to determine the expression profile of EphA4 protein in the relevant cell types of the knee joint and the effect of an intraarticular knee injury on the EphA4 expression profile within the knee joint. Figure 2 shows that articular chondrocytes, subchondral osteoblasts, osteocytes, meniscal cells, and synoviocytes of the uninjured tibial joint under basal conditions all expressed some levels of EphA4 immunostaining (Fig. 2a, c, e, g, and i, respectively), which was greatly enhanced by an intraarticular tibial plateau injury, as the EphA4 immunostaining level in the relevant cell types of the injured joint at 5 weeks post-injury was much more intense immunostaining (brownish color) than those in the contralateral uninjured joint (Fig. 2b, d, f, h, j). At higher magnifications, highly immunoreactive staining was seen on articular chondrocytes (Fig. 2d), subchondral osteoblasts (Fig. 2f), osteocytes (Fig. 2h), and meniscal and synovial cells (Fig. 2j).

Immunohistochemical staining of EphA4 in the injured joints (b, d, f, h, j) and the corresponding contralateral uninjured (a, c, e, g, i) at 5 weeks after the intraarticular tibial plateau injury. All slides were counter-stained with hematoxylin. a and b Full view of the femoral-tibial gap. c and d Emphasize the articular chondrocyte layers. Some of the EphA4-expressing articular chondrocytes are indicated by white arrows. e and f Focus on the subchondral bone layer, and some of the EphA4-expressing osteoblasts are identified by green arrows while EphA4-expressing osteoclasts are depicted by black *. g and h EphA4-expressing osteocytes, and some of the EphA4-expressing osteocytes are indicated by white *. i and j Mensicus and synovium, and EphA4-expressing synoviocytes are indicated by black arrows. Immunoreactive EphA4 was stained in brown color. k Nonimmune IgG staining, which showed no staining in any of the samples. Scale bars = 200 µm
The EphA4 signaling is activated by autophosphorylation of two juxtamembrane tyrosines, tyr-596 (Y596) and Y602 [6]. To determine whether synovial inflammation in response to the joint injury would activate the EphA4 signaling in these cell types, we compared the relative level of the protein-tyrosine phosphorylated (pY)-596 species of EphA4 in the injured joint with that in contralateral uninjured joint. The basal expression levels of total EphA4 (Fig. 3, top left panel) and pY596-EphA (Fig. 3, bottom left panel) in articular chondrocytes, osteoclasts, osteoblasts, osteocytes, and synoviocytes of the uninjured knee joint were confirmed to be low. However, at 5 weeks after the injury, there were large increases in both the levels of immunoreactive total EphA4 (Fig. 3, top right panel) and pY596-EphA4 (Fig. 3, bottom right panel) in all these cells. Thus, the tibial plateau injury not only upregulates the expression of the EphA4 protein but also activates the EphA4 signaling in all relevant cells inside the injured joint.

Immunohistochemical staining of the total (top) and phosphorylated (bottom) species of EphA4 protein in the contralateral uninjured (left) and the injured (right) joints of wild-type littermate mice (a), and comparison of immunohistochemical staining of phosphorylated species of EphA4 of WT littermate mice and Epha4 KO mice (b). The slide was counter-stained with hematoxylin. Immunoreactive EphA4 was stained in brown color. Scale bars = 200 µm
The EphA4 Signaling is a Negative Regulator in Synovium
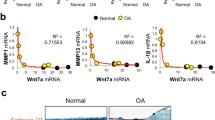
During synovial inflammation, large numbers of monocytes infiltrate into the inflamed synovium, where they differentiate into activated M1 macrophages [38]. which then secrete pro-inflammatory cytokines to induce synoviocytes to take on an aggressive and invasive phenotype to break down the articular cartilage through increased secretion and activation of degradative enzymes, such as MMPs and ADAMTSs, whilst the increased production of secretory factors, e.g., soluble RANKL, would promote osteoclast differentiation, survival, and resorptive activity, contributing to erosion of perichondral and subchondral bones. To determine whether EphA4 has a regulatory role in the synovial secretion of pro-inflammatory cytokines and degradative enzymes, we compared the mRNA levels of several pro-inflammatory cytokines and matrix degradative enzymes in synovial membranes of Epha4 KO mice with those in synovial membranes of age- and sex-matched WT littermates. Figure 4a shows that the level of IL-1β and TNFα mRNAs (the two major pro-inflammatory cytokines in OA), but not IL-6, was each increased five- to tenfold in the Epha4 KO synovial membranes compared to WT synovial membranes. The expression levels of Mmp9 and Mmp13 (but not Mmp3) were 45- to 150-fold greater, respectively, in Epha4 KO synovial membranes than in WT synovial membranes. To confirm a suppressive role of the EphA4 signaling on synovial secretion of IL-1β and Mmp9 proteins, we treated the isolated synovial membranes from WT mice with 10 ng/mL of EfnA4-fc for 24 h to activate the EphA4 signaling and then measured the levels of IL-1β and Mmp9 protein released into conditioned media (CM). The EfnA4-fc treatment reduced the released levels of Mmp9 protein by ~ 25% (Fig. 4b, top panel) and IL-1β protein to an undetectable level (Fig. 4b, bottom panel). (The CM Mmp13 protein level was not measured in this experiment). Thus, it appears that the EphA4 signaling has a negative regulatory role in inflamed synovia with respect to their releases of pro-inflammatory cytokines and matrix proteases.

