
Abstract
A high-performance liquid chromatography method with fluorescence detection was developed for the quantification of riboflavin (RF), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD) and their photodegradation products, lumichrome (LC) and lumiflavin (LF), in liquid milk and milk products. Both sample preparation and chromatographic separation were studied to avoid acidic conditions that proved to affect flavin stability and degrade FAD into FMN. The sample preparation includes centrifugal skimming and ultrafiltration steps and is suitable for routine application. Linear response was obtained for individual flavins in the respective concentration ranges of interest and relative standard deviation (RSD) was lower than 5%, except for FAD (RSD 11%). The recovery ranged between 80–100%. The proposed method proved to be suitable for assessing flavins in commercial liquid milk and fermented milk products, and for monitoring the degradation of FAD, FMN and RF and the formation of LF and LC in bottled milk exposed to light during shelf storage.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Vitamins are essential nutrients in the human diet. Riboflavin (vitamin B2, RF) is a water-soluble vitamin with an important function for the cell growth and for the biological redox reactions [1]. Other flavin compounds include the RF coenzymes, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), and other minor compounds, such as hydroxyethylflavin, formylmethylflavin, cyclodehydroriboflavin, hydroxy-FMN, riboflavin-α-d-glucoside [2, 3]. FMN and FAD play a role in cellular metabolism of other water-soluble vitamins [4] and act as electron carriers in several biological processes [5]. Meat, eggs, cereals and bakery products, some green vegetables and dairy products are the main sources of RF [6]. In many foods, FAD is the major flavin, whereas eggs and milk are the richest sources of RF [1]. Riboflavin and flavins in general are relatively stable to food thermal processing, dehydration and usual storage conditions [7, 8]. In contrast, these compounds are extremely sensitive to oxygen and visible or UV light. In particular, RF shows a complex photochemistry due to the property of being easily reduced and oxidized [9]. Beside the loss of nutritional value [10], photooxidative reactions involving RF in milk may lead to sensorial changes [11], including those usually described as sunlight flavor [12]. Shuping and et al. [13] demonstrated that the light-induced polyhydroxy-containing ribityl group of triplet-excited RF is easily cleaved, originating several compounds. Among these, lumichrome (LC) and, to a lesser extent, lumiflavin (LF) have been well characterized [2], the latter being preferentially derived at alkaline pH. These compounds are in turn involved in UV–visible light absorption, acting as potential photosensitizers, thus causing undesirable impairment of sensorial properties of foods.
Several analytical methods have been proposed for the detection of RF and its derivatives in milk and dairy products, particularly in infant formula. Most of these methods are based on the chromatographic separation and detection by either UV [14, 15], fluorescence [3, 16,17,18,19,20] or mass spectrometry [21]. In particular, this last proved to be a successful technique for simultaneous detection of multiple vitamins in infant formula [22, 23] and in human milk [24, 25]. The use of capillary electrophoresis [26], microbial growth-based assays [7], fluorometry [27], molecularly imprinted solid phase extraction followed by fluorometry [28], front-face spectroscopy [29, 30] were also proposed. In chromatographic methods, milk is usually clarified by acidification to precipitate casein. However, depending on the pH, flavin compounds are present in different forms (cationic, neutral and anionic) having different fluorescence intensity [31]. Furthermore, both FMN and FAD hydrolyse into RF at pH below 5 [18] and thus the total RF concentration including the parent flavins is usually reported.
The determination of the individual flavins and their derivatives in milk, particularly those which may form as a consequence of light exposure, has been assuming increasing importance due to the interest of manufacturers in preserving the nutritional value and sensory properties of milk and milk products during shelf storage. Although the simultaneous determination of RF, FAD and FMN in milk has already been proposed [21, 25], to the best of our knowledge, no analytical method is available that allows also LF and LC to be detected in milk in the same run. This study aimed to develop such a method and high-performance liquid chromatography (HPLC) with fluorescence detection was chosen as an appropriate technique for making it suitable for routine application. In this respect, we also developed a sample preparation protocol that avoids inducing any chemical change of the analytes during sample manipulation, thus allowing the quantification of natural levels of individual flavins in milk and milk products and understanding their fate during milk processing, fermentation, and storage. The proposed method was also used to conduct a preliminary survey of the levels of RF and the other flavins in commercial milk and dairy products.
