
Abstract
Osteogenesis imperfecta (OI) is a group of inherited disorders characterized by recurrent fragile fractures. Serpin peptidase inhibitor, clade F, member 1 (SERPINF1) is known to cause a distinct, extremely rare autosomal recessive form of type VI OI. Here we report, for the first time, the detection of SERPINF1 mutations in Chinese OI patients. We designed a novel targeted next-generation sequencing panel of OI-related genes to identify pathogenic mutations, which were confirmed with Sanger sequencing and by co-segregation analysis. We also investigated the phenotypes of OI patients by evaluating bone mineral density, radiological fractures, serum bone turnover markers, and pigment epithelium-derived factor (PEDF) concentration. Six patients with moderate-to-severe bone fragility, significantly low bone mineral density, and severe deformities of the extremities were recruited from five unrelated families for this study. Six pathogenic mutations in SERPINF1 gene were identified, five of which were novel: (1) a homozygous in-frame insertion in exon 3 (c.271_279dup, p.Ala91_Ser93dup); (2) compound heterozygous mutations in intron 3 (c.283 + 1G > T, splicing site) and exon 5 (c.498_499delCA, p.Arg167SerfsX35, frameshift); (3) a homozygous frameshift mutation in exon 8 (c.1202_1203delCA, p.Thr401ArgfsX); (4) compound heterozygous missense mutation (c.184G > A, p.Gly62Ser) and in-frame insertion (c.271_279dup, p.Ala91_Ser93dup) in exon 3; and (5) a heterozygous nonsense mutation in exon 4 (c.397C>T + ?, p.Gln133X + ?). Serum PEDF levels were barely detectable in almost all subjects. We identified five novel mutations in SERPINF1 and confirmed the diagnostic value of serum PEDF level for the first time in Chinese patients with the extremely rare OI type VI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) is a molecularly and phenotypically heterogeneous connective tissue disorder characterized by recurrent fractures, progressive skeletal deformities, and growth deficiency [1]. Extra-skeletal manifestations include blue sclera, early-onset hearing loss, dentinogenesis imperfecta, and joint hypermobility. According to the Sillence system of classification, OI patients are classified into four types (types I–IV), based on the severity of the disease [2]. Although most cases of OI have an autosomal dominant inheritance pattern, typically caused by mutations in genes encoding type I collagen such as COL1A1 or COL1A2, OI type VI (MIM #610968) is a unique autosomal recessive form that is caused by mutations in SERPINF1 [3, 4].
OI type VI was first described as an extremely rare autosomal recessive disorder with moderate-to-severe bone fragility [5]. Type VI OI patients typically present with early-onset recurrent fractures and usually without extra-skeletal features [3, 4, 6]. Bone histology reveals distinctive osteoid accumulation, delayed bone mineralization, and a “fish-scale” lamellar appearance [4–7].
The candidate gene of OI type VI, SERPINF1, is located on chromosome 17p13.3 and encodes the 50-kDa secreted glycoprotein pigment epithelium-derived factor (PEDF) [3]. It has been revealed that the absence of circulating PEDF levels is specific for OI type VI [8]. PEDF belongs to the serpin superfamily and is involved in angiogenesis, tumorigenesis, neuroprotection, and fat metabolism [9–12]. In bone, PEDF is mainly secreted from osteoblasts, osteocytes, and a few chondrocytes and osteoclasts [13, 14]. It has been reported that PEDF contributes to the regulation of osteoblast and osteoclast function and possibly plays an important role in bone mineralization. In Serpinf1 −/− mice, excessive unmineralized bone matrix accumulation was detected, which resembles the phenotypes of OI type VI [15]. PEDF enhances osteoblast differentiation and increases matrix mineralization by modulating the expression of osteocyte-related factors, such as sclerostin and matrix extracellular phosphoglycoprotein [16]. PEDF also upregulates expression of osteoprotegerin, a decoy RANKL receptor that is synthesized in osteoblasts, which inhibits osteoclast differentiation and decreases bone resorption activity [17]. Moreover, PEDF binds to type I collagen, which may also be indirectly associated with the pathogenesis of OI [18, 19]. So far, about 32 individuals with OI type VI have been reported, and 22 unique mutations in SERPINF1 have been identified (http://www.le.ac.uk/ge/collagen/). However, detailed information about the phenotypes and gene mutations of OI type VI in the Chinese population is still needed.
