
Abstract
The main congenital conditions of hypophosphatemia expressed in adulthood include several forms of hereditary hypophosphatemic rickets and a congenital disorder of vitamin D metabolism characterized by osteomalacia and hypophosphatemia in adult patients. Hypophosphatemia in adults is defined as serum phosphate concentration < 0.80 mmol/L. The principal regulators of phosphate homeostasis, as is well known, are parathyroid hormone (PTH), activated vitamin D, and Fibroblast Growth Factor 23 (FGF23). Differential diagnosis of hypophosphatemia is based on the evaluation of mechanisms leading to this alteration, such as high PTH activity, inadequate phosphate absorption from the gut, or renal phosphate wasting, either due to primary tubular defects or high FGF23 levels. The most common inherited form associated to hypophosphatemia is X-linked hypophosphatemic rickets (XLH), caused by PHEX gene mutations with enhanced secretion of the FGF23. Until now, the management of hypophosphatemia in adulthood has been poorly investigated. It is widely debated whether adult patients benefit from the conventional treatments normally used for pediatric patients. The new treatment for XLH with burosumab, a recombinant human IgG1 monoclonal antibody that binds to FGF23, blocking its activity, may change the pharmacological management of adult subjects with hypophosphatemia associated to FGF23-dependent mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital conditions of hypophosphatemia are part of the disorders that cause defective bone mineralization, manifesting with rickets and osteomalacia in childhood and osteomalacia in adulthood [1,2,3,4]. The main congenital conditions of hypophosphatemia expressed in adulthood are subdivided into several different forms of hypophosphatemic rickets, including X-linked hypophosphatemic rickets (XLH), autosomal-dominant hypophosphatemic rickets (ADHR), autosomal recessive hypophosphatemic rickets (ARHR), hereditary hypophosphatemic rickets with hypercalciuria (HHRH), X-linked recessive hypophosphatemic rickets (XLRH), hypophosphatemic rickets with hyperparathyroidism (HRH), and a congenital disorder of vitamin D metabolism, such as hereditary 1,25-dihydroxyvitamin D-resistant rickets (HVDRR). These disorders are called forms of rickets as they tend to occur in childhood, but they can also occur in adulthood. Osteomalacia is caused by the disruption of mineral deposition of newly formed osteoid and, unlike rickets, a disorder of the growth plates affecting only pediatric subjects, osteomalacia can affect both children and adults with XLH [5].
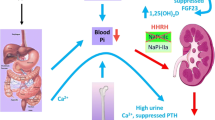
Hypophosphatemia is defined as serum phosphate concentration < 0.80 mmol/L in adults, and < 0.30 mmol/L is classified as severe hypophosphatemia [2]. The principal regulators of phosphate homeostasis are parathyroid hormone (PTH), 1,25-dihydroxyvitamin D [1,25(OH)2D3], and Fibroblast Growth Factor 23 (FGF23) [6]. Under physiological conditions, an increase of serum inorganic phosphate levels induces secretion of FGF23 and PTH, with an increase of urinary phosphate excretion in the proximal renal tubule, due to reduction of expression of the sodium-dependent phosphate co-transporters (NaPi-2a and NaPi-2c) [7].
In case of hypophosphatemia, indices of phosphate excretion, such as tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR), are required to discriminate between renal or non-renal mediated forms. Medical history, physical examination, and other biochemical exams of bone metabolism, help exclude several causes [2]. Further tests, such as estimation of FGF23 and genetic testing, are needed to discriminate between the potential causes.
Most congenital conditions of hypophosphatemia are diagnosed in early childhood, with the most common diagnosis being XLH, whereas some conditions, such as ADHR, have a variable phenotype that may not manifest until adulthood [8]. The management of congenital hypophosphatemia in adulthood is currently poorly investigated [4]. It is widely debatable whether adult patients with congenital hypophosphatemia benefit from the conventional treatments usually administered to pediatric patients (phosphate supplements and activated vitamin D) [4]. The new drug, burosumab for XLH, a recombinant human IgG1 monoclonal antibody that binds to FGF23, blocking its activity, may modify the pharmacological treatment of adults with hypophosphatemia associated to FGF23-dependent mechanisms [9, 10]. The effectiveness of burosumab has been shown in both children [10] and adults for clinical manifestations, such as stiffness, rickets severity, fracture healing, quality of life, and biochemical values, such as TmP/GFR and 1,25(OH)2D3 [11,12,13,14].
Hypophosphatemic Rickets
Hypophosphatemic rickets is a group of disorders with similar biochemical alterations, including excessive renal phosphate wasting and low serum phosphate [15]. These disorders can be divided into FGF23-associated and non-associated hypophosphatemic rickets, where the first are mostly due to an alteration of the regulation of the FGF23 synthesis as the primary factor causing the excessive renal phosphate wasting. The most common inherited form is FGF23-associated XLH, caused by PHEX gene mutations, with an incidence of 3.9 per 100,000 live births and a prevalence of 4.8 per 100,000 (children and adults) [16,17,18].
X-linked Hypophosphatemic Rickets
X-linked hypophosphatemic rickets (OMIM#307,800) is a rare congenital disease characterized by renal phosphate wasting and consequent hypophosphatemia [19]. It is an X-linked dominant disorder caused by mutations in the PHEX gene, located at Xp22.1, encoding a cell-surface-bound protein-cleavage enzyme (phosphate-regulating neutral endopeptidase PHEX). There are over 170 different mutations in the PHEX gene, and a clear genotype–phenotype correlation has not yet been described [20]. This disorder seems to be completely penetrant; however, its severity varies widely even among family members [21, 22], and other genetic and environmental factors may influence its clinical manifestation [3].
The pathogenesis of XLH is not yet fully understood; however, animal studies show that loss of Phex function results in enhanced FGF23 secretion, mainly by osteocytes [23]. PHEX is predominantly expressed in osteoblasts and codes for an enzyme that degrades local small integrin-binding ligands, N-linked glycoproteins (SIBLING proteins), particularly osteopontin (OPN) [24], besides suppressing the secretion of serum levels of the FGF23. Therefore, downregulation of PHEX in XLH patients increases skeletal OPN deposition, contributing to local inhibition of mineralization [24]. Moreover, high serum FGF23 concentrations increase renal excretion of phosphate by downregulating renal sodium–phosphate transporters (NaPi-2a and NaPi-2c) and reduce intestinal phosphate absorption by inhibiting active vitamin D synthesis [25].
Regarding clinical manifestations in adult patients, typical findings of XLH can include short stature, osteomalacia with bone pain, pseudofractures and fragility fractures, bone deformities, degenerative joint disease, early osteoarthritis, joint stiffness, decreased joint mobility due to enthesopathy (calcified ligaments and teno-osseous junctions), and other extra-osseus calcifications, including enthesophytes and spinal stenosis, muscle pain and weakness, and dental disease, such as periodontitis, inflammation of the gums, tooth decay, and dental abscesses [27,28,29,30]. In adult patients, pseudofractures are frequently reported [27, 28, 31]. Adult patients with XLH, as well as other hypophosphatemic rickets, can also manifest variable hearing loss [33], due to mild-to-severe sensorineural hearing loss. X-ray exams show generalized osteosclerosis and thickening of the petrous bone, with narrowed internal auditory meatus [33]. The symptoms described above can cause diminished quality of life, including psychosocial impact and disability, impairing ability to work [34].
Hypophosphatemia is the primary link between high FGF23 levels and the pathophysiology of XLH disease; however, it has been proposed that FGF23 also contributes to this disease with other molecular mechanisms [26, 35].