Materials and methods
Chemicals and reagents
Riboflavin (RF), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), methanol, ammonium acetate and trichloroacetic acid (TCA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lumiflavin (LF) and lumichrome (LC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All the chemicals were of analytical grade, at least. HPLC grade water was obtained by a Milli-Q system (Millipore Corp., Bedford, MA, USA).
Samples
The commercial samples considered in this study and the respective packages were as follows: raw milk (n = 4) in 1-L clear glass bottles; full-fat pasteurized milk (n = 4) in 1-L clear PET bottles; full-fat UHT-sterilized milk (n = 1), skimmed UHT-sterilized milk (n = 2), full-fat pasteurized goat milk (n = 2), full-fat UHT-sterilized goat milk (n = 1), pasteurized soymilk (n = 1), fermented milk (n = 2), all in 1-L carton bricks with aluminum foil; UHT-sterilized liquid infant formula (n = 1) in 500-mL light-excluding high density polyethylene (HDPE) bottle; skimmed plain yogurt (n = 1) in 125-mL clear glass pot. All samples were collected at local market, except raw milk samples, which were taken at four different alpine pastures in Italy. All samples were refrigerated (5 ± 1 °C) and protected from light during transportation to the laboratory, then immediately frozen (− 20 °C) until analysis. Moreover, a set of milk bottles was kept at the display conditions in commercial markets. With this aim, five 1 L bottles (clear PET) of full-fat pasteurized milk were taken at the filling machine of an industrial manufacturing plant and brought to the laboratory under refrigerated (5 ± 1 °C) and light-protected conditions. The unopened bottles were stored in a refrigerated (5 ± 1 °C) display case under light exposure (fluorescent lamp, Philips TL-D super 58W/840, Italy). One bottle out of five was taken after 0, 5, 28, 45 and 76 h of storage and the milk analysed after careful mixing by repeated gentle inversion of the bottle. All samples in this study were analysed in triplicate.
Sample preparation
The optimized protocol for sample preparation was as follows: 2 mL of sample were centrifuged at 18,000g for 30 min at 5 °C using a benchtop centrifuge (Hettich, Tuttlingen, Germany). The upper layer of fat was removed with a spatula and the skimmed sample was ultrafiltered by centrifugation at 14,000g for 40 min at 20 °C using a disposable 10 kDa cut-off membrane Microcon (Millipore, Billerica, MA). The permeate was filtered through a 0.22 µm PVDF filter (Millipore) prior to HPLC separation. Samples were protected from light during preparation.
The sample acidification conditions described by Severo Silva Jr et al. [18] for sample preparation were tested on standard water solutions containing either all of the flavins considered in this study (concentration level 3, see “Method validation”) or FAD (440 µg/L) only. Briefly, the standard water solution (500 µL) was added with 30% (w/v) TCA (750 µL, final TCA concentration 12%, pH 1 ± 0.5) and analysed by HPLC under the chromatographic conditions described at section “Chromatographic and quantitation conditions”.
Chromatographic and quantitation conditions
A Waters Alliance 2695 chromatograph (Milford, MA, USA) equipped with a VWR Hitachi L2480 fluorescence detector (Milan, Italy) set at 420 nm for excitation and 530 nm for emission, and a Hypersil ODS chromatographic column (100 × 3 mm, 3 µm particle size) (CPS Analitica, Milan, Italy) set at 40 °C was used. The samples were kept at 15 °C in the autosampler. The injection volume was 50 µL. The elution solvents were: (A) 50 mM ammonium acetate buffer, pH 6, and (B) methanol, and the flow rate was 0.6 mL/min. Elution conditions were as follows: 15% B for 7 min, from 15 to 18% B in 5 min, from 18 to 35% B in 3 min and from 35 to 50% B in 3 min. The column was then washed with 100% B for 3.5 min and re-equilibrated for 8.5 min before the next injection. Chromatographic data were processed using Empower2 software (Waters). FAD, FMN, RF, LF and LC were quantified using the external standard method. Five-level calibration curves were obtained with standard solutions containing the analytes at the respective concentrations spanning the expected ranges in milk.