Deficient EphA4 expression enhanced expression levels of pro-inflammatory cytokines (red bars) and matrix proteases (cyan bars) in synovial membranes (a), and the EfnA4-fc-mediated activation of the EphA4 signaling suppressed the synovial secretion of Mmp-9 and IL-1β proteins (b) in vitro. The EfnA4-fc-mediated activation of the EphA4 signaling in mouse synoviocytes also reduced the mRNA expression of certain inflammatory cytokines (IL-1β and IL-10) and Mmp9 and Mmp13 (c). a Synovial membranes were isolated from the knee joints of 12 weeks old male EphA4 KO mice and age- and gender-matched WT littermates (n = 6–9 synovial membranes per group). The relative level of each test gene was measured with RT-qPCR. The results are shown as mean ± SEM. The relative level of each gene in the EphA4 KO synovial membranes are reported as relative fold of the corresponding mRNA level in WT synovial membranes. *P < 0.05 and **P < 0.01. b Synovial membranes isolated from 12 weeks old male C57BL/6J mice and treated with or without 10 ng/mL of EfnA4-fc for 24 h to activate the EphA4 signaling. The concentration of Mmp9 and IL-1β protein in the conditioned media was determined with corresponding ELISA and normalized against respective cellular protein levels. Results are shown as mean ± SEM (n = 6 for group). c Synoviocytes were isolated from 12 weeks old male C57BL/6J mice and treated with or without 10 ng/mL of EfnA4-fc for 24 h to activate the EphA4 forward signaling. The mRNA expression levels of the test pro-inflammatory cytokines and matrix proteinases were determined by RT-qPCR. Results are shown as percentage of the mean of the corresponding mRNA level of respective vehicle-treated cells (mean ± SEM, n = 3 for each group). The solid line indicates the 100% level of respective mRNA of the controls. Statistical significance was determined with the two-tailed Student’s t-test. *P < 0.05 and **P < 0.01
The two major cell types in the synovial membrane are synovial macrophages and synoviocytes (also known as synovial fibroblasts) [39]. We have previously shown that the EphA4 forward signaling in cells of the monocyte–macrophage lineage, including macrophages and osteoclasts has potent suppressive actions of their cellular functional activities [22]. Unlike macrophages, synoviocytes are cells of mesenchymal origin and exhibit many characteristics common fibroblasts. Table 2 shows that with the exception of EphB1, which was very low (ΔCt > 33 cycles), primary mouse synoviocytes under basal conditions expressed abundant amounts of mRNA of all test members of Eph receptors (including EphA4) and Efn ligands. We next evaluated whether the EphA4 signaling is also a negative regulator of cellular functions of synoviocytes by determining the effects of the activation of EphA4 signaling (through the EfnA4-fc treatment) on the expression of pro-inflammatory cytokine genes and key Mmps in primary mouse synoviocytes by RT-qPCR. Under basal conditions, mouse synoviocytes expressed low (but detectable) levels of mRNA of inflammatory cytokines (i.e., IL-1β, IL-1α, IL-4, IL-6, TNF-α, and IL-13), but considerably higher levels of mRNA for Mmp9 and Mmp13, and to a lower level, Mmp3 (data not shown). The 24-h EfnA4-fc treatment of synoviocytes suppressed the expression of IL-1β, IL-10, Mmp9 and Mmp13 mRNA by ~ 65%, ~ 70%, ~ 90%, and ~ 80%, respectively (Fig. 4c). Thus, similar to that in osteoclasts/macrophages, the EphA4 signaling has suppressive actions in synoviocytes.
EphA4 is a Positive Regulator of the Functional Activity of Articular Chondrocytes
We next determined the potential regulatory role of the EphA4 signaling on the functions of articular cartilage. We compared the relative expression levels of several chondrogenic marker genes in isolated articular cartilage layer of Epha4 KO mice with those of age-matched WT littermates (Fig. 5a). Deficient Epha4 expression in articular cartilage layer reduced the expression levels of several key chondrogenic marker genes [i.e., aggrecan (Acan), type-2 collagen (Col2α1), type-10 collagen (Col10α1), and Sox9], but it did not alter the expression levels of the ligands of EphA4 receptor, i.e., Efna4 and Efnb2 as well as lubricin (Prg4), Conversely, the 24-h EfnA4-fc treatment of WT articular cartilage layers significantly upregulated the expression levels of Acan, Prg4, Col2α1, and Sox9, while it had no effects on the expression levels of Col10α1, Epha4, or its ligands, Efna4 and Efnb2 (Fig. 5b).

Deficient EphA4 expression in articular cartilage layers of Epha4 KO mice downregulated (a), while treatment of articular cartilage layers of WT mice with 10 ng/mL EfnA4-fc chimeric protein for 24 h upregulated (b), articular chondrogenic genes, such as aggrecan (Acan), type II collagen (Col2α1), type X collagen (Col10α1), Sox9, and lubricin (Prg4). Neither conditions altered the expression of ligand genes of EphA4, such as Epha4, Efna4, and Efnb2. Results are shown as relative fold of the corresponding mRNA level in EphA4 KO articular cartilage layers of age- and gender-matched WT littermates (mean ± SEM, n = 3 per group). *P < 0.05, **P < 0.01, and ***P < 0.001
To confirm whether these EphA4-dependent changes in chondrogenic gene expression would alter the functional activity of primary articular chondrocytes, we examined the effect of the 4 weeks of the EfnA4-fc treatment on the ability of articular chondrocytes to form acidic proteoglycan-producing colonies in culture. For comparison, we also treated the same articular chondrocytes with 10 ng/mL of EphA4-fc chimeric protein, which binds and activates the reverse signaling of the cell surface Efn ligands. The EfnA4-fc (but not EphA4-fc) treatment significantly increased the number (Fig. 6a, bottom left) and average size (Fig. 6a, bottom right) of the acidic proteoglycan-producing colonies compared with vehicle-treated articular chondrocytes. The EfnA4-fc treatment in Epha4 KO articular chondrocytes did not have an effect (Fig. 6b), indicating that the anabolic effect of EfnA4-fc in articular chondrocytes was mediated through the signaling of EphA4.