Regarding bone mineralization, it is known that hypophosphatemia is the primary mechanism by which elevated serum FGF23 impairs bone development, inhibiting apoptosis of hypertrophic chondrocytes and mineralization, and leading to rickets and osteomalacia [34]. However, studies conducted on Hyp osteocytes have also described abnormal mineralization in a phosphate-normal in vitro environment [36]. Thus, it has been suggested that there may be hypophosphatemia-independent autocrine/paracrine effects of FGF23 involving calcitriol and tissue non-specific alkaline phosphatase (TNAP) [34].
Recently, a study has shown a link between autocrine/paracrine roles of locally produced calcitriol and the FGF23-mediated regulation of chondrocyte differentiation and bone mineral deposition [37]. This study showed that, also in case of hypophosphatemia and low serum calcitriol, mice with high FGF23 levels did not have skeletal abnormalities when CYP24A1 concentrations were repressed [37]. Therefore, it was suggested that mineralization in the control animals was disrupted by FGF23-mediated activation of CYP24A1 degrading locally produced calcitriol [37]. Moreover, calcitriol appeared to directly affect expression of OPN (inhibitor of hydroxyapatite crystal formation), although no conclusive studies are currently available [38]. The SIBLING protein OPN contains an ASARM peptide motif cleavage, which releases phosphorylated ASARM (pASARM) peptides (potent inhibitors of mineralization) [34, 38, 39]. The pASARM peptides are directly cleaved by PHEX with an FGF23-independent mechanism, contributing to bone abnormalities in XLH [40]. However, this process is also exacerbated by FGF23-induced upregulation of OPN [39], probably in addition to other mechanisms that are still poorly understood [41]. Recent evidence suggests that accumulation of pyrophosphate (PPi), a known mineralization inhibitor, may be another factor with a role in the impairment of mineralization in XLH patients [34].
Regarding ectopic calcifications, XLH patients can present calcifications at joints and bone attachments of tendons (enthesopathies), as well as nephrocalcinosis.
Enthesopathies have also been reported in untreated patients. Although a clear pathogenesis of enthesopathy has not yet been described, it seems that FGF23 could have a specific role, given recent evidence of expression of FGFR and Klotho in sites where enthesopathy develops [18, 30], and enthesopathy does not appear to be influenced by conventional XLH treatment [3].
On the other hand, nephrocalcinosis has not been reported in untreated XLH patients; this is widely considered to be the result of conventional therapy associated with active vitamin D dosage [4, 33]. This has also suggested a potential role for FGF23 in enhancing renal calcium reabsorption in XLH disease through the transient receptor potential cation channel subfamily V member 5 (TRPV5) channel, promoting calcifications [42]. In FGF23-blocking clinical trials without the use of active vitamin D, nephrocalcinosis has not been described until now; however, there are not long-term data [43].
There are very few reports that describe cardiovascular complications, hypertension, and left ventricular hypertrophy in patients affected by XLH, and they appear to be side effects of conventional treatment and/or associated to increased renal sodium reabsorption due to FGF23 [34]. Significant associations between high levels of FGF23 and impaired flow-mediated dilation, arterial stiffness, atherosclerosis, and left ventricular hypertrophy have been described in chronic kidney disease (CKD) patients [34]. The patients affected by XLH, unlike CKD patients, have lower FGF23 levels, low phosphate levels, and concomitant standard treatment with vitamin D, which may prevent FGF23-driven left ventricular hypertrophy [34, 44].
XLH patients also usually have muscle weakness associated with FGF23-induced hypophosphatemia, with lower muscle density and lower peak muscle force and power compared to age- and gender-matched controls [34]. The expression of PHEX in myocytes suggests a potential direct role for FGF23 in muscular weakness in these patients. Moreover, it has been described that FGF23 induces senescence in mesenchymal stem cells derived from skeletal muscle [34, 45].
Craniosynostosis has classically been associated with hypophosphatemic rickets, but the relationship is still not clear [34]. Effects of blocking FGF23 on the development of craniosynostosis during clinical trials have not yet been described [11].
Severe dental manifestations, including dental abscesses, periodontopathies and malocclusion, have been described in most untreated XLH patients; however, the molecular mechanisms that cause these complications are poorly understood [34, 46].
Reports of hearing loss frequency in XLH patients are quite variable. A number of causes have been identified, including conductive hearing loss, sensorineural hearing loss, and cochlear dysfunction. However, a more standardized approach for assessing hearing loss is clearly necessary for future investigations [34].
Typical biochemical alterations associated with XLH include hypophosphatemia, renal phosphate wasting with reduced TmP/GFR ratio (tubular maximal reabsorption of phosphate adjusted for glomerular filtration rate), and high serum alkaline phosphatase (ALP) activity and FGF23 levels [33, 47]. Serum 1,25(OH)2D3 levels tend to be inappropriately low or normal in the setting of hypophosphatemia, while serum 25-vitamin D [25-(OH)2D3] is usually normal [48]. Serum calcium concentrations are generally in the lower normal range, and urinary calcium excretion is tendentially low, due to impaired 1,25(OH)2D3 synthesis [51]. Secondary hyperparathyroidism is common, both without phosphate treatment and as a potential consequence of phosphate treatment [3]. In XLH, high PTH concentrations in the absence of phosphate treatment could be due to relative deficiency in 1,25(OH)2D3, attributable to FGF23 excess [33]. The direct effect of FGF23 on PTH is not yet clarified. Experimental studies have shown an inhibitory effect of FGF23 on both PTH production and secretion. In particular, preclinical studies have shown that recombinant FGF23 increased parathyroid Klotho levels and activated MAPK signaling, suppressing PTH expression over short time courses [49]. On the other hand, a study conducted on wistar rats treated with intravenous recombinant FGF23 and inhibition of the FGF receptor showed that FGF23 rapidly inhibited PTH secretion in normocalcemia, but in case of acute hypocalcemia, when increased PTH secretion is needed to restore the calcium homeostasis, this effect was not described [50].
The plasma levels of intact FGF23 are usually high in this disease; however, XLH diagnosis cannot be excluded in case of normal levels of FGF23, because normal concentrations are also inappropriate in hypophosphatemia [52]. Moreover, FGF23 levels are most informative in untreated patients, because they are influenced by phosphate intake and vitamin D therapy [53].
In adult patients, the diagnosis of XLH should be considered in the presence or history of clinical and/or radiological signs of osteomalacia, pseudofractures, and/or lower limb deformities, early osteoarthritis and enthesopathies associated with serum concentrations of phosphate below the age-related reference range, and renal phosphate wasting [53]. The medical history of these patients may reveal rickets, dental problems, and delayed growth in childhood, and physical examination may show lower limb varus or valgus deformities, small stature, an unusual gait, and variable degrees of motor disability. The mutational analysis of the PHEX gene is recommended [53], although sporadic cases (about 20% to 30% of cases) are common, and in some cases a mutation cannot be detected [3]. In cases where genetic analysis is not available, elevated plasma intact FGF23 concentrations and/or a positive family history for XLH may support the diagnosis [53]. Regardless of the age of the patient, an early diagnosis followed by optimal treatment is crucial to control the clinical manifestations, prevent complications, and improve quality of life [53].