The unknown peak eluting at 11.1 min in the chromatograms of both fermented milk and yogurt samples was identified by UPLC-MS/MS. These samples were prepared following the proposed procedure (Paragraph 2.3). An Acquity (Waters) UPLC module coupled to a high-resolution exactive (Thermo Scientific, San Jose, CA, USA) mass spectrometer and an HESI-II probe for electrospray ionization was used. The operative conditions were: spray voltage + 3.0 kV, sheath gas flow 60, auxiliary gas flow 20, capillary temperature 360 °C, capillary + 95 V, tube lens + 170 V, skimmer + 38 V, and heater temperature 300 °C. The chromatographic column was the HSS T3 (150 × 2.1 mm, 1.8 µm particle size) (Waters) kept at 50 °C. The eluents: (A) 0.1% formic acid and (B) acetonitrile were used with the following elution gradient: 10% B for 4 min, from 10 to 15% B in 0.1 min, and then 15% B for 10 min. The flow rate was 0.45 mL/min and the injection volume was 5 µL. Data acquisition was performed in full-scan mode in the range [m/z]+ 100–900 u with an isolation window of ± 2 ppm. The AGC target, injection time, mass resolution and energy in the collision cell were 1 × 106, 100 ms, 50,000 and 10 V, respectively. Data were processed using Xcalibur software (Thermo Scientific) and the peak was identified considering both the accurate mass and the fragments obtained in the collision cell. Standard solution of RF (324 µg/L) gave a [m/z]+ 377.1464 and daughter ions 243.0882 and 172.0874. The unknown peak gave a [m/z]+ 538.1816 and daughter ions 377.1463 and 243.0881.
Method validation
The in-house validation of the method was carried out in terms of selectivity, linearity, limit of detection (LOD), limit of quantification (LOQ), precision, recovery and accuracy [32]. The concentration levels of the five target flavins in both standard water solutions and spiked milk samples used in the validation procedure were: (level 1) 1.1 µg/L FAD, 1.1 µg/L FMN, 81.0 µg/L RF, 0.28 µg/L LF and 1.1 µg/L LC; (level 2) 27.5 µg/L FAD, 27.5 µg/L FMN, 324 µg/L RF, 7.0 µg/L LF and 27.5 µg/L LC; (level 3) 440 µg/L FAD, 440 µg/L FMN, 1296 µg/L RF, 112 µg/L LF and 440 µg/L LC.
The selectivity of the method was evaluated by analyzing the flavins both in absence and presence of possible interferences originating from the milk matrix. With this aim, a standard water solution at the concentration level 3 and a milk sample spiked with the five analytes at the same concentration were used.
Linearity was tested on five concentration levels within the intervals reported in Table 1 and samples were analysed in triplicate. The equations of the calibration curves and the correlation coefficients (r) were obtained by the linear regression analysis.
The values of LOD and LOQ were calculated as the lowest concentration of analyte in a sample that resulted in a signal-to-noise ratio of 3 and 10, for LOD and LOQ, respectively. Values were measured in eight independent replicates.
The recovery (%) in spiked milk was calculated according to the following formula [32]:
where Cf concentration of the analyte in spiked milk sample; Ci initial concentration in milk sample; Cs concentration added from the standard solution.
The initial concentration in non-spiked milk and the concentration after spiking at the levels 2 and 3 above specified were determined in triplicate. The recovery was also calculated for the standard solutions at levels 2 and 3 to evaluate the ultrafiltration efficiency. The accuracy was evaluated by means of recovery assay of replicate analyses for both standard solution and milk spiked at levels 2 and 3 (n = 6).
Precision was expressed as relative standard deviation (RSD) of the analytical response. Both the standard solutions and the spiked milk at the three levels of concentration were analysed. Three independent replicates were carried out on three different days (n = 9). For the instrumental repeatability, duplicate injections were carried out from the same vial at the three concentration levels on three different days (n = 18). The repeatability of the retention times was also evaluated over a total of 24 injections carried out on three different days.
Statistical analysis
Once the hypotheses of normality and homoscedasticity were accepted, data of FMN, RF, LF, and LC at different light exposure times were analysed by one-way analysis of variance (ANOVA). Differences among means were checked using the Bonferroni’s test. Data of the standard solution were analysed against the data of the solution after acidification by Student’s t test. Differences among RF concentrations determined in milk samples of different origin and exposed to light were analysed by Student’s t test. Statistical treatment of data was performed by means of SPSS Win 12.0 program (SPSS Inc., Chicago, IL) and a p < 0.05 was assumed as the significance limit.