The EfnA4-fc-mediated activation of the EphA4 signaling in the WT articular chondrocytes (a), but not in the EphA4 KO articular chondrocytes (b), increased the number and the average size of acidic proteoglycan-producing colonies after 4 weeks in culture in vitro. Top panels show representative Alcian blue staining of acidic proteoglycan-producing colonies of vehicle-treated control and EfnA4-fc-treated WT (a) or EphA4 KO (b) articular chondrocytes. Bottom panels show the quantitative analyses of the number and average area of the colonies. Significance was determined by two-tailed Student’s t-test. *P < 0.05, n = 4 per group. P = N.S. (P is not significant), when P > 0.05
To determine whether the anabolic effects of the EphA4 signaling on the cellular activities of articular chondrocytes were due to increased cell proliferation and/or reduced cell apoptosis, we assessed the effects of the 24-h treatment of primary articular chondrocytes with 10 ng/mL of EfnA4-fc or vehicle on cell viability/proliferation and apoptosis. The EfnA4-fc treatment did not alter the cell number as indicated by MTT assay or apoptosis (Fig. 7a) of WT articular chondrocytes or those of EphA4 KO articular chondrocytes (Fig. 7b). Since the cell number in a given culture is determined by the balance of cell proliferation rate and cell apoptosis rate, and since there was no significant difference in the apoptosis rate in either the EfnA4-fc-treated or untreated WT or EphA4 KO articular chondrocytes, we conclude that the EfnA4-fc treatment also did not affect the proliferation of WT or EphA4 KO articular chondrocytes.

Activation of the EphA4 signaling by a 24-h treatment with 10 ng/mL of EfnA4-fc did not affect the survival/proliferation or apoptosis of articular chondrocytes of WT mice (a) or those of EphA4 KO articular chondrocytes (b). The survival (total cell number) was measured by the MTT-based Roche Cell Proliferation Kit I and apoptosis was determined with the Roche Cell Death Detection ELISAPLUS kit. Cells treated with 10 ng/mL of EphA4-fc were included for comparison. Results are shown as mean ± SEM with n = 4–8 per groups. Significance was determined by two-tailed Student’s t-test. N.S. not significant, i.e., P > 0.05. Total cell number (survival) is determined by the balance of cell proliferation rate and the apoptosis rate. Since there were no differences in apoptosis in either case. The total cell number determined by the MTT assay can be a relative indicator of cell proliferation
The EphA4 Signaling Has Opposite Contrasting Effects on the Vav/Rac Signaling vs. Ephexin/Rho Signaling in Synoviocytes Compared to Those in Articular Chondrocytes
Previous studies have indicated that the Rho family of small GTPases are key mediators of the EphA4 forward signaling [40,41,42,43]. Our previous studies have suggested that the molecular mechanism of the EphA4 signaling to suppress the osteoclast activity is in part mediated through differential actions of the EphA4 signaling on the Vav/Rac signaling versus the Ephexin/Rho signaling [30]. To determine whether the EphA4 signaling has similar differential regulatory effects on the Vav/Rac versus Ephexin/Rho signaling in synoviocytes and articular chondrocytes, we compared the effects of the stimulation the EphA4 signaling (with the EfnA4-fc treatment) on activation status of Rho versus that of Rac in synoviocytes with those in articular chondrocytes using respective GTP-Rac or GTP-Rho pulldown activation assay. As shown in Fig. 8a, stimulation of the EphA4 signaling by the EfnA4-fc treatment reduced the amount of the pull-down GTP-Rac protein and increased that of the pull-down GTP-Rho protein from extracts of pooled synoviocytes from three mice. Surprisingly, the same EfnA4-fc treatment had exactly opposite effects on these two members of small G protein family in pooled articular chondrocytes in that it increased the amounts of pull-down GTP-Rac protein but reduced the amounts of pull-down GTP-Rho proteins (Fig. 8b).

The EfnA4-fc-induced activation of the EphA4 signaling has opposite differential effects on the Ephexin1/RhoA activation versus the Vav1/Rac1 activation in synoviocytes when compared to those in articular chondrocytes. a and b The activation status of Rac1 and RhoA determined by relative amounts of GTP-bound Rac or Rho to total Rac or Rho, respectively, in synoviocytes and in articular chondrocytes with or without the 24-h treatment with 10 ng/mL EfnA4-fc. GTP-bound RhoA or Rac1 of each cell type were assayed with respective commercial kits. Total Rac or Rho levels were determined by western blots. Top panels in a and b show the western of GTP-bound and total RhoA and Rac1, respectively. Each pull-down assay required approximately 1 mg cellular protein. Thus, it was necessary to pool samples from three preparations for each pull-down assay. Each assay was performed twice. Bottom panels in a and b summarize the relative ratio of GTP-RhoA (or Rac1) to total RhoA (or Rac1) in the synoviocytes and in articular chondrocytes, respectively. c The pY87-Ephexin1 and total Ephexin1 levels in the EfnA4-fc-treated or vehicle-treated synoviocytes or articular chondrocytes were assayed by western immunoblots using respective specific antibodies. Top panels in c show respective western immunoblots of the pY87-Ephexin1 and total Ephexin1 bands, and bottom panels in c summarize the relative ratio of pY87-Ephexin1/total Ephexin1 and that of the Ephexin1/actin ratio in each test cell type. Results are shown as mean ± SEM, n = 3–4 for each. d The pY174-Vav1 and total Vav1 levels in the EfnA4-fc-treated or vehicle-treated synoviocytes or articular chondrocytes were assayed by western immunoblots using respective specific antibodies. Top panels in d show respective western immunoblots of the pY174-Vav1 and total Vav1 bands, and bottom panels in d summarize the relative ratio of pY175-Vav1/total Vav1 in and that of the total Vav1/actin in each test cell type. The dotted lines in c and d represent the 100% of the respective vehicle-treated controls. Results are shown as mean ± SEM, n = 3–4 for each
Rho and Rac are activated by respective Rho GEFs (GTP exchange factors), e.g., Ephexin, and RacGEF, e.g., Vav, respectively [40, 44]. Both GEFs are activated in part by protein-tyrosine phosphorylation [45, 46]. To evaluate whether the contrasting effects of the EphA4 signaling on synoviocytes as opposed to those on articular cartilage were mediated partly by an opposite effect of the EphA4 signaling on the activation of Ephexin and Vav in these two cell types, we determined the effects of the 24-h treatment of EfnA4-fc on the ratio of pY87-Ephexin1/total Ephexin1 and that of pY174-Vav1/total Vav1 in these two cell types. Consistent with suppression of Rac1 activation and RhoA activation in synoviocytes, the EfnA4-fc treatment significantly upregulated Ephexin1 (Fig. 8c) and downregulated Vav1 (Fig. 8d), respectively, in synoviocytes. However, the EfnA4-fc treatment suppressed Ephexin1 (Fig. 8c) and upregulated Vav1 (Fig. 8b) in articular chondrocytes, which is in accordance with the observed activation of Rac1 and suppression of RhoA in articular chondrocytes. Accordingly, the activation of the EphA4 signaling yielded opposing biological effects in synoviocytes vs. articular chondrocytes in that the EfnA4-fc-induced activation of EphA4 signaling upregulated the Ephexin1/RhoA signaling in synoviocytes but an upregulation of the Vav1/Rac1 signaling in articular chondrocytes.