Patients should be controlled at regular intervals by expert multidisciplinary teams, with knowledge of metabolic bone diseases, as suggested by recent guidelines [53]. The clinical XLH course varies widely from patient to patient; consequently, treatment and monitoring should be assessed on a case-by-case basis according to their medical history. The recent XLH guidelines recommend a clinical follow-up of adult patients to evaluate: height, weight, BMI, history of headaches, oral manifestations, musculoskeletal pain, pseudofractures, fatigue, and level of physical function [53]. In adults with recent oral manifestations, a dental orthopantomogram is suggested and should be repeated according to clinical needs [53]. In some studies, hypertension, left ventricular hypertrophy and/or electrocardiogram alterations have been described in patients with XLH [54, 55]; therefore, a yearly blood pressure measurement is suggested [54]. Regarding biochemical follow-up, serum ALP level is a reliable biomarker of osteomalacia in adult patients; however, given that ~ 50% of circulating ALP originates from hepatocytes, bone-specific ALP is preferred [33]. In undertreated patients, ALP concentrations are high and urinary calcium excretion is usually low, while when the rickets are healed, ALP levels tend to normalize, and urinary calcium excretion tends to increase [47]. PTH levels should be monitored regularly, given that oral phosphate stimulates the secretion of PTH, causing secondary hyperparathyroidism [56]. Regular measurements of serum and urinary calcium levels are required to evaluate the safety of active vitamin D [53].
Since low phosphate concentrations and inappropriately low levels of calcitriol contribute to many XLH-associated symptoms, conventional treatment includes supplementation with oral phosphate and calcitriol or alfacalcidol. An early standard treatment can correct lower limb deformities, improve oral health, and promote growth [33], but insufficiently corrects the other clinical manifestations as discussed below, and can further increase serum FGF23 concentrations [25, 34]. Moreover, conventional treatment has been associated with adverse effects, such as secondary hyperparathyroidism, nephrocalcinosis, nephrolithiasis, and cardiovascular abnormalities, as is discussed in the paragraph dedicated to treatment [34, 53].
Autosomal-dominant Hypophosphatemic Rickets
Autosomal-dominant hypophosphatemic rickets (OMIM#193,100) is a rare disease with clinical and biochemical findings similar to XLH [57, 58]. ADHR is caused by a missense mutation in the FGF23 gene itself that stabilizes the protein product, leading to increased FGF23 activity, which in turns leads to phosphate wasting at the level of the proximal tubules [19]. The age at presentation of ADHR can vary between childhood and adulthood, due to incomplete penetrance. The clinical expression varies widely and, in some cases, despite carrying the gene mutation, hypophosphatemia never develops [57, 58], and the disease does not manifest (incomplete penetrance), while some affected patients spontaneously normalize [57, 58].
Typical biochemical alterations in patients with active disease include hypophosphatemia, phosphaturia, and inappropriately normal or low 1,25(OH)2D3 concentrations similar to XLH [58]. In adulthood, clinical expression can include osteomalacia, bone pain, weakness, and fractures/pseudofractures [57, 58]. It has been reported that most patients who develop the disease in adulthood are women and, in some cases, the onset of clinically evident disease is correlated with a physiological stress, such as pregnancy [57, 58]. It is not well known why patients with ADHR only sometimes generate inappropriate FGF23 and hypophosphatemia. Some investigations in ADHR patients and in a mouse model of ADHR have suggested that low iron status results in increased FGF23 mRNA [58,59,60]. It has been suggested that the low iron stores, due to pregnancy or any other stressor, may lead to clinical manifestation or exacerbation later in life. The mutant intact FGF23 is resistant to cleavage, and this leads to exacerbation of disease at times of low iron stores when there is increased expression of FGF23 [61]. Iron deficiency drives an increase in FGF23 gene expression, causing hypophosphatemia, while normalization of iron in ADHR patients has been associated with the normalization of the biochemical and skeletal phenotype [60].
Autosomal Recessive Hypophosphatemic Rickets
Autosomal recessive hypophosphatemic rickets is a rare hereditary renal phosphate wasting disorder, characterized by hypophosphatemia, rickets and/or osteomalacia and slow growth, clinically similar to XLH and ADHR with the exception of a relatively higher bone mineral density (BMD), [19, 62]. Clinical manifestations include dental defects, sclerotic bone lesions, and enthesopathies [19].
This disease has been subdivided into ARHR type 1 linked to inactivating mutations in the DMP1 gene (OMIM#241,520), and ARHR type 2 in the ENPP1 gene (OMIM#613,312) [63]. In case of DMP1 mutations, the phenotype is similar to XLH, whereas ENPP1 mutations are usually associated with generalized arterial calcification in infancy [64]. The effect of loss of function of DMP1 is not yet clear; however, it is known that it causes an increased transcription of FGF23 by osteocytes, and is involved in the regulation of transcription in undifferentiated osteoblasts [65]. The gene product of ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1) is a cell-surface enzyme responsible for generating inorganic pyrophosphate that is able to inhibit bone [63].
Hereditary hypophosphatemic rickets with hypercalciuria
Hereditary hypophosphatemic rickets with hypercalciuria (OMIM#241,530) is an autosomal recessive hereditary disorder resulting from specific transporter mutations due to a mutation in the SLC34A3 gene, encoding the NaPi-2c renal phosphate cotransporter [66,67,68]. It is characterized by hypophosphatemia, renal phosphate wasting, hypercalciuria with circulating PTH usually low to low-normal, associated with childhood rickets and, in adulthood, with osteopenia/osteomalacia [67, 68]. The reduced PTH-dependent calcium reabsorption in the distal renal tubules leads to the development of kidney stones and/or nephrocalcinosis in approximately half of the subjects with HHRH [69]. Heterozygous NaPi-2c mutations are often associated with isolated hypercalciuria, and increased risk of kidney stones or nephrocalcinosis threefold in affected subjects compared with the general population [69]. In this disorder, the FGF23 level is not increased, the hypophosphatemia causes an increase of 1,25(OH)2D3 and, consequently, an increase of intestinal calcium absorption and hypercalciuria [68].
Dent’s Disease, X-linked Recessive Hypophosphatemic Rickets
Dent’s disease (X-linked recessive hypophosphatemic rickets; OMIM#300,554) is an X-linked disorder due to a mutation in the voltage-gated chloride channel gene (CLCN5) [70, 71]. This disease is characterized by renal proximal tubular dysfunction leading to hypophosphatemia, with hypercalciuria, and low molecular weight proteinuria. These patients develop rickets or osteomalacia and nephrolithiasis or nephrocalcinosis with progressive renal failure in early adulthood [70, 71]. Therefore, in contrast to patients affected by XLH, these subjects manifest hypercalciuria, high serum 1,25(OH)2D3 levels, and proteinuria of up to 3 g/day [70, 71].
Hypophosphatemic Rickets with Hyperparathyroidism
A syndrome characterized by both hypophosphatemic rickets and hyperparathyroidism due to parathyroid hyperplasia has been described (Hypophosphatemic rickets with hyperparathyroidism; OMIM#612,089) [72].
Brownstein CA et al. described a 23-year-old woman with hypophosphatemic rickets and hyperparathyroidism who presented at 13 months of age with rachitic skeletal alterations [72]. Biochemical evaluation reported hypophosphatemia and elevated serum PTH levels with inappropriate renal phosphate wasting. The patient was treated with calcitriol and oral phosphate salt supplementation with improvement of bone pain, and she began to walk shortly before her second birthday. At age 7 she developed hypercalcemia, persistent hypophosphatemia and hyperparathyroidism, and underwent surgical removal of 3.5 hyperplastic parathyroid glands (histology showed benign multigland hyperplasia). In adulthood, at age 19, she again reported hypercalcemia and underwent 75% removal of the then-enlarged parathyroid remnant (benign hyperplasia). At age 23, Arnold-Chiari I malformation was also found [72].