Herein, we investigate the phenotypes and the pathogenic mutations in SERPINF1 in Chinese OI type VI patients, and evaluate the serum levels of PEDF among OI type VI patients, compared to patients with other types of OI and healthy control subjects.
Materials and Methods
Subjects
The initial clinical diagnosis of OI was made by endocrinology department of Peking Union Medical College Hospital (PUMCH) according to the following criteria: recurrent fractures under mild trauma with or without extra-skeletal manifestations such as blue sclera, hearing loss, dentinogenesis imperfecta, and hypermobility of joints [1]. There were more than 300 families in our center, nearly 100 of whom had a definite molecular diagnosis. Six OI patients with SERPINF1 mutations were recruited. Age- and sex-matched OI patients with COL1A1/COL1A2 mutations were also included. The control group included six individuals who had been assessed in PUMCH because they had diseases such as short stature with unknown origin or dyspepsia.
Phenotypes Evaluation
Medical history was collected, and physical examination was performed. Serum biochemical parameters, including calcium, phosphate, alkaline phosphatase (ALP, assayed as a marker of bone formation), alanine aminotransferase, and creatinine levels, were measured using standard procedures in the central clinical laboratory of PUMCH. Serum levels of beta cross-linked carboxytelopeptide of type I collagen (β-CTX, assayed as a marker of bone resorption) and 25-hydroxyvitamin D (25OHD, assayed as a marker of vitamin D nutrition status) were measured using an automated electrochemiluminescence system (E170; Roche Diagnostics, Switzerland).
The new fractures were inferred by examining the medical history and confirmed by a radiologist using X-ray films. Bone mineral density (BMD) in the lumbar spine 2–4 (LS), femoral neck (FN), and total hip (TH) was measured using dual-energy X-ray absorptiometry (DXA, Lunar Prodigy, GE Healthcare, Madison WI, USA). In order to exclude the influence of age on BMD, the Z scores of BMD were calculated according to the age- and sex-matched normal ranges of Chinese and Indian children [20–22].
Pathogenic Mutation Identification
Peripheral venous blood was collected. Genomic DNA was extracted from leukocytes using a QIAamp DNA Mini Kit (Qiagen, Germany). A targeted next-generation sequencing (NGS) capture panel was created to examine OI-related genes. The pathogenic OI genes in this panel included COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, SP7, TMEM38B, WNT1, and PLOD2. All exons, splice sites, and immediate flanking intron sequences of all the OI genes of interest were amplified in patients 1, 2, 3, and 4. DNA samples were sheared into fragments of 200–300 bases and purified using an AMPre XP beads kit. Ends of the fragments were repaired, and an “A” base was added to the 3’ end with Klenow. After ligation of the Illumina adapters to the ends of DNA fragments, the sequencing library was constructed. After clonal amplification by emulsion polymerase chain reaction (PCR), the library was hybridized with the capture panel. Finally, the captured fragments were enriched by PCR and loaded onto a flow cell, followed by sequencing and imaging on Illumina HiSeq2000 platform according to the standard protocol.
The primary sequencing data were mapped to the human genome (NCBI37/hg19) using Burrows Wheeler Aligner software, and single nucleotide variants were identified using SOAP SNP software. Then, the biological information was compared to databases including ExAC, dbSNP, HapMap, 1000 Genomes Asian, ESP6500, Cosmic, and HGMD. Insertion and deletion mutations were detected using the GATK software package (version 3.5). Nonsense mutations were predicted by PolyPhen-2 and SIFT software. Splicing sites were predicted by Human Splicing Finder and NetGene2 Server. Transmission promoters were predicted by BDGP Neural Network Promoter Prediction software. In comparison with the type I collagen database, we were able to identify the novel variants according to the American College of Medical Genetics and Genomics (ACMG) recommended standards [23].