Discussion
A major function of articular cartilage of the joint surface and synovial fluids is to provide cushion and lubrication for facilitating the natural joint motion [47]. Injuries to the joint as well as chronic or increased joint loading can lead to acute and chronic synovial inflammation, which activates synoviocytes to release pro-inflammatory cytokines that promote the local releases of MMPs and other matrix degradative enzymes to break down extracellular matrices of articular cartilage [48, 49] that can cause lesions in the articular cartilage and/or synovium, severe joint pain and swelling, muscle weakness, impaired physical function, limited joint movement, and eventually OA. Understanding of the molecular mechanism(s) regulating the local release of cytokines and proteases from synoviocytes and those responsible for the inability of articular chondrocytes to regenerate the damaged cartilage would not only yield important insights into the pathogenesis of OA but could also offer novel potential therapeutic targets for the development of therapies of OA.
In this report, we presented strong in vitro evidence that the EphA4 signaling in synoviocytes and in articular chondrocytes could play a regulatory role in OA. Specifically, we showed that an intraarticular tibial plateau injury, which eventually developed PTOA [31], rapidly and drastically upregulated the expression and activation of EphA4 in the relevant cell types in the injured knee joint, such as articular chondrocytes, subchondral osteoblasts, osteocytes, meniscal cells, and synoviocytes. More importantly, we showed for the first time that the EphA4 signaling exerts opposite biological responses on synoviocytes versus articular chondrocytes. Accordingly, we found that activation of the EphA4 signaling in synovium (and synoviocytes) of WT mice suppressed, whereas deficient expression of Epha4 in synovium of Epha4 KO mice significantly enhanced, the expression and release of inflammatory cytokines (e.g., IL-1β and TNFα) and matrix degradative enzymes (e.g., Mmp9 and Mmp13). During OA development, activated synoviocytes in inflamed synovium would promote a major catabolic action of OA by enhancing the local releases of proinflammatory cytokines and matrix proteases to carry out the degradation of articular cartilage surface. Thus, it follows that the EphA4 signaling has anti-catabolic actions on synovium and synoviocytes. Consistent with an anti-catabolic function of the EphA4 signaling in catabolic cells, we have previously shown that the EphA4 signaling in mature osteoclasts is a potent negative regulatory mechanism of their bone resorption activity [22]. On the other hand, we show in this study that activation of the EphA4 signaling in articular chondrocytes enhanced the expression of key cartilaginous genes, such as Acan, Prg4, Col2α1, and Col10α1 and promoted their cartilage biosynthesis activity. The fact that the EfnA4-fc treatment in Epha4 KO articular chondrocytes was unable to upregulate the formation of acidic proteoglycan-producing cell colony and their activity to produce cartilage supports our premise that the pro-anabolic action in articular chondrocytes was mediated by the EphA4 signaling. These findings indicate that contrary to synoviocytes and osteoclasts, the EphA4 signaling in articular chondrocytes is of pro-anabolic nature. However, the molecular mechanism responsible for the opposite biological effects of the EphA4 signaling on synoviocytes vs. chondrocytes remains unclear.
Mechanistically, the cellular actions of EphA4 in many cell types are in a large part mediated through differential regulation of various members of the Rho family of small GTPases, which serve as molecular switches to regulate a number of cellular functions [45]. For example, in neurons [50,51,52], osteoclasts [30], and vascular smooth muscle cells [42], the EphA4 signaling enhances the phosphorylation and activation of a Rho GTP exchange factor (GEF), such as Ephexin, Vsm-RhoGEF, which in turn activates Rho by exchanging GDP on Rho with GTP; but at the same time, it also inhibits the activity of Rac, Cdc42, or Rap, through the suppression of a Rac GEF, such as Vav [43, 50]. We have previously proposed a molecular model, which we termed the “EphA4 phosphorylation switch” mechanism [30]. to explain the opposite regulation of Ephexin/Rho and Vav/Rac by the EphA4 signaling in osteoclasts. This model was based on the findings that Vav [53] and Ephexin [45, 51] each binds through their plekstrin-homology domain [42, 54] to the same structural domain of the EphA4 receptor that contains the Y596/Y602 residues [55, 56]. We postulated that the phosphorylated EphA4 receptor has a higher binding affinity for Ephexin over Vav; a speculation based on the findings that the binding of Vav [53], but not that of Ephexin, to EphA4 [51] in neurons requires the phosphorylation of Y596/Y602. Hence, when the EphA4 receptor is activated and phosphorylated, the binding site of EphA4 assumes the conformation favoring the binding of Ephexin over Vav. Conversely, when Y596/Y602 residues are dephosphorylated or under-phosphorylated, the receptor would prefer Vav over Ephexin for binding.