HRH has been associated to a de novo translocation with a breakpoint adjacent to alpha-Klotho, encoding a beta-glucuronidase, implicated in aging and regulation of FGF signaling [72]. High plasma alpha-Klotho levels and beta-glucuronidase activity were found in the affected patient; unexpectedly, high levels of the circulating FGF23 level were also described. These findings suggested that a high alpha-Klotho concentration mimics a normal response to hyperphosphatemia and implicates a role of alpha-Klotho in the selective regulation of phosphate levels and in the regulation of parathyroid mass and function [72].
Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets
Hereditary 1,25-dihydroxyvitamin D-resistant rickets (OMIM #277,440), also known as vitamin D-resistant rickets type 2A, is a rare monogenic disease caused by mutation in the VDR gene [73, 74]. Patients with HVDRR often have consanguinity in the family [75]. Mutation in the VDR gene leads to partial or complete resistance to 1,25-dihydroxyvitamin D-resistant leading to hypocalcemia, secondary hyperparathyroidism, consequent hypophosphatemia, and elevated ALP [3]. Moreover, there are significant high serum 1,25(OH)2D3 levels, unlike 1α-hydroxylase deficiency, also known as vitamin D-dependent rickets type 1A [76]. Multiple mutations in the VDR gene have been reported to cause HVDRR [73]. In adulthood, many patients have near normal PTH concentrations and normal bone mineral density (BMD), although with persistent elevated serum 1,25(OH)2D3 levels, suggesting residual target organ resistance. Most HVDRR patients during and after puberty and into adulthood are able to maintain normal serum calcium concentrations with modest oral calcium supplements, or without them altogether [74].
Differential diagnosis
The differential diagnoses of congenital conditions of hypophosphatemia is based on the evaluation of mechanisms leading to hypophosphatemia: high PTH activity, inadequate phosphate absorption from the gut or renal phosphate wasting (either primary tubular defects or high levels of circulating FGF23) [2, 53, 77, 78].
The evaluation of clinical manifestations, the onset of symptoms, radiographic and biochemical examinations, and extended molecular genetic analysis can be used to establish the diagnosis in unclear cases of hypophosphatemia [2]. Moreover, an accurate history and physical examination are necessary to exclude several causes of genetic or acquired of hypophosphatemia [2]. The indices of tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) are fundamental to determine if hypophosphatemia is caused by renal loss [2]. The measurement of serum PTH, 25(OH)2D3, and sometimes serum 1,25(OH)2D3, may be necessary to investigate the other genetic or acquired forms of hypophosphatemia. In the case of hypophosphatemia due to primary hyperparathyroidism, the differential diagnosis is based on the analysis of the typical biochemical picture of primary hyperparathyroidism and, in particular, hypophosphatemia is usually less severe than renal phosphate wasting, because the levels of calcitriol are higher. On the other hand, low serum concentration of 1,25(OH)2D3 with adequate levels of 25(OH)2D3 should raise suspicion of a FGF23-mediated hypophosphatemia [2].
In patients with XLH, plasma FGF23 levels are high, but quite variable [79], and an increased concentration of FGF23 is not specific for XLH, even in the setting of hypophosphatemia, since ADHR, ARHR, tumor-associated osteomalacia (TIO) and fibrous dysplasia (FD) all cause FGF23-mediated hypophosphatemia [79, 80]. On the other hand, low concentrations of FGF23 associated to hypophosphatemia could be associated to different diseases, such as Fanconi syndrome, dietary phosphate deficiency, or malabsorption [79, 80]. Urine amino acids, protein and glucose excretion indicate a proximal tubular disorder, such as Fanconi syndrome, whereas low urinary phosphate levels could be associated with dietary phosphate deficiency or impaired bioavailability or, in case of hypercalciuria before starting treatment, HHRH should be considered [2, 8, 53]. Nutritional rickets and XLH might sometimes coexist, and diagnosis of XLH should be considered if, in case of vitamin D or calcium, an adequate supplementation does not improve the serum levels of phosphate [53].
Several FGF23 immunoassays are available to date, although they are often reserved only for research [3]. They can detect intact FGF23 (iFGF-23) or both iFGF-23 and C-terminal FGF23 (cFGF-23) portions, and most assays include a double antibody sandwich ELISA with colorimetric reading. It is not yet clear if the cFGF-23 assay provides comparable sensitivity for iFGF-23 in subjects with different stages of renal function [3]. Recently, a new automated assay for iFGF-23 on the DiaSorin Liaison platform has been approved for clinical use [81]. This assay is reserved for the measurement of iFGF-23 in EDTA plasma and uses three monoclonal antibodies (one coated on microparticles and directed against the N-terminal portion of the iFGF-23; another, labeled with fluorescein, directed against the C-terminal fragment; and the third, bound with isoluminol, directed against fluorescein). It has shown excellent analytical characteristics, greatly improving the evaluation of this analyte [81].
Finally, the pathological and pharmacological medical history allows exclusion of other acquired causes of hypophosphatemia in the differential diagnosis [2, 3].
Table 1 summarizes the biochemical findings in selected genetic hypophosphatemic disorders.
Treatments of Congenital Conditions of Hypophosphatemia
Conventional treatment of hypophosphatemia due to phosphate wasting genetic disorders is essentially based on treatment of the XLH with phosphate supplements and calcitriol or alfacalcidol, given the similarities in pathophysiology between the disorders [3, 53].
Treatment is usually initiated at the time of diagnosis of XLH and continued at least until growth completion [82]. The doses vary according to age and severity of phenotype [53]. In pediatric patients, the main goal of treatment is to prevent rickets before they develop bone changes and improve growth. Therefore, therapy should be started early, otherwise if started late in adolescence or in adulthood the treatment is not equally effective [53]. Healing of rickets is usually associated to the normalization of ALP levels and radiological signs. In childhood, the conventional therapy lowers the ALP level to the upper limit of normal within about 1 year and increases height by about 1 standard deviation [82]. Another goal of treatment in pediatric patients includes dental health, improving dental mineralization, and decreasing the number of dental abscesses [53, 82]. Phosphate supplements should be given frequently (for example, 4–6 times per day), since serum phosphate levels increase rapidly but return to baseline levels within 1.5 h. Oral phosphate supplements should always be assumed together with active vitamin D, because it is known that phosphate alone can cause secondary hyperparathyroidism and thereby renal phosphate wasting [53]. Large doses of active vitamin D promote both growth and bone healing but may cause hypercalciuria and nephrocalcinosis and titration to optimal doses and compliance represent usually important challenges [53]. No long-term clinical investigations have been conducted so far assessing the impact of the standard treatment on the incidence of complications such as enthesopathies, hearing impairments, fatigue, Chiari syndrome, and craniosynostosis.
In adulthood, the conventional treatment has long been debated and currently it is recommended only in symptomatic patients with XLH, including pseudofractures, musculoskeletal pain, dental issues, planned orthopedic or dental surgery, or osteomalacia with high serum levels of bone-specific ALP [53]. Treatment if needed is also suggested in physiological conditions, such as pregnancy and breastfeeding [53]. Dosage reduction of active vitamin D is recommended in case of long-term immobilization, to prevent hypercalciuria and hypercalcemia due to increased bone resorption, and to stop phosphate supplements in patients with markedly increased parathyroid hormone levels [53]. In case of secondary hyperparathyroidism, it is suggested that active vitamin D might be given without phosphate supplements [53]. Treatment is also recommended before and 3–6 month after surgical, orthopedic or dental, interventions to promote bone mineralization [33, 83].