Sanger sequencing was performed in patients 5 and 6, as well as for NGS verification in the rest of the subjects. Based on the NGS preliminary results, fragments containing SERPINF1 exons 3, 4, 5, 8 and their exon–intron junctions were amplified by PCR. PCR primers were designed using the Primer 3 program (Supplementary table). PCR was conducted under the following conditions: denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 30 s, annealing at 59–60 °C for 30 s, and extension at 72 °C for 60 s. The direct nucleotide sequencing of PCR products was completed by BigDye Terminators Cycle Sequencing Ready Reaction Kit (Applied Biosystems, version 3.1) and analyzed by ABI 3130 automatic sequencer (Applied Biosystems). Finally, patient sequences were interpreted by Chromas software (version 2.4.1) and referenced to the NCBI reference sequence NM_002615.5 (SERPINF1).
Measurement of Serum PEDF
All serum samples were well preserved at −80 °C. Serum PEDF levels were measured with a DuoSet ELISA Kit (R&D Systems, Minneapolis, MN) in OI type VI patients SERPINF1 mutation carriers, sex- and age- matched OI patients with COL1A1/COL1A2 mutations, and the control patients without bone diseases. Briefly, 100 μL standards in reagent diluent or samples with a final dilution of 1:8000 were added to the wells that were precoated with the capture antibody. The following steps were performed, according to the general double-antibody sandwich ELISA protocol and the manufacturer’s instructions. Then the optical density (OD) was determined immediately, using a microplate reader set to 450 and 540 nm. Duplicate readings were taken for each sample, and the average zero standard OD was subtracted from every reading. Finally, we adjusted our readings by subtracting the readings at 540 nm from the readings at 450 nm. The intra-assay and inter-assay coefficients of variation were 7.7 and 8.1%, respectively. The specificity was very high, and no significant cross-reactivity was observed.
Results
Phenotypes of OI Type VI
All patients were born by full-term spontaneous vaginal delivery without prenatal fractures. Fragile fractures usually began at the age of 7–18 months. The predilection sites of the recurrent fractures included the humerus, femur, tibia, radius, clavicle, costal bones, and vertebrae. Particularly, long bones in the extremities were the most common sites of fracture. The number of fractures patients sustained varied from 1.3 to 14 per year. The moderate-to-severe bone fragility resulted in short stature, decreased mobility, and bone deformities (such as curved long bones, severe scoliosis, and kyphosis). Patients 2, 3, and 4 had hypermobility of the joints. Patients 3 and 5 had severe sarcopenia of the limb muscles (Table 1). No other extra-skeletal manifestations were found, such as blue sclera or hearing impairments.
One of patient 3’s sisters had multiple fractures during childhood and died at eight years old. The other patients had no relevant family history.
Serum levels of calcium and phosphorus of all patients fluctuated within the normal range. The serum ALP levels of all patients were elevated, except for patients 1 and 6. Slightly elevated β-CTX levels were observed in patients 1, 2, and 4. Serum 25OHD levels of patients were all low, which indicated vitamin D deficiency (Table 2). Patients had extremely low BMD Z scores, especially in the lumbar spine, which ranged from −2.88 to −8.35 (Table 2). Radiological findings revealed extensive osteoporosis, thin long bone with the thin cortex, metaphyseal flaring, and multiple vertebral compressions. Notably, wormian bone in the skull was observed in two patients (Fig. 1).