Our previously proposed “EphA4 phosphorylation switch” model could provide a potential mechanism for the observed EphA4-dependent upregulation of the Ephexin1/RhoA signaling and downregulation of the Vav1/Rac1 signaling in synoviocytes. However, this model would not explain the opposite effects of the EphA4 signaling on the Ephexin/RhoA signaling vs. the Vav/Rac1 seen in articular chondrocytes. It follows that the apparent discrepancy seen in articular chondrocytes vs. synoviocytes would indicate that the determination of the relative binding affinity of a given Rho GEF to the EphA4 receptor is more complicated than that depending merely on the phosphorylation status of the EphA4 receptor as proposed in our “EphA4 phosphorylation switch” model [30] and may involve some type of cell-type-specific components. There is strong evidence that members of the Rho GEF family, including Vav and Ephexin, are dynamically associated with molecular chaperones, such as Hsc70, Hsp90 [57,58,59]. This association would stabilize the conformation of the Rho GEFs and also prevents the ubiquitin-mediated degradation of Rho GEFs [60, 61]. It is possible that the association of a specific molecular chaperones could also alter their binding affinity of a given Rho GEF for the EphA4 receptor. There is also evidence for cell-type-specific variations in the induced expression of many of these chaperones [62]. Thus, it is conceivable that these Rho GEFs could dynamically associated with different certain yet-to-be-determined cell-type-specific chaperones in different skeletal cell types. For the purpose of description of our revised model, we will refer these cell-type-specific chaperones in synoviocytes and chondrocytes to as “chaperone X” and “chaperone Y”, respectively. We further speculate that the conformation of the Ephexin-“X” complex would assume a greater binding affinity than the Vav-“X” complex for the phosphorylated EphA4 in synoviocytes, whereas the binding affinity of Ephexin-“Y” in chondrocytes has a significantly lower binding affinity for the phosphorylated EphA4 than Vav-“Y”. Accordingly, when the EphA4 receptor is activated and their Y596/Y602 residues are phosphorylated in synoviocytes, the EphA4 protein would favor the binding of the Ephexin-“X” complex over the Vav-“X” complex, which then leads to the subsequent upregulation of the Ephexin/RhoA signaling and downregulation of the Vav/Rac1 signaling (Fig. 9a). The Vav/Rac1 pathway is essential for the cellular secretion of inflammatory cytokines, such as IL-1β [63], and extracellular matrix proteases, e.g., MMPs [64]. Thus, the downregulation of the Vav/Rac1 signaling in synoviocytes would suppress the release of inflammatory cytokines and matrix degradative enzymes and thereby reduces the risk of OA. However, the RhoA signaling plays important roles in multiple cellular processes, such as cell growth, transformation, adhesion, and cytoskeleton remodeling, in many cell types, including synoviocytes [65,66,67]. Therefore, the EphA4-induced activation of the Ephexin/RhoA signaling promotes cellular activities of the activated synoviocytes, including increased cell adhesion and reduced migration.

A proposed model of mechanism of action of the EphA4 signaling to differentially regulate Ephexin/RhoA vs. Vav/Rac1 in synoviocytes as opposed to that in articular chondrocytes. See text for description of the model
In contrast to synoviocytes, an activation of the EphA4 signaling in articular chondrocytes would upregulate the Vav/Rac1 signaling and downregulates the Ephexin1/RhoA/ROCK signaling (Fig. 9b). In the context of OA, there is strong evidence that the RhoA/ROCK signaling plays a key role in the pathogenesis of OA [68, 69]. For example, RhoA/ROCK signaling promotes the various OA pathological factors, such as TGFα [70, 71], Wnt/β-catenin [69], and leptin [72], to induce the degradation of chondrocytes extracellular matrices. Additionally, the RhoA-mediated activation of ROCK in chondrocytes is essential for the activation of Sox9, which in turn promotes chondrocyte hypertrophy [37, 73]. The combined effects of the RhoA/ROCK-dependent enhanced chondrocyte hypertrophy and increased degradation of the articular cartilage matrices would lead to OA. It follows that the EphA4 signaling-dependent downregulation of the Ephexin/RhoA/ROCK signaling in articular chondrocytes could thereby reduce the OA risk (Fig. 9b). The regulatory role of the Vav/Rac1 signaling in OA pathology is less clear. Nevertheless, there is evidence that targeted disruption of Rac gene in chondrocytes reduced cartilage formation and resulted in chondrodysplasia, suggesting that the Rac1 pathway is important in cartilage development [74]. Moreover, the Rac signaling enhances the expression of the cartilage growth factor gene, CTGF/CCN2, expression via TGFβ/Smad signaling in chondrocytes [75]. It has also been reported that a single injection of recombinant CTGF/CCN2 in gelatin hydrogel into the joint cavity of monosodium iodoacetate (MIA)-induced OA model rats possessed the ability to repair damaged articular cartilage [76]. Furthermore, Rac1 promotes endochondral bone growth through increasing chondrocyte proliferation via upregulation of the inducible nitric oxide synthase–nitric oxide signaling [77]. As a result, it is conceivable that the EphA4-dependent upregulation of the Vav/Rac1 signaling in articular chondrocytes could lead to enhanced biosynthesis of cartilage.
We should, however, emphasize that the various members of the Rho small GTPase family can be activated by several different GEFs. For example, it has been reported that a vascular smooth muscle Rho GEF, Vsm-RhoGEF, can mediate the GDP-GTP exchange on RhoA [42]. Therefore, the possibilities that different members of tissue-specific Rho GEFs may mediate the GDP-GTP exchange and activation of different members of Rho small GTPases in synoviocytes vs. chondrocytes and that the binding affinity of these different members of tissue-specific Rho GEFs on EphA4 receptor could also be different should not be ignored. In any event, we should note that there is currently no direct evidence supporting either the cell type-specific chaperones or cell type-specific Rho GEFs. Our future work will address these alternative hypotheses.