Standard treatment can partially improve bone pain and osteomalacia in adult patients [53]. Spontaneous insufficiency fractures are more likely to heal faster with therapy [33, 83]. On the other hand, this treatment does not prevent or improve hearing loss or enthesopathies, and it does not modify osteoarthrosis-related pain [53, 83]. Regarding dental manifestations, treatment is beneficial for dentin mineralization and dental health, reducing the frequency of dental abscesses, especially if started in early childhood, but this has not been thoroughly evaluated in periodontal defects [83]. Hearing difficulties/loss and metabolic complications can manifest during adulthood, but it is not clear if treatment can prevent occurrence [32, 83]. Moreover, conventional treatment tends to increase FGF23 levels in humans with XLH. The clinical consequences of this FGF23 increase are uncertain, but may have negative effects on health, in particular on metabolism and cardiac functions [83].
The doses of calcitriol or alfacalcidol usually prescribed in adult patients range from 0.50 to 0.75 μg daily for calcitriol and 0.75–1.5 μg daily for alfacalcidol, and phosphate tablets range from 750–1,600 mg daily (based on elemental phosphorus) in 2–4 divided doses, as recommended by the latest guidelines [53]. The dose of phosphorus should be increased gradually in order to avoid gastrointestinal adverse effects [53]. There is no evidence to recommend monitoring calcitriol concentrations for adjustment of the dose of active vitamin D derivatives, and 25(OH)D deficiency should be adequately corrected [83].
In adulthood the goal of treatment is to relieve XLH symptoms, and not to normalize serum phosphate concentrations [83]. It is important to remember that, in adulthood, serum ALP of the bone-specific isoenzyme is not a major marker of treatment efficacy but altered concentrations could indicate extensive osteomalacia [83]. In long-term treatment, inappropriate or excessive treatment may cause hyperparathyroidism and hypercalciuria with consequent nephrocalcinosis and renal insufficiency. Therefore, it is necessary to carefully monitor plasma calcium, PTH, creatinine and 24-h urinary calcium excretion [33]
The challenges often faced by using conventional treatment are variable compliance and poor tolerability, including gastrointestinal symptoms, such as diarrhea and abdominal cramps [53]. The balance between optimization of skeletal symptomatology and minimization of side effects is challenging, and usually requires frequent medical visits. In absence of specific symptoms, little evidence suggests that starting or continuing treatment can improve outcomes, in addition to having potential adverse effects [53].
In addition to conventional treatment, thanks to the identification of the role of FGF23 excess in the pathophysiology of XLH, much effort has been directed to develop new pharmacological treatment targeting the FGF23 pathway. Recently, burosumab (formerly KRN23), a fully human monoclonal IgG1 antibody-neutralizing FGF23, was approved by health authorities for the treatment of patients with XLH in the European Union and the USA. In particular, it is now licensed for children older than 1 year of age in Europe and for both children and adults in the USA [53, 83]. Burosumab binds the amino-terminal domain of FGF23, preventing FGF-23 from binding to its receptor [83, 84].
In childhood, two open-label uncontrolled trials investigated the use of burosumab in a total of 65 children affected by severe XLH (aged 1–12 years), showing that burosumab treatment (12–16 months) resulted in: a statistically significant increase in TmP/GFR and raised serum phosphate concentrations into the lower end of the age-related normal range, with increased 1,25(OH)2D3 levels; a significant reduction in the severity of rickets (as measured by the Rickets Severity Score and the Radiographic Global Impression of Change); a significant improvement in physical ability (as measured by walking distance in total distance walked in 6 min); and a significant reduction in patient-reported pain and functional disability (evaluated with the use of the Pediatric Orthopedic Society of North America Outcomes Data Collection Instrument) [85,86,87,88]. Radiographic improvements were associated to improvements in ALP and small improvements in height Z‐score (+ 0.15) [86]. These trials lacked an active comparator or a placebo group but did suggest benefit from switching from conventional treatment to burosumab in these growing children [85, 86]. The most common adverse reactions described with burosumab were injection-site reactions, headache and pain in the extremities [85, 86]. The EMA and FDA authorizations approved a starting dose of 0.4 mg/kg body weight and 0.8 mg/kg body weight, respectively, given every 2 weeks [53, 87, 88]. Burosumab treatment, if available, is recommended in children with XLH ≥ 1 year and in adolescents with growing skeletons in case of radiographic evidence of overt bone disease and disease refractory to conventional treatment; or clinical complications associated to conventional therapy; or poor compliance to conventional therapy [53]. Recently, the first randomized, active-controlled, phase 3 trial of burosumab in children (aged 1–12 years) with XLH at 16 clinical sites has been published [89]. Sixty-one patients were enrolled, of these, 32 were randomly assigned to continue receiving conventional therapy and 29 to receive burosumab. Both interventions lasted 64 weeks. Patients treated with burosumab showed significantly greater improvement in Radiographic Global Impression of Change global score than did patients in the conventional therapy group (p < 0·0001) [89]. Three serious adverse events occurred in each group, but all resulted unrelated to treatment and resolved. Significantly greater clinical improvements were described in rickets severity, growth, and biochemistries among pediatric patients with XLH treated with burosumab compared with those continuing conventional treatment at 64 weeks. Therefore, it could have the potential to prevent long-term complications associated with XLH, however, in childhood it remains to be evaluated what the long‐term effect, if any, would be from prolonged burosumab treatment on achieved height at the end of growth, and craniosynostosis, as well as, the effect of burosumab on dental health [89]. At last, there is not any data to clarify what approach might be necessary in adolescents transitioning to adulthood.
The use of burosumab in adult patients with skeletal pain associated with XLH and/or osteomalacia has been investigated through one open-label, uncontrolled trial and one randomized, double blind, placebo-controlled study (24 weeks), including a total of 148 patients [14, 87, 88]. All clinical trials have included moderately to severely affected XLH patients, therefore there are no results regarding patients with mild clinical forms [14, 87, 88].
In adulthood, short-term treatment with burosumab has shown the following effects: significantly increased TmP/GFR with raised levels of serum phosphate in the lower normal range and increased 1,25(OH)2D3 levels, as reported in pediatric trials; accelerated healing of active fractures and pseudofractures, and healed osteomalacia; and significantly reduced stiffness, measured with the Western Ontario and the McMaster Universities Osteoarthritis Index (WOMAC) stiffness subscale [53]. Improvement of osteomalacia with burosumab may be reflected in increases in bone formation and resorption markers, that was reported in the clinical trials and a normalization of bone remodeling would favor mechanism fracture healing [14]. Stiffness may have multiple causes in adults with XLH, and reduction of stiffness in patients treated with burosumab may partially be due to improvements in muscle function with this treatment [14]. The reduction of pain and improvement of physical function was reported but without statistical significance relative to placebo [14]. Burosumab with its effects above described improve pain in adults, but do not treat at least in the short term, every cause of pain or physical impairment, osteoarthritis or enthesopathy-related pain [14].
Recently, results derived by a 24‐week burosumab treatment continuation period after 24 weeks double‐blind placebo‐controlled trial in 134 adults [14] have been published [90]. After 24 weeks, all patients received open-label burosumab until week 48. Burosumab treatment from weeks 24–48 showed that serum phosphorus levels remained normal in most of patients (about 80%), and by week 48, more than half of baseline fractures/pseudofractures healed fully with burosumab, compared with 35.2% with burosumab after placebo [90]. Both in patients who already took burosumab and those who switched from placebo to burosumab, the treatment resulted in clinically significant and sustained improvement from baseline to week 48 regarding stiffness, pain, physical function, and total distance walked in 6 min. No serious adverse events were reported and rates of adverse events were similar for burosumab and placebo. At last, nephrocalcinosis scores did not change from baseline by more than one grade at either week 24 or 48 [90].