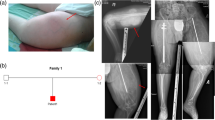
Radiological data of osteogenesis imperfecta type VI patients. a Anteroposterior (AP) view of right femoral bone in patient 3 showed severe osteoporosis and “popcorn” epiphysis (white arrow). b AP view of right humeral bone of patient 3 showed proximal fracture, delayed healing, and bowed bones. c AP view of upper limb of patient 2 showed proximal comminuted humeral fracture and thin long bone with thin cortex. d The lower limbs of patient 2 showed multiple remodeling femoral fractures and metaphyseal flaring. e Wormian bone in the occipital bone of patient 2 was shown with the white arrow
Mutations in SERPINF1
The mean sequencing depth of NGS was 288X, and the SERPINF1 gene was completely covered. Six different SERPINF1 mutations were found in the six probands from five unrelated families, five of which were novel mutations (http://oi.gene.le.ac.uk/variants) (Table 3). A homozygous in-frame duplication (c.271_279dup, p.Ala91_Ser93dup) was identified in patient 1 (Fig. 2a). Sanger sequencing revealed that his father didn’t carry the c.271_279dup mutation and his mother was a heterozygous carrier. The possibilities of the Sanger sequencing result of patient 1 were nonpaternity or partial de novo mutation. Due to ethical reasons, his parents refused to do further tests. Compound heterozygous mutations of intron 3 (c.283 + 1G > T) and exon 5 (c.498_499delCA, p.Arg167Serfs) were detected in patient 2 (Fig. 2b). The splice mutation (c.283 + 1G > T) was predicted to activate another potential splicing site downstream, leading to the addition of intron 3 sequence to the original exon 3 sequence. Patient 3 was found to carry a homozygous frameshift mutation in exon 8 (c.1202_1203delCA, p.Thr401ArgfsX) in SERPINF1, and her parents were both heterozygous carriers (Fig. 2c). Patient 4 carried a heterozygous in-frame duplication (c.271_279dup, p.Ala91_Ser93dup) and missense mutation (c.184G > A, p.Gly62Ser) in exon 3 (Fig. 2d). For c.184G > A, the SIFT score was 0 and Polyphen-2 score was 1, which indicated that it might be a pathogenic mutation. Patients 5 and 6 were siblings from the same nonconsanguineous family, and both of them had a heterozygous nonsense mutation (c.397C>T, p.Gln133X) (Fig. 2e). In the Sanger sequencing result of this pedigree, we could also see the c.390 T > C in the two patients and their mother. The c.390 T > C was proved to be an SNP according to the public databases (rs8074840). It can result in p.Thr130Thr, a synonymous mutation whose total population allele frequency is 0.3161. So we didn’t identify it as a pathogenic mutation for patients 5 and 6. No other pathogenic mutations were detected in this pedigree according to NGS results. It is possible that the other pathogenic mutation located in deep intron regions which could be neglected. And the epigenetic changes, which would also participate in the pathogenesis of an autosomal recessive inherited disease, could also be invisible in NGS. Co-segregation analysis showed that healthy parents were heterozygous carriers of SERPINF1. No mutations were identified in the remaining regions of SERPINF1 or in other known pathogenic genes of OI. Among the novel mutations, the p.Gly62Ser mutation was highly conserved throughout the species (Fig. 3). The five kinds of novel mutations were absent in the 100 unrelated control subjects and were not classed as polymorphisms in all public databases (Fig. 4).



Pedigrees of the five families and Sanger sequencing chromatograms of the SERPINF1 gene. In the pedigrees, black arrows represent the proposituses. The black symbols indicate OI patients, while the shadow symbols indicate carriers. a In patient 1, a homozygous mutation was identified as c.271_279dup (p.Ala91_Ser93dup) in exon 3. b In patient 2, novel compound heterozygous mutations were identified as c.283 + 1G > T in intron 3 and c.498_499delCA (p.Arg167SerfsX35) in exon 5. c In patient 3, novel homozygous mutation was identified as c.1202_1203delCA (p.Thr401ArgfsX) in exon 8. d In patient 4, novel heterozygous mutations were identified as c.184G > A (p.Gly62Ser) in exon 3, and a reported mutation of c.271_279dup (p.Ala91_Ser93dup) in the same exon. e In patients 5 and 6, a novel heterozygous mutation was identified as c.397C > T (p.Gln133X) in exon 4

Locations of predicted protein consequences for SERPINF1 mutations in PEDF molecular. Upper Schematic representation of pigment epithelium-derived factor (PEDF, NP_002606). Lower Amino acid alignment of the PEDF sequence among species. The novel missense c.184G > A is indicated by the red arrow. p.Gly62Ser and p.Ala91_Ser93dup affected highly conserved motifs in the PEDF sequence (Color figure online)

Sites of known unique mutations and six novel mutations in SERPINF1. Blue and red boxes indicate the exons and introns of SERPINF1, respectively. Arrows indicate the sites of unique mutations on the exons or introns or exon–intron junctions. The five novel mutations found in this study are presented in red arrows (Color figure online)
PEDF Protein Concentration
The mean serum PEDF level was extremely low in OI type VI patients as compared to OI patients with COL1A1/COL1A2 mutations and controls without bone diseases (P = 6.34 × 10−5, 1.35 × 10−5, respectively, Fig. 5a, b). However, the serum PEDF level of patient 4 was 3.93 µg/ml. PEDF levels in the SERPINF1 mutation carriers ranged from 2.44 to 7.95 µg/ml. Serum PEDF levels ranged from 4.61 to 10.87 µg/ml in OI patients with COL1A1/COL1A2 mutations. The control group without bone diseases had PEDF serum concentrations between 5.99 and 10.07 µg/ml, which was similar to patients with COL1A1/COL1A2 mutations.