In summary, we have demonstrated for the first time that the EphA4 signaling in synoviocytes yielded anti-catabolic actions through the suppression of local release of proinflammatory cytokines and matrix proteases, but yet pro-anabolic actions in articular chondrocytes via the promotion of the biosynthesis of cartilaginous tissues. In conclusion, the fact that the EphA4 signaling appears to have both anti-catabolic and pro-anabolic actions in the context of OA raises the exciting possibility that the EphA4 signaling could be an attractive and novel therapeutic target for the development of an effective therapy for OA/PTOA.
Data Availability
All research data of this study and materials used in this study are stored in the laboratory of Dr. Kin-Hing William Lau, and they may be available to others upon request in writing and approval from the Research Office of the Loma Linda VA Healthcare system.
References
Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson DG, Pawson T, Davis S, Yancopoulos GD (1996) Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 17:9–19
Pasquale EB (1997) The Eph family of receptors. Curr Opin Cell Biol 9:608–615
Poitz DM, Ende G, Stutz B, Augstein A, Friedrichs J, Brunssen C, Werner C, Strasser RH, Jellinghaus S (2015) EphrinB2/EphA4-mediated activation of endothelial cells increases monocyte adhesion. Mol Immunol 68:648–656
Zimmer G, Rudolph J, Landmann J, Gerstmann K, Steinecke A, Gampe C, Bolz J (2011) Bidirectional ephrinB3/EphA4 signaling mediates the segregation of medial ganglionic eminence- and preoptic area-derived interneurons in the deep and superficial migratory stream. J Neurosci 31:18364–18380
Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, Vearing C, Geleick D, Feldheim DA, Boyd AW, Henkemeyer M, Nikolov DB (2004) Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci 7:501–509
Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F (2001) Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 106:745–757
Zisch AH, Pazzagli C, Freeman AL, Schneller M, Hadman M, Smith JW, Ruoslahti E, Pasquale EB (2000) Replacing two conserved tyrosines of the EphB2 receptor with glutamic acid prevents binding of SH2 domains without abrogating kinase activity and biological responses. Oncogene 19:177–187
Kullander K, Klein R (2002) Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol 3:475–486
Pasquale EB (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133:38–52
Kuijper S, Turner CJ, Adams RH (2007) Regulation of angiogenesis by Eph-ephrin interactions. Trends Cardiovasc Med 17:145–151
Genander M, Frisen J (2010) Ephrins and Eph receptors in stem cells and cancer. Curr Opin Cell Biol 22:611–616
Pasquale EB (2005) Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol 6:462–475
Rundle CH, Xing W, Lau KW, Mohan S (2016) Bidirectional ephrin signaling in bone. Osteoporos Sarcopenia 2:65–76
Edwards CM, Mundy GR (2008) Eph receptors and ephrin signaling pathways: a role in bone homeostasis. Int J Med Sci 5:263–272
Compagni A, Logan M, Klein R, Adams RH (2003) Control of skeletal patterning by ephrinB1-EphB interactions. Dev Cell 5:217–230
Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, Adams RH, Wieacker P (2004) Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet 74:1209–1215
Allan EH, Hausler KD, Wei T, Gooi JH, Quinn JM, Crimeen-Irwin B, Pompolo S, Sims NA, Gillespie MT, Onyia JE, Martin TJ (2008) EphrinB2 regulation by PTH and PTHrP revealed by molecular profiling in differentiating osteoblasts. J Bone Miner Res 23:1170–1181
Kaur A, Xing W, Mohan S, Rundle CH (2019) Changes in ephrin gene expression during bone healing identify a restricted repertoire of ephrins mediating fracture repair. Histochem Cell Biol 151:43–55
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K (2006) Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4:111–121
Cheng S, Zhao SL, Nelson B, Kesavan C, Qin X, Wergedal J, Mohan S, Xing W (2012) Targeted disruption of ephrin B1 in cells of myeloid lineage increases osteoclast differentiation and bone resorption in mice. PLoS ONE 7:e32887
Irie N, Takada Y, Watanabe Y, Matsuzaki Y, Naruse C, Asano M, Iwakura Y, Suda T, Matsuo K (2009) Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem 284:14637–14644
Stiffel V, Amoui M, Sheng MH, Mohan S, Lau KH (2014) EphA4 receptor is a novel negative regulator of osteoclast activity. J Bone Miner Res 29:804–819
Kwan Tat S, Pelletier JP, Amiable N, Boileau C, Lajeunesse D, Duval N, Martel-Pelletier J (2008) Activation of the receptor EphB4 by its specific ligand ephrin B2 in human osteoarthritic subchondral bone osteoblasts. Arthritis Rheumatol 58:3820–3830
Kwan Tat S, Pelletier JP, Amiable N, Boileau C, Lavigne M, Martel-Pelletier J (2009) Treatment with ephrin B2 positively impacts the abnormal metabolism of human osteoarthritic chondrocytes. Arthritis Res Ther 11:R119
Tonna S, Poulton IJ, Taykar F, Ho PW, Tonkin B, Crimeen-Irwin B, Tatarczuch L, McGregor NE, Mackie EJ, Martin TJ, Sims NA (2016) Chondrocytic ephrin B2 promotes cartilage destruction by osteoclasts in endochondral ossification. Development 143:648–657
Kwan Tat S, Lajeunesse D, Pelletier JP, Martel-Pelletier J (2010) Targeting subchondral bone for treating osteoarthritis: what is the evidence? Best Pract Res Clin Rheumatol 24:51–70
Valverde-Franco G, Pelletier JP, Fahmi H, Hum D, Matsuo K, Lussier B, Kapoor M, Martel-Pelletier J (2012) In vivo bone-specific EphB4 overexpression in mice protects both subchondral bone and cartilage during osteoarthritis. Arthritis Rheumatol 64:3614–3625