Results and discussion
Analytical method development
The chromatographic conditions were first optimized using the standard water solution at the concentration level 3 (see Paragraph 2.5). A milk sample, spiked at the same level and prepared using our ultrafiltration (UF) protocol, was analysed in parallel to detect the possible presence of interfering peaks in the chromatogram. The best chromatographic separation was obtained using 50 mM of ammonium acetate buffer at pH 6 as eluting solvent. Depending on pH, RF exists under different forms having different fluorescence spectra and the maximum fluorescence values were observed in the range of 4–8 [31]. The pH also affects the fluorescence intensity of both FAD and FMN [20, 29]. Adoption of an eluent buffered at pH 6 allowed preserving the chemical equilibrium among flavins present in the milk. Setting the column temperature at 40 °C resulted in the best compromise between peak resolution and retention time of the investigated compounds. Although a few small unidentified peaks were present in the milk chromatogram, the optimized conditions allowed the target flavins to elute as interference-free peaks that were reliably identified (Fig. 1a). These conditions were thus retained for further optimisation.

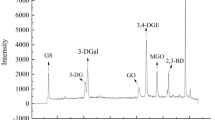
a HPLC-FLD chromatogram of the investigated flavins in standard solution (a) and spiked milk (concentration level 3); b) analysed with the proposed method. b Chromatographic patterns of 440 µg/L standard solution of FAD (flavin adenine dinucleotide) (pH 6.6); c) and the same solution added with 30% trichloroacetic acid (pH 1); d) and corrected for the dilution factor (2.5). Peak identity: 1: FAD, flavin adenine dinucleotide; 2: FMN, flavin mononucleotide; 3: RF, riboflavin; 4: LF, lumiflavin; 5: LC, lumichrome
Previously reported methods for RF quantification in milk usually imply protein precipitation by addition of a strong acid, most often TCA [15, 17, 18], which brings the sample to pH around 1. However, some flavins are reported to be unstable at acidic pH. In particular, FAD in water solution at strongly acidic pH is hydrolyzed into FMN, even at room temperature, and its fluorescence response rises when pH drops from 4 to 2.5, then decreases once more [33,34,35]. The fluorescence response of the target flavins was evaluated by analyzing the standard water solution (concentration level 3) either at the normal pH (6.5 ± 0.1) or after addition of TCA to a final concentration of 12% (pH 1 ± 0.5), as provided by the method of Severo Silva Jr et al. [18] (Table 2). While no differences were observed for RF, LF and LC, fluorescence responses were significantly higher in the acidified solution for both FAD (p = 0.0011) and FMN (p = 0.0021), supporting previously reported results. To understand the reason of these differences, a water solution of FAD (440 µg/L) with and without the addition of TCA was analysed by our chromatographic method (Fig. 1b). Under acidic condition, the analytical response of FAD dramatically increased, despite the correction of the peak area for the dilution factor of 2.5, and a significant amount of FMN formed, but no RF was detected. Contrary to the observation of Severo Silva Jr et al. [18], the addition of 30% TCA did not lead to the complete hydrolysis of FAD to FMN. Capo-chichi et al. [35] reported that FAD in blood is stable in presence of TCA concentration as low as 3%. Such a low concentration, however, is not effective in milk deproteination. Furthermore, injecting a sample having a very low pH has a negative effect on the peak shape. This was evident in the chromatogram of the acidified FAD solution (Fig. 1b) where peaks broadened, making the separation less selective with respect to that of the solution at neutral pH. To overcome the multiple drawbacks of sample acidification, Gliszczynska-Swigło and Koziołowa [3] proposed an extraction procedure using ammonium acetate solution at pH 6, which, however, did not seem to guarantee an effective clarification of the sample such as that required for routine application of the method. Other authors proposed the extraction of vitamins and vitamers using organic solvents to precipitate proteins [19, 23, 25]. However, these methodologies usually imply subsequent steps of evaporation and reconstitution of the extract or, alternatively, the extract has to be diluted with an aqueous buffer. Recently, Koop and et al. [36] adopted the specific binding of flavins to a recombinant bacterial protease as a tool for their selective extraction from milk. However, the authors reported that most flavins (ca. 75%) in milk are unspecifically associated with proteins, including proteins present in the milk fat globule membrane, and thus are not scavenged by the protease, unless a preliminary treatment with TCA is carried out. Considering the above knowledge, we developed a sample preparation procedure based on the ultrafiltration (UF) of milk previously skimmed by centrifugation at 5 °C. The low temperature minimizes the hydrophobic interactions that are responsible for flavins association with milk components [37]. A preliminary assay was carried out to select both the type and the cut off of UF filter (data not shown). Using disposable UF filters with a 10 kDa cut-off membrane, proteins were effectively removed from milk without any modification of pH, which was maintained around 6.6 ± 0.1, while sample manipulation was minimized. The subsequent filtration on 0.22 µm filter allows further clarification of the sample.