PEDF serum levels in individual participants and mean values in OI type VI patients and control groups. a PEDF serum levels in OI type VI patients are significantly decreased as compared with patients with COL1A1/COL1A2 mutations and normal controls. PEDF serum levels in SERPINF1 mutation carriers are between the above. b Mean PEDF serum levels of OI type VI patients were significantly lower than OI patients with COL1A1/COL1A2 mutations and normal controls (P = 6.34 × 10−5, 1.35 × 10−5, respectively)
Discussion
OI type VI is an extremely rare and autosomal recessive form of OI that is caused by SERPINF1 mutations [4–6, 24]. We reported novel pathogenic mutations in the SERPINF1 gene for the first time and described the associated phenotypes in six patients with SERPINF1 gene mutations from the Chinese population. Specifically, we identified five novel pathogenic variants in six OI patients and confirmed a previously reported mutation in two probands. We also demonstrated that PEDF levels were extremely low in Chinese OI patients with SERPINF1 gene mutations, but found no clear association between PEDF levels and bone fracture rate, bone turnover markers, or BMD at baseline.
None of the patients received iliac bone biopsies, as it is a relatively invasive procedure. For patients 1, 2, 3, and 4, the diagnosis of OI type VI was made on the basis of clinical manifestations and sequence analysis of SERPINF1 gene. Patients 5 and 6 had typical clinical features of OI and undetectable serum PEDF levels. Since they were both confirmed of only one heterozygous mutation in SERPINF1 gene, the diagnosis of OI type VI could be regarded as strongly suspected but “inconclusive” [25].
Until now, 22 unique pathogenic variants have been reported in SERPINF1 to cause type VI OI (http://www.le.ac.uk/ge/collagen/). The majority of SERPINF1 mutations are frameshift and nonsense mutations that lead to loss of PEDF function due to protein truncation (http://www.le.ac.uk/ge/collagen/). We identified one patient in our cohort with a frameshift mutation [c.283 + 1G > T] + [c.498_499delCA], which could probably produce a premature stop codon and lead to nonsense-mediated decay (NMD). This patient had severe skeletal deformities and was demonstrated delayed healing after orthopedic surgery. Another frameshift mutation c.1202_1203delCA located in exon 8 was identified in patient 3. Since the functional studies hadn’t been performed, the mechanism of this homozygous mutation leading to undetectable serum PEDF level still remained unclear. Two siblings from one nonconsanguineous family were found to carry a novel heterozygous nonsense mutation (c.397C > T). Although the younger brother had not yet developed severe bone deformities, both brothers had extremely low LS-BMD Z scores, delayed healing after long bone fractures, and dependent ambulation. This nonsense mutation could probably also lead to NMD, which might account for the severe bone phenotypes. In our study, we found two patients carried c.271_279 dup, an in-frame duplication in SERPINF which adds three amino acids to PEDF. The addition of the amino acids was deduced to affect the stability of PEDF and thus result in OI [7]. A recent study showed that the c.271_279 dup mutation in SERPINF1 had a negative impact on MC3T3-E1 osteoblasts by interfering with collagen deposition and mineralization [24]. Previous studies reported a patient with the c.271_279 dup in SERPINF1 who had a less severe skeletal phenotype [7]. This is consistent with one proband in our cohort (patient with [c.184G > A] + [c.271_279 dup] in SERPINF1); however, the other proband (homozygous c.271_c.279dup in SERPINF1) in the cohort was wheelchair-bound and could not walk independently.