Burosumab is generally a well-tolerated drug, with a good safety profile. The described adverse events included: injection-site reactions, headache and pain in the extremities, like those observed in the pediatric trials [53]. Forty-eight weeks of burosumab treatment was not associated with hyperparathyroidism or renal or cardiac ectopic mineralization [14, 90]. There was no evidence of hyperphosphatemia or ectopic mineralization in the trials and no immunogenicity has been associated with burosumab treatment so far [53].
In the USA, the FDA approved a dose of burosumab of 1 mg/kg body weight, with a maximum dose of 90 mg, given subcutaneously every 4 weeks in adulthood [87]. If available, it is recommend to consider burosumab treatment in adult patients affected by XLH with the following clinical features: persistent bone and/or joint pain associated with XLH and/or osteomalacia limiting daily activities; presence of pseudofractures or fractures due to osteomalacia; and insufficient response or refractory, and/or complications related to conventional therapy [53].
Burosumab should be used only at referral centers, and a monitoring of fasting serum phosphate levels is suggested 7–11 days after the last injection of burosumab to avoid inadvertently causing hyperphosphatemia [53]. After reaching a steady state (presumably about 3 months), measuring of serum phosphate levels is suggested, preferably during the last week before a subsequent injection, to analyze an eventual underdosing [53]. In case of fasting serum phosphate levels above the upper limit of normal, burosumab should be discontinued, and subsequently can be restarted at approximately half of the previous dose, once serum phosphate concentrations are below the normal range [53].
To date, no long-term data are available on burosumab given its recent introduction, especially regarding the incidence of hyperparathyroidism, renal function impairment, and rheumatic complications [91]. Limitations regarding burosumab treatment include the unfeasibility of adjusting the dosage according to the PTH and/or the calcium urinary excretion levels [91]. Moreover, burosumab treatment is a very expensive therapy, and therefore should be weighed against the cost of managing the complications due to XLH [91]. Various aspects of burosumab treatment are yet to be explored, including whether specific groups of patients with XLH respond better than others and whether such response can be predicted [82]. There is no data on the safety of burosumab during pregnancy or lactation. In the future, the long-term effect of burosumab on complications related to XLH in adult patients should be analyzed. New clinical studies will be necessary in order to study disease burden and treatment options in adults, the effect of burosumab on prevention of dental complications, craniosynostosis and muscle weakness, and long-term safety [82]. Further, since symptoms related to XLH are not only due to alterations in phosphate and FGF23, other drug targets will need to be investigated. At last, the utility of burosumab for FGF23‐related skeletal diseases other than XLH is yet to be explored but may prove beneficial.
Like XLH, both ADHR and ARHR can be treated with phosphate supplementation combined with calcitriol or other active vitamin D analogs [33]. However, in case of ADHR, iron supplementation to normalize iron stores may be a more appropriate therapy, considering studies indicating that iron deficiency appears to be a key pathologic mechanism in ADHR, but no studies have tested the hypothesis of treatment with iron yet [60, 92]. Preliminary data derived from a pilot clinical trial of oral iron therapy for patients with ADHR showed normalization of FGF23 and phosphate levels [93]. On the other hand, certain forms of intravenous iron can precipitate acute increases in intact FGF23 [58]. Therefore, intravenous iron (especially iron polymaltose and iron carboxymaltose) should be avoided in treating these iron‐deficient ADHR subjects, as these could cause profound hypophosphatemia [58].
Regarding patients affected by HHRH, the standard therapy requires long-term medical therapy with phosphate supplements [94, 95], and not supplementation with vitamin D, as for nutritional rickets [96], or if confused with XLH. The addition of calcitriol to phosphate therapy may lead to hypercalcemia, hypercalciuria, nephrocalcinosis, and possibly renal insufficiency [97]. Oral phosphate supplementation quickly improves rachitic bone disease in HHRH. However, the long-term safety of this treatment is completely unknown with respect to renal complications, such as renal calcifications, and whether hyperparathyroidism or enthesopathies can develop, as in XLH [98]. Moreover, it is not clear whether the renal phosphate-leak persists life-long or therapy may be stopped, as in ADHR [99], and whether HHRH tends to accelerate bone loss in adulthood, as reported in some subjects affected by NaPi-2a mutations [100, 101].
Regarding XLRH, current treatments/interventions include low sodium diet and thiazide diuretics to reduce hypercalciuria, and the use of supplementations for hypophosphatemia, hypokalemia, and acidosis. Treatment with vitamin D should be used carefully to avoid exacerbation of hypercalciuria [102].
Finally, the treatment of HVDRR in adulthood often consists of modest oral calcium supplements, or no supplementation at all, since they can maintain normal serum calcium levels and report near normal PTH [75]. In cases of partial resistance to 1,25(OH)2D3, the administration of pharmacological doses of vitamin D showed clinical and radiologic improvement [75].
Conclusion
The main congenital conditions of hypophosphatemia expressed in adults include several forms of hereditary hypophosphatemic rickets/osteomalacia caused by different pathogenetic mechanisms. These disorders are rare and chronic diseases, and their clinical manifestations and complications can alter the quality of life until adulthood. They are multisystem diseases and, therefore, require adequate multidisciplinary care, involving endocrinologists, orthopedists, physiotherapists, and dentists. Differential diagnosis of hypophosphatemia is of fundamental importance among the various potential causes. Unfortunately, there are few investigations reported in literature on the long-term management of hypophosphatemia in adulthood. Most of the published studies concern the treatment of XLH with standard treatment. Studies have recently been conducted on burosumab, an innovative therapy for XLH, and in the future, data on this new treatment will emerge. In addition to burosumab, other drugs that aim at the inhibition of the effects of high FGF23 concentrations are under development that might enter clinical use in the future.