Consistently with its hydrophilicity, only 6% of RF is retained in the cream from raw milk and only 2% is bound to fat globule in market milk [37], likely due to the homogenization process. Thus, the preliminary skimming of samples was considered not to affect the RF recovery. Nevertheless, lacking specific literature data at this regard, the possible loss of the other flavins when removing fat was assessed. The same recovery (as peak area) was obtained by analyzing three milk samples that were spiked with the five flavins (levels 1 and 3) before and after skimming (< 5%).
In principle, this analytical approach looked to be suitable to perform the evaluation of the actual levels of RF and the related flavins in milk and milk products since no changes in their natural status are induced during the whole analytical process. The described analytical procedure was further processed for validation.
Validation of the analytical method
The linearity was assessed for each flavin by calculating five-point calibration curves. The obtained r values were all higher than 0.99 (Table 1). The LOD and LOQ values obtained for RF, FAD and FMN were comparable to those obtained by Cataldi et al. [26] using capillary zone electrophoresis and lower than those reported by Capo-chichi et al. [35] and Gliszczynska-Swigło and Rybicka [38] for the respective HPLC-FL methods. Differences in LOD and LOQ observed for both FNM and RF are likely due to the strong pH dependence of fluorescence intensity of RF, FAD and FMN [19, 33, 34]. To the authors’ knowledge, no LOD and LOQ values are reported in the literature for both LC and LF.
The average values of percent recovery were all higher than 80%. Very similar values were obtained by analyzing the standard solutions and milk spiked with the analytes, thus a matrix effect could be excluded (Table 1).
The precision of the method was calculated as RSD of the analytical response (peak area) obtained by analyzing both standard solutions and spiked milk samples. The average RSD values obtained for the five flavins as pure molecules in water solutions ranged between 2.3 and 9.3%, whereas values obtained for spiked milk samples were all lower than 5%, except for FAD (average RSD: 11%) (Table 1). Both the instrumental repeatability (n = 18) and the repeatability of retention times (n = 24), estimated on three different days, were negligible. Overall, these data confirm the reliability of the analytical method here proposed and emphasize the need of avoiding strong acidic conditions in both sample preparation and analysis. Based on these results, we decided that the use of an internal standard could be omitted. Addition of an internal standard solution would inevitably cause an unwanted dilution of the sample.
Overall, these data confirm the reliability of the analytical method here proposed and emphasize the need of avoiding strong acidic conditions in both sample preparation and HPLC analysis.
Riboflavin and its derivatives in commercial samples of liquid milk and milk products
The proposed method was applied for the determination of FAD, FMN, RF, LC and LF in commercial samples of milk of different origin (cow, goat, soy) and submitted to different processing conditions (raw, pasteurized, UHT-sterilized), as well as in selected liquid milk products having different pH value (infant formula vs yogurt and fermented milk) (Table 3). The analysed samples also differed in the type of packaging (clear glass bottle, PET bottle, carton bricks with aluminum foil, HDPE bottle) and package volume (from 125 mL to 1 L). FAD was detected in raw cow’s milk (3.5–7.5 µg/L), but not in the heat-treated milk products, including the infant milk formula, irrespective of the severity of the thermal process they were submitted to. An effect of the high pH value (7.1 ± 0.1) of the infant formula could not be excluded [39]. Consistently, the levels of FAD were much higher in pasteurized goat milk (509–886 µg/L) than in UHT goat milk (41 µg/L). FMN showed an opposite behavior in both types of milk, suggesting a possible heat-induced degradation of FAD into FMN. The literature is very scarce on the thermal stability of FAD and FMN. Cataldi et al. [26] also observed a decrease of FAD and an increase of FMN in UHT milk with respect to fresh milk, although of minor entity, but gave no explanation for these data. Therefore, this aspect needs to be further investigated. All samples contained RF as the dominant flavin compound, regardless of the thermal treatment and type of packaging. Levels in raw milk (2.62–3.20 mg/L) were significantly higher than in pasteurized milk samples (1.87–1.99 mg/L; p = 0.001) and in UHT milk (2.53–2.69 mg/L; p = 0.046), while RF level did not result significantly different