In our study, serum PEDF levels were barely detectable in five OI type VI patients, which was consistent with the previous results [4, 8]. Serum PEDF levels in patients with COL1A1/COL1A2 mutations, controls without bone diseases, and SERPINF1 mutation carriers were similar to previous studies [8, 26]. Our results further demonstrate the diagnostic value of serum PEDF level in OI type VI. Recently, researchers discovered that restoration of serum PEDF could not correct the abnormalities of Serpinf1 −/− mice [27]. However, overexpression of PEDF in vitro, along with increased bone gamma-carboxyglutamate, alkaline phosphate, and type I collagen, could contribute to excessive bone mineralization [27]. Interestingly, another study showed PEDF restoration could increase bone mass under Wnt3a exposure, which suggested a new relationship between PEDF and WNT signaling in osteoblast differentiation and bone mineralization [28]. Further studies of PEDF and OI are needed to elucidate the exact mechanism of SERPINF1 mutation leading to OI.
There are several limitations to our study. Firstly, we did not perform bone biopsy, because it was an invasive procedure, and the combination of genetic and clinical diagnosis would be sufficient to establish a firm diagnosis in most of the patients. Secondly, the lack of functional studies made it difficult to explain the measurable serum PEDF level in patient 4 and that how all the mutations function on the transcript level. Thirdly, the sequence analysis of patients 5 and 6 only revealed one heterozygous mutation in SERPINF1 gene, and no second sequence abnormality was detected. So the diagnosis of OI type VI could not be completely confirmed. At last, our study sample size may be too small to analyze the association between phenotype and genotype in OI type VI. However, OI type VI is an extremely rare disease, and our cohort is by far the largest cohort studied in China.
Conclusion
By utilizing next-generation sequencing, we successfully identified five novel SERPINF1 mutations in six Chinese patients with OI type VI and confirmed a recurrent SERPINF1 mutation in two patients. We further analyzed the phenotypes in detail and confirmed the important diagnostic role of serum PEDF level in OI type VI patients.
References
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387(10028):1657–1671
Sillence DO, Senn A, Danks DM (1979) Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16(2):101–116
Becker J, Semler O, Gilissen C et al (2011) Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 88(3):362–371
Homan EP, Rauch F, Grafe I et al (2011) Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res 26(12):2798–2803
Glorieux FH, Ward LM, Rauch F et al (2002) Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res 17(1):30–38
Venturi G, Gandini A, Monti E et al (2012) Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type I collagen. J Bone Miner Res 27(3):723–728
Tucker T, Nelson T, Sirrs S et al (2012) A co-occurrence of osteogenesis imperfecta type VI and cystinosis. Am J Med Genet A 158A(6):1422–1426
Rauch F, Husseini A, Roughley P, Glorieux FH, Moffatt P (2012) Lack of circulating pigment epithelium-derived factor is a marker of osteogenesis imperfecta type VI. J Clin Endocrinol Metab 97(8):E1550–E1556
Dawson DW, Volpert OV, Gillis P et al (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285(5425):245–248
Becerra SP, Notario V (2013) The effects of PEDF on cancer biology: mechanisms of action and therapeutic potential. Nat Rev Cancer 13(4):258–271
Sanchez A, Tripathy D, Yin X et al (2012) Pigment epithelium-derived factor (PEDF) protects cortical neurons in vitro from oxidant injury by activation of extracellular signal-regulated kinase (ERK) 1/2 and induction of Bcl-2. Neurosci Res 72(1):1–8
Borg ML, Andrews ZB, Duh EJ et al (2011) Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes 60(5):1458–1466
Quan GM, Ojaimi J, Li Y et al (2005) Localization of pigment epithelium-derived factor in growing mouse bone. Calcif Tissue Int 76(2):146–153
Tombran-Tink J, Barnstable CJ (2004) Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: possible mediators of angiogenesis and matrix remodeling in the bone. Biochem Biophys Res Commun 316(2):573–579
Bogan R, Riddle RC, Li Z et al (2013) A mouse model for human osteogenesis imperfecta type VI. J Bone Miner Res 28(7):1531–1536