Valverde-Franco G, Lussier B, Hum D, Wu J, Hamadjida A, Dancause N, Fahmi H, Kapoor M, Pelletier JP, Martel-Pelletier J (2016) Cartilage-specific deletion of ephrin-B2 in mice results in early developmental defects and an osteoarthritis-like phenotype during aging in vivo. Arthritis Res Ther 18:65
Kuroda C, Kubota S, Kawata K, Aoyama E, Sumiyoshi K, Oka M, Inoue M, Minagi S, Takigawa M (2008) Distribution, gene expression, and functional role of EphA4 during ossification. Biochem Biophys Res Commun 374:22–27
Lau KH, Amoui M, Stiffel V, Chen ST, Sheng MH (2015) An osteoclastic transmembrane protein-tyrosine phosphatase enhances osteoclast activity in part by dephosphorylating EphA4 in osteoclasts. J Cell Biochem 116:1785–1796
Stiffel V, Rundle CH, Sheng MH-C, Das S, Lau K-HW (2019) A mouse noninvasive intraarticular tibial plateau compression loading-induced injury model of posttraumatic osteoarthritis. Calcif Tissue Int 106:158–171
Zhao J, Ouyang Q, Hu Z, Huang Q, Wu J, Wang R, Yang M (2016) A protocol for the culture and isolation of murine synovial fibroblasts. Biomed Rep 5:171–175
Senolt L, Grigorian M, Lukanidin E, Simmen B, Michel BA, Pavelka K, Gay RE, Gay S, Neidhart M (2006) S100A4 is expressed at site of invasion in rheumatoid arthritis synovium and modulates production of matrix metalloproteinases. Ann Rheum Dis 65:1645–1648
Vickers SM, Johnson LL, Zou LQ, Yannas IV, Gibson LJ, Spector M (2004) Expression of alpha-smooth muscle actin by and contraction of cells derived from synovium. Tissue Eng 10:1214–1223
Steenvoorden MM, Tolboom TC, van der Pluijm G, Lowik C, Visser CP, DeGroot J, Gittenberger-DeGroot AC, DeRuiter MC, Wisse BJ, Huizinga TW, Toes RE (2006) Transition of healthy to diseased synovial tissue in rheumatoid arthritis is associated with gain of mesenchymal/fibrotic characteristics. Arthritis Res Ther 8:R165
Jonason JH, Hoak D, O’Keefe RJ (2015) Primary murine growth plate and articular chondrocyte isolation and cell culture. Methods Mol Biol 1226:11–18
Haudenschild DR, Chen J, Pang N, Lotz MK, D’Lima DD (2010) Rho kinase-dependent activation of SOX9 in chondrocytes. Arthritis Rheumatol 62:191–200
Kinne RW, Stuhlmuller B, Burmester GR (2007) Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther 9:224
Edwards JC (1982) The origin of type A synovial lining cells. Immunobiology 161:227–231
Kim K, Lee SA, Park D (2019) Emerging roles of ephexins in physiology and disease. Cells 8(2):87
Winning RS, Ward EK, Scales JB, Walker GK (2002) EphA4 catalytic activity causes inhibition of RhoA GTPase in Xenopus laevis embryos. Differentiation 70:46–55
Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, Mochizuki N (2003) EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ Res 93:23–31
Richter M, Murai KK, Bourgin C, Pak DT, Pasquale EB (2007) The EphA4 receptor regulates neuronal morphology through SPAR-mediated inactivation of Rap GTPases. J Neurosci 27:14205–14215
Vedham V, Phee H, Coggeshall KM (2005) Vav activation and function as a rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol Cell Biol 25:4211–4220
Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O’Connell S, Cowan CW, Hu L, Goldberg JL, Debant A, Corfas G, Krull CE, Greenberg ME (2005) Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron 46:191–204
Lopez-Lago M, Lee H, Cruz C, Movilla N, Bustelo XR (2000) Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol Cell Biol 20:1678–1691
Martin JA, Buckwalter JA (2006) Post-traumatic osteoarthritis: the role of stress induced chondrocyte damage. Biorheology 43:517–521
Strobel S, Loparic M, Wendt D, Schenk AD, Candrian C, Lindberg RL, Moldovan F, Barbero A, Martin I (2010) Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther 12:R34
Lotz MK, Kraus VB (2010) New developments in osteoarthritis. Posttraumatic osteoarthritis: pathogenesis and pharmacological treatment options. Arthritis Res Ther 12:211
Noren NK, Pasquale EB (2004) Eph receptor-ephrin bidirectional signals that target Ras and Rho proteins. Cell Signal 16:655–666
Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME (2001) EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 105:233–244
Chen F, Liu Z, Peng W, Gao Z, Ouyang H, Yan T, Ding S, Cai Z, Zhao B, Mao L, Cao Z (2018) Activation of EphA4 induced by EphrinA1 exacerbates disruption of the blood–brain barrier following cerebral ischemia-reperfusion via the Rho/ROCK signaling pathway. Exp Ther Med 16:2651–2658
Fang WB, Brantley-Sieders DM, Hwang Y, Ham AJ, Chen J (2008) Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J Biol Chem 283:16017–16026
Bustelo XR (2002) Regulation of Vav proteins by intramolecular events. Front Biosci 7:d24–30
Wegmeyer H, Egea J, Rabe N, Gezelius H, Filosa A, Enjin A, Varoqueaux F, Deininger K, Schnutgen F, Brose N, Klein R, Kullander K, Betz A (2007) EphA4-dependent axon guidance is mediated by the RacGAP alpha2-chimaerin. Neuron 55:756–767
Takeuchi S, Yamaki N, Iwasato T, Negishi M, Katoh H (2009) Beta2-chimaerin binds to EphA receptors and regulates cell migration. FEBS Lett 583:1237–1242
DeGeer J, Kaplan A, Mattar P, Morabito M, Stochaj U, Kennedy TE, Debant A, Cayouette M, Fournier AE, Lamarche-Vane N (2015) Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J Cell Biol 210:817–832
Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA (2013) Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev Cell 24:573–585
Gulbranson DR, Davis EM, Demmitt BA, Ouyang Y, Ye Y, Yu H, Shen J (2017) RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc Natl Acad Sci USA 114:E8224–E8233
Nethe M, Hordijk PL (2010) The role of ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci 123:4011–4018