References
Marcucci G, Cianferotti L, Beck-Peccoz P, Capezzone M, Cetani F, Colao A, Davì MV, degli Uberti E, Del Prato S, Elisei R, Faggiano A, Ferone D, Foresta C, Fugazzola L, Ghigo E, Giacchetti G, Giorgino F, Lenzi A, Malandrino P, Mannelli M, Marcocci C, Masi L, Pacini F, Opocher G, Radicioni A, Tonacchera M, Vigneri R, Zatell MC, Brandi ML (2015) Rare diseases in clinical endocrinology: a taxonomic classification system. J Endocrinol Invest 38:193–259
Manghat P, Sodi R, Swaminathan R (2014) Phosphate homeostasis and disorders. Ann Clin Biochem 51:631–656
Marcucci G, Masi L, Ferrarì S, Haffner D, Javaid MK, Kamenický P, Reginster JY, Rizzoli R, Brandi ML (2018) Phosphate wasting disorders in adults. Osteoporos Int 29:2369–2387
McKenna MJ, Martin-Grace J, Crowley R, Twomey PJ, Kilbane MT (2019) Congenital hypophosphataemia in adults: determinants of bone turnover markers and amelioration of renal phosphate wasting following total parathyroidectomy. J Bone Miner Metab 37:685–693
Penido M, Alon US (2012) Phosphate homeostasis and its role in bone health. Pediatr Nephrol 27:2039–2048
Quarles LD (2012) Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol 8:276–286
Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M (2009) FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Ren Physiol 297:F282–F291
Imel EA, Econs MJ (2012) Approach to the hypophosphatemic patient. J Clin Endocrinol Metab 97:696–706
Zhang X, Imel EA, Ruppe MD et al (2016) Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending-dose trial treating adults with X-linked hypophosphatemia. J Clin Pharmacol 56:176–185
Collins M (2018) Burosumab: at long last, an effective treatmen for FGF23-associated hypophosphatemia. J Bone Miner Res 33:1381–1382
Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M (2014) Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Investig 124:1587–1597
Imeal EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA, Insogna K, Peacock M, Carpenter TO (2015) Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab 100:2565–2573
Ruppe MD, Zhang X, Imel EA, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA, Insogna K, Peacock M, Carpenter TO (2016) Effect of four monthly doses of a human monoclonal anti-FGF23 antibody (KRN23) on quality of life in X-linked hypophosphatemia. Bone Rep 5:158–162
Insogna KL, Briot K, Imel EA, Kamenický P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, Imanishi Y, Ito N, Lachmann RH, Tanaka H, Perwad F, Zhang L, Chen CY, Theodore-Oklota C, Mealiffe M, San Martin J, Carpenter TO (2018) A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res 33:1383–1393
Bastepe M, Juppner H (2008) Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord 9:171–180
Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK (2009) Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol 160:491–497
Endo I, Fukumoto S, Ozono K, Namba N, Inoue D, Okazaki R, Yamauchi M, Sugimoto T, Minagawa M, Michigami T, Nagai M, Matsumoto T et al (2015) Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J 62:811–816
Rafaelsen S, Johansson S, Ræder H, Bjerknes R (2016) Hereditary hypophosphatemia in Norway: a retrospective population- based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol 174:125–136
Carpenter TO (2012) The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 30:1–9
David V, Martin A, Hedge AM, Drezner MK, Rowe PS (2011) ASARM peptides: PHEX-dependent and -independent regulation of serum phosphate. Am J Physiol Renal Physiol 300:F783–F791
Chesher D, Oddy M, Darbar U, Sayal P, Casey A, Ryan A, Sechi A, Simister C, Waters A, Wedatilake Y, Lachmann RH, Murphy E (2018) Outcome of adult patients with X- linked hypophosphatemia caused by PHEX gene mutations. J Inherit Metab Dis 41:865–976
Whyte MP, Schranck FW, Armamento-Villareal R (1996) X- Linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab 81:4075–4080
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291:E38–E49
Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, McKee MD (2013) Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Min Res 28:688–699
Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ (2010) Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab 95:1846–1850
Bai X, Miao D, Xiao S, Qiu D, St-Arnaud R, Petkovich M, Gupta A, Goltzman D, Karaplis AC (2016) CYP24 inhibition as a therapeutic target in FGF23-mediated renal phosphate wasting disorders. J Clin Invest 126:667–680
Beck-Nielsen SS, Brusgaard K, Rasmussen LM, Brixen K, Brock-Jacobsen B, Poulsen MR, Vestergaard P, Ralston SH, Albagha OM, Poulsen S, Haubek D, Gjørup H, Hintze H, Andersen MG, Heickendorff L, Hjelmborg J, Gram J (2010) Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int 87:108–119
Che H, Roux C, Etcheto A, Rothenbuhler A, Kamenicky P, Linglart A, Briot K (2016) Impaired quality of life in adults with X- linked hypophosphatemia and skeletal symptoms. Eur J Endocrinol 174:325–333
Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, Briot K, Linglart A, Chaussain C (2017) Phosphate and vitamin D prevent periodontitis in X- linked hypophosphatemia. J Dent Res 96:388–395
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M (1991) Effects of therapy in X- linked hypophosphatemic rickets. N Engl J Med 325:1843–1848
Berndt M, Ehrich JH, Lazovic D, Zimmermann J, Hillmann G, Kayser C, Prokop M, Schirg E, Siegert B, Wolff G, Brodehl J (1996) Clinical course of hypophosphatemic rickets in 23 adults. Clin Nephrol 45:33–41
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prié D, Rothenbuhler A, Wicart P, Harvengt P (2014) Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 3:R13–R30
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL (2011) A clinician’s guide to X- linked hypophosphatemia. J Bone Miner Res 26:1381–1388
Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, de Lucas CC, Schnabel D, Jandhyala R, Mäkitie O (2019) FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis 14(1):58
Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG (2016) Excessive osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp Mice. PLoS Biol 14:e1002427
Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD (1998) Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol 275(4 Pt 1):E700–E708
Bai X, Miao D, Xiao S, Qiu D, St-Arnaud R, Petkovich M et al (2016) CYP24 inhibition as a therapeutic target in FGF23-mediated renal phosphate wasting disorders. J Clin Invest 126:667–680
Addison WN, Azari F, Sorensen ES, Kaartinen MT, McKee MD (2007) Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up- regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem 282:15872–15883
Addison WN, Masica DL, Gray JJ, McKee MD (2010) Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Min Res 25:695–705
Staines KA, MacRae VE, Farquharson C (2012) The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodelling. J Endocrinol 214:241–255
Yuan Q, Jiang Y, Zhao X, Sato T, Densmore M, Schüler C et al (2014) Increased osteopontin contributes to inhibition of bone mineralization in FGF23- deficient mice. J Bone Miner Res Off J Am Soc Bone Miner Res 29:693–704
Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R et al (2014) FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J 33:229–246
Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I et al (2009) Therapeutic effects of Anti-FGF23 antibodies in hypophosphatemic rickets/ osteomalacia. J Bone Miner Res 24:1879–1888
Haffner D, Leifheit-Nestler M (2017) Extrarenal effects of FGF23. Pediatr Nephrol 32:753–765
Sato C, Iso Y, Mizukami T, Otabe K, Sasai M, Kurata M et al (2016) Fibroblast growth factor-23 induces cellular senescence in human mesenchymal stem cells from skeletal muscle. Biochem Biophys Res Commun 470:657–662
Connor J, Olear EA, Insogna KL, Katz L, Baker S, Kaur R et al (2015) Conventional therapy in adults With X-Linked hypophosphatemia: effects on enthesopathy and dental disease. J Clin Endocrinol Metab 100:3625–3632
Pavone V, Testa G, Gioitta Iachino S, Evola FR, Avondo S, Sessav G (2015) Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol 25:221–226
Carpenter TO, Olear EA, Zhang JH, Ellis BK, Simpson CA, Cheng D, Gundberg CM, Insogna KL (2014) Effect of paricalcitol on circulating parathyroid hormone in X- linked hypophosphatemia: a randomized, double- blind, placebo- controlled study. J Clin Endocrinol Metab 99:3103–3111
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V et al (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Mace ML, Gravesen E, Nordholm A, Olgaard K, Lewin E (2018) Fibroblast growth factor (FGF) 23 regulates the plasma levels of parathyroid hormone in vivo through the FGF receptor in normocalcemia, but not in hypocalcemia. Calcif Tissue Int 102:85–92
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full- length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C, Olear E, Gundberg CM (2010) Circulating levels of soluble klotho and FGF23 in X- linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab 95:E352–E357