Li F, Song N, Tombran-Tink J, Niyibizi C (2015) Pigment epithelium derived factor suppresses expression of Sost/Sclerostin by osteocytes: implication for its role in bone matrix mineralization. J Cell Physiol 230(6):1243–1249
Akiyama T, Dass CR, Shinoda Y et al (2010) PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem Biophys Res Commun 391(1):789–794
Sekiya A, Okano-Kosugi H, Yamazaki CM, Koide T (2011) Pigment epithelium-derived factor (PEDF) shares binding sites in collagen with heparin/heparan sulfate proteoglycans. J Biol Chem 286(30):26364–26374
Marini JC, Reich A, Smith SM (2014) Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 26(4):500–507
Xu H, Zhao Z, Wang H et al (2013) Bone mineral density of the spine in 11,898 Chinese infants and young children: a cross-sectional study. PLoS ONE 8(12):e82098
Khadilkar AV, Sanwalka NJ, Chiplonkar SA, Khadilkar VV, Mughal MZ (2011) Normative data and percentile curves for dual energy X-ray Absorptiometry in healthy Indian girls and boys aged 5–17 years. Bone 48(4):810–819
Tan LJ, Lei SF, Chen XD et al (2007) Establishment of peak bone mineral density in Southern Chinese males and its comparisons with other males from different regions of China. J Bone Miner Metab 25(2):114–121
Richards CS, Bale S, Bellissimo DB et al (2008) ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 10(4):294–300
Al-Jallad H, Palomo T, Roughley P et al (2015) The effect of SERPINF1 in-frame mutations in osteogenesis imperfecta type VI. Bone 76:115–120
Bardai G, Moffatt P, Glorieux FH, Rauch F (2016) DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int. doi:10.1007/s00198-016-3709-1
Al-Jallad H, Palomo T, Moffatt P et al (2014) Normal bone density and fat mass in heterozygous SERPINF1 mutation carriers. J Clin Endocrinol Metab 99(11):E2446–E2450
Rajagopal A, Homan EP, Joeng KS et al (2016) Restoration of the serum level of SERPINF1 does not correct the bone phenotype in Serpinf1 null mice. Mol Genet Metab 117(3):378–382
Belinsky GS, Sreekumar B, Andrejecsk JW et al (2016) Pigment epithelium-derived factor restoration increases bone mass and improves bone plasticity in a model of osteogenesis imperfecta type VI via Wnt3a blockade. FASEB J 30(8):2837–2848
Caparros-Martin JA, Valencia M, Pulido V et al (2013) Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype–phenotype correlations. Am J Med Genet A 161A(6):1354–1369
Cho SY, Ki CS, Sohn YB et al (2013) Osteogenesis imperfecta type VI with severe bony deformities caused by novel compound heterozygous mutations in SERPINF1. J Korean Med Sci 28(7):1107–1110
Minillo RM, Sobreira N, de Faria Soares MDF et al (2014) Novel deletion of SERPINF1 causes autosomal recessive osteogenesis imperfecta type VI in two Brazilian families. Mol Syndromol 5(6):268–275
Ward L, Bardai G, Moffatt P et al (2016) Osteogenesis imperfecta type VI in individuals from Northern Canada. Calcif Tissue Int 98(6):566–572
Stephen J, Girisha KM, Dalal A et al (2015) Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet 58(1):21–27
Acknowledgements
This study is supported by Grants from the National Natural Science Foundation of China (81570802) and National Key Program of Clinical Science (WBYZ2011-873). We appreciate our patients and their families for their participation.
Author’s Contributions
All listed authors have made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; participated in drafting the manuscript or revising it critically for content; and have approved the final version of the submitted manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jian-yi Wang, Yi Liu, Li-jie Song, Fang Lv, Xiao-jie Xu, A San, Jian Wang, Huan-ming Yang, Zi-ying Yang, Yan Jiang, Ou Wang, Wei-bo Xia, Xiao-ping Xing, and Mei Li declare that there is no conflict of interests regarding the publication of this paper.
Human and Animal Rights
The study protocol was approved by the Ethics Committee of PUMCH.
Informed Consent
Written informed consent was obtained from the parents of the patients before they were enrolled in the study.
Additional information
Yi Liu and Li-jie Song contributed equally to this work and should be considered as co-first author.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Jy., Liu, Y., Song, Lj. et al. Novel Mutations in SERPINF1 Result in Rare Osteogenesis Imperfecta Type VI. Calcif Tissue Int 100, 55–66 (2017). https://doi.org/10.1007/s00223-016-0201-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-016-0201-z