Kamynina E, Kauppinen K, Duan F, Muakkassa N, Manor D (2007) Regulation of proto-oncogenic dbl by chaperone-controlled, ubiquitin-mediated degradation. Mol Cell Biol 27:1809–1822
Oehler R, Pusch E, Zellner M, Dungel P, Hergovics N, Homoncik M, Eliasen MM, Brabec M, Roth E (2001) Cell type-specific variations in the induction of hsp70 in human leukocytes by feverlike whole body hyperthermia. Cell Stress Chaperones 6:306–315
Schotte P, Denecker G, Van Den Broeke A, Vandenabeele P, Cornelis GR, Beyaert R (2004) Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J Biol Chem 279:25134–25142
Zhuge Y, Xu J (2001) Rac1 mediates type I collagen-dependent MMP-2 activation. Role in cell invasion across collagen barrier. J Biol Chem 276:16248–16256
Tkach V, Bock E, Berezin V (2005) The role of RhoA in the regulation of cell morphology and motility. Cell Motil Cytoskelet 61:21–33
Shen DW, Pouliot LM, Gillet JP, Ma W, Johnson AC, Hall MD, Gottesman MM (2012) The transcription factor GCF2 is an upstream repressor of the small GTPAse RhoA, regulating membrane protein trafficking, sensitivity to doxorubicin, and resistance to cisplatin. Mol Pharm 9:1822–1833
Chi X, Wang S, Huang Y, Stamnes M, Chen JL (2013) Roles of rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci 14:7089–7108
Deng Z, Jia Y, Liu H, He M, Yang Y, Xiao W, Li Y (2019) RhoA/ROCK pathway: implication in osteoarthritis and therapeutic targets. Am J Transl Res 11:5324–5331
Zhu S, Liu H, Wu Y, Heng BC, Chen P, Liu H, Ouyang HW (2013) Wnt and Rho GTPase signaling in osteoarthritis development and intervention: implications for diagnosis and therapy. Arthritis Res Ther 15:217
Pritchard S, Votta BJ, Kumar S, Guilak F (2008) Interleukin-1 inhibits osmotically induced calcium signaling and volume regulation in articular chondrocytes. Osteoarthr Cartil 16:1466–1473
Appleton CT, Usmani SE, Mort JS, Beier F (2010) Rho/ROCK and MEK/ERK activation by transforming growth factor-alpha induces articular cartilage degradation. Lab Investig 90:20–30
Liang J, Feng J, Wu WK, Xiao J, Wu Z, Han D, Zhu Y, Qiu G (2011) Leptin-mediated cytoskeletal remodeling in chondrocytes occurs via the RhoA/ROCK pathway. J Orthop Res 29:369–374
Woods A, Wang G, Beier F (2005) RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem 280:11626–11634
Wang G, Woods A, Agoston H, Ulici V, Glogauer M, Beier F (2007) Genetic ablation of Rac1 in cartilage results in chondrodysplasia. Dev Biol 306:612–623
Woods A, Pala D, Kennedy L, McLean S, Rockel JS, Wang G, Leask A, Beier F (2009) Rac1 signaling regulates CTGF/CCN2 gene expression via TGFbeta/Smad signaling in chondrocytes. Osteoarthr Cartil 17:406–413
Nishida T, Kubota S, Kojima S, Kuboki T, Nakao K, Kushibiki T, Tabata Y, Takigawa M (2004) Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor). J Bone Miner Res 19:1308–1319
Wang G, Yan Q, Woods A, Aubrey LA, Feng Q, Beier F (2011) Inducible nitric oxide synthase-nitric oxide signaling mediates the mitogenic activity of Rac1 during endochondral bone growth. J Cell Sci 124:3405–3413
Acknowledgements
The work is supported in part by a Grant from the Department of Defense, the US Army Medical Research and Materiel Command (USAMRMC) under Grant No. W81XWH-14-1-0207 and a Merit Review Grant (IO1 BX002964) from the Biomedical Laboratory Research and Development Service, Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development. The views, opinions and/or findings contained in this report are those of the authors and should not be construed as an official position, policy or decision of the US Army, the Department of Veterans Affairs, or the US Government, unless so designated by other documentation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. In conducting research using animals, the investigators adhered to the Animal Welfare Act Regulations and other Federal statutes relating to animals and experiments involving animals and the principles set forth in the current version of the National Institutes of Health (NIH) Guidelines for Research Involving Recombinant DNA Molecules. Dr. Lau is also the recipient of a Research Career Scientist Award (1K6 BX002782) from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
Author information
Authors and Affiliations
Contributions
K-HWL, VS, and MS contributed to the conception and design of the study. Material preparation and data collection were performed by VS, AT, and CR. Data analyses were performed by K-HWL and VS. The first draft of the manuscript was written by K-HWL. All authors commented on previous versions of the manuscript, read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors (Virginia Stiffel, Alexander Thomas, Charles H. Rundle, Matilda H.-C. Sheng, and Kin-Hing William Lau) have no conflict of interest to declare.
Human and Animal Rights
All animal protocols were reviewed and approved by the Animal Care and Use Committee of the Pettis Memorial VA Medical Center and the Animal Care and Use Review Office (ACURO) of US Army Medical Research and Materiel Command of the Department of Defense. In conducting research using animals, the investigators adhered to the Animal Welfare Act Regulations and other Federal statutes relating to animals and experiments involving animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Stiffel, V.M., Thomas, A., Rundle, C.H. et al. The EphA4 Signaling is Anti-catabolic in Synoviocytes but Pro-anabolic in Articular Chondrocytes. Calcif Tissue Int 107, 576–592 (2020). https://doi.org/10.1007/s00223-020-00747-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00747-7