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A (2019) Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 15:435–455
Alon US, Monzavi R, Lilien M, Rasoulpour M, Geffner ME, Yadin O (2003) Hypertension in hypophosphatemic rickets—role of secondary hyperparathyroidism. Pediatr Nephrol 18:155–158
Nakamura Y, Takagi M, Takeda R, Miyai K, Hasegawa Y (2017) Hypertension is a characteristic complication of X- linked hypophosphatemia. Endocr J 64:283–289
Schmitt CP, Mehls O (2004) The enigma of hyperparathyroidism in hypophosphatemic rickets. Pediatr Nephrol 19:473–477
Econs MJ, McEnery PT (1997) Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab 82:674–681
Imel EA, Biggin A, Schindeler A, Munns CF (2019) FGF23, Hypophosphatemia, and Emerging Treatments. JBMR Plus 13(3):e10190
Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ (2011) Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab 96:3541–3549
Bai XY, Miao D, Goltzman D, Karaplis AC (2003) The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 278:9843–9849
Huang X, Jiang Y, Xia W (2013) FGF23 and phosphate wasting disorders. Bone 28:120–132
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278
Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nürnberg P (2003) Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet 34:379–381
Gattineni J, Baum M (2010) Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): implications for disorders of phosphate metabolism. Pediatr Nephrol 25:591–601
Alon US (2011) Clinical practice. Fibroblast growth factor (FGF)23: a new hormone. Eur J Pediatr 170:545–554
Jaureguiberry G, Carpenter TO, Forman S, Harald Jüppner H, Bergwitz C (2008) A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am J Physiol Renal Physiol 295:F371–F379
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM (2006) Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78:193–201
Bergwitz C, Miyamoto KI (2019) Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch 471:149–163
Haito-Sugino S, Ito M, Ohi A, Shiozaki Y, Kangawa N, Nishiyama T, Aranami F, Sasaki S, Mori A, Kido S, Tatsumi S, Segawa H, Miyamoto KI (2012) Processing and stability of type IIc sodium-dependent phosphate cotransporter mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria. Am J Physiol Cell Physiol 302:C1316–C1330
Devuyst O, Thakker RV (2010) Dent’s disease. Orphanet J Rare 5:28
Wrong OM, Norden AG, Feest TG (1994) Dent’s disease; a familial proximal renal tubular syndrome with low-molecular- weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM 87:473–493
Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, Nabeshima Y, Reyes-Mugica M, Carpenter TO, Lifton RP (2008) A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A 4(105):3455–3460
Malloy PJ, Feldman D (2010) Genetic disorders and defects in vitamin D action. Endocrinol Metab Clin N Am 39:333–346
Tiosano D, Gepstein V (2012) Vitamin D action: lessons learned from hereditary 1,25-dihydroxyvitamin-D-resistant rickets patients. Curr Opin Endocrinol Diabetes Obes 19:452–459
Isojima T, Ishizawa M, Yoshimura K, Tamura M, Hirose S, Makishima M, Kitanaka S (2015) Hereditary 1,25-dihydroxyvitamin D-resistant rickets (HVDRR) caused by a VDR mutation: a novel mechanism of dominant inheritance. Bone Rep 7(2):68–73
Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S (1998) Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 338:653–661
Goldsweig BK, Carpenter TO (2015) Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep 13:88–97
Tiosano D, Hochberg Z (2009) Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab 27:392–401
Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, Minagawa M, Sugimoto T, Yamauchi M, Michigami T, Matsumoto T (2008) Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemicpatients: proposal of diagnostic criteria using FGF23 measurement. Bone 42:1235–1239
Igaki JM, Yamada M, Yamazaki Y, Koto S, Izawa M, Ariyasu D, Suzuki E, Hasegawa H, Hasegawa Y (2011) High iFGF23 level despite hypophosphatemia is one of the clinical indicators to make diagnosis of XLH. Endocr J 58:647–655
SouberbiellePrié JCD, Piketty ML, Rothenbuhler A, Delanaye P, Chanson P, Cavalier E (2017) Evaluation of a new fully automated assay for plasma intact FGF23. Calcif Tissue Int 101:510–518
Saraff V, Nadar R, Högler W (2020) New developments in the treatment of X-linked hypophosphataemia: implications for clinical management. Paediatr Drugs. 22(2):113–121
Lecoq AL, Brandi ML, Linglart A, Kamenický P (2020) Management of X-linked hypophosphatemia in adults. Metabolism 103S:154049
Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H et al (2008) Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J Bone Miner Res Off J Am Soc Bone Miner Res 23:1509–1518
Whyte MP, Carpenter TO, Gottesman GS et al (2019) Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol 7:189–199
Carpenter TO, Whyte MP, Imel EA et al (2018) Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med 378(21):1987–1998
US Food & Drug Administration. CRYSVITA (prescribing information) (2018) FDA.gov
European Medicines Agency. Crysvita. Annex I — summary of product characteristics. EMA (2018)
Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Högler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M, Chen CY, Skrinar A, San Martin J, Portale AA (2019) Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 15(393):2416–2427
Portale AA, Carpenter TO, Brandi ML, Briot K, Cheong HI, Cohen-Solal M, Crowley R, Jan De Beur S, Eastell R, Imanishi Y, Imel EA, Ing S, Ito N, Javaid M, Kamenicky P, Keen R, Kubota T, Lachmann R, Perwad F, Pitukcheewanont P, Ralston SH, Takeuchi Y, Tanaka H, Weber TJ, Yoo HW, Zhang L, Theodore-Oklota C, Mealiffe M, San Martin J, Insogna K (2019) Continued Beneficial Effects of Burosumab in Adults with X-Linked Hypophosphatemia: Results from a 24-Week Treatment Continuation Period After a 24-Week Double-Blind Placebo-Controlled Period. Calcif Tissue Int 105(3):271–284
Lambert AS, Zhukouskaya V, Rothenbuhler A, Linglart A (2019) X-linked hypophosphatemia: Management and treatment prospects. Joint Bone Spine 86:731–738
Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, Robling AG, Stayrook KR, Jideonwo V, Magers MJ, Garringer HJ, Vidal R, Chan RJ, Goodwin CB, Hui SL, Peacock M, White KE (2011) Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A 108:E1146–E1155
Imel EA, Liu Z, Coffman M, Acton D, Econs MJ (2018) Oral Iron Therapy Normalizes Fibroblast Growth Factor 23 (FGF23) in Patients with Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res 33:S1–S56
Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA (1985) Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 312:611–617
Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, Maor J, Weissgarten J, Averbukh Z, Cohen N et al (1987) “Idiopathic” hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect. N Engl J Med 316:125–129
Reginato AJ, Coquia JA (2003) Musculoskeletalmanifestations of osteomalacia and rickets. Best Pract Res Clin Rheumatol 17:1063–1080
Kremke B, Bergwitz C, Ahrens W, Schutt S, Schumacher M, Wagner V, Holterhus PM, Jüppner H, Hiort O (2009) Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/NaPi-IIc can be masked by vitamin D deficiency and can be associated with renal calcifications. Exp Clin Endocrinol Diabetes 117:49–56
HYP-Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136
ADHR Consortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Karim Z, Gerard B, Bakouh N, Alili R, Leroy C, Beck L, Silve C, Planelles G, Urena-Torres P, Grandchamp B, Friedlander G, Prie D (2008) NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med 359:1128–1135
Prie D, Huart V, Bakouh N, Planelles G, Dellis O, Gerard B, Hulin P, Benque-Blanchet F, Silve C, Grandchamp B, Friedlander G (2002) Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodiumphosphate cotransporter. N Engl J Med 347:983–991
Ehlayel AM, Copelovitch L (2019) Update on Dent Disease. Pediatr Clin North Am 66:169–178
Author information
Authors and Affiliations
Contributions
GM contributed to conceptualization of the study, performed the literature research and wrote the article. ML Brandi contributed to conceptualization of the study. All authors critically revised the article for intellectual content and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
GM declares that she has no conflict of interests. MLB has received honoraria from Amgen, Bruno Farmaceutici, Calcilytix, Kyowa Kirin; academic grants from and/or was speaker for Abiogen, Alexion, Amgen, Bruno Farmaceutici, Eli Lilly, Kyowa Kirin, MSD, NPS, Servier, Shire, SPA; and has been consultant for Alexion, Bruno Farmaceutici, Kyowa Kirin, Servier, Shire.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marcucci, G., Brandi, M.L. Congenital Conditions of Hypophosphatemia Expressed in Adults. Calcif Tissue Int 108, 91–103 (2021). https://doi.org/10.1007/s00223-020-00695-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00695-2