between pasteurized and UHT milk (p = 0.239). Comparing these data may be incorrect since only few milk samples of different origins were analysed. Consistently with our results, however, Cataldi et al. [26], Schmidt et al. [40] and Sunaric et al. [41] reported higher levels of RF in UHT milk than in less severely heated milk. Considering that RF is relatively heat stable [7], interactions described between flavins and milk components [36, 37] suggest that technological parameters other than the heating conditions can lead to the differences between RF levels in pasteurized and UHT milk. Levels comparable to those reported in the literature were observed in the samples of soymilk (3.34 mg/L) [42] and infant formula (3.75 mg/L), the latter being fortified with RF. Lowest levels of RF were found in heat-treated goat milk (1.16–1.41 mg/L), in accordance to data of Cataldi et al. [26] and Sunaric et al. [41]. Flavin levels were highly variable in the fermented milk products including yogurt. This situation is supported by the ability of lactic acid bacteria to produce and uptake flavins at the same time [43]. It must be underlined that, as expected, these products had low pH values (Table 3). Such an acidic environment might have promoted the partial conversion of FAD into FMN during either the fermentation or processing steps. A further peak was detected at 11 min in the chromatograms of these samples (Fig. 2). The UPLC-MS/MS analysis showed this compound to be a glycosidic form of RF. Other authors [3] previously reported the presence of the galactoside form of RF, a product of the bacterial metabolism of specific strains in yogurt and in sour milk. Capo-chichi et al. [35] proposed galactosyl-RF to be used as an internal standard in HPLC-FL of flavins in blood.

HPLC-FLD chromatogram of the investigated flavins in milk exposed to light for 76 h (a), fermented milk 1 (b), and soymilk (c) analysed with the proposed method. Peak identity: 1: FAD, flavin adenine dinucleotide; 2: FMN, flavin mononucleotide; 3: glycoside-RF; 4: RF, riboflavin; 5: LF, lumiflavin; 6: LC, lumichrome
Both LC and LF are products of photoreduction (side-chain cleavage) of RF. Lumichrome was detected in all of the samples packaged in clear packages, including raw and pasteurized milk samples (2.88–6.68 µg/L) and yogurt (13.7 µg/L). Likely, these samples were exposed to light for a certain time, either during collection (samples Milk 1–4) or during storage on the shelves at the market (samples Milk 5–8). The lack of LC in all samples packaged in light-protecting containers supports this hypothesis. No LF was detected in any of the analysed samples, consistently with the knowledge that this compound typically forms at neutral and alkaline pH [2]. To further confirm the reliability of the analytical method here proposed for detecting the products of RF photodegradation in milk, bottles of pasteurized milk were directly taken at the filling plant and protected from light until the start of the experiment. A controlled exposure to fluorescent light for 76 h induced a sharp decrease of both RF and FMN, while LC increased up to 92 µg/L (Table 4). Remarkably, changes in contents of these molecules were already significant (p < 0.05) after 5 h of exposure. Amounts up to 1.2 µg/L of LF were only observed when light exposure progressed, confirming that this compound is a minor product of RF photodegradation. Hall et al. [16] reported that RF was no more detectable in milk after 9 h of exposure to light. However, surface exposure to the light and sample thickness were not comparable to ours, since exposure trials were conducted on 10 mL aliquots of milk in glass vials.
Despite the increasing interest in this topic, available literature is still scarce. Overall, photodegradation of RF may follow different pathways whose mechanisms and kinetics, however, were mainly investigated in aqueous solutions [2, 44].
Conclusions
Riboflavin and the related flavins are unstable under acidic conditions and their fluorescence response is strongly pH dependent. The most important feature of the analytical method here developed and validated is that no acidic conditions were adopted in both sample preparation and chromatographic separation. Therefore, the actual levels of RF and its derivatives can be effectively evaluated allowing to study their behavior in liquid milk and milk products upon processing or storage. After preliminary skimming, the sample is clarified by centrifugal UF with disposable filters that allowed accuracy levels higher than 80% to be achieved for all the compounds of interest. The subsequent HPLC conditions were also designed to avoid using aggressive eluents and, for the first time, to elute LF and LC in the same run as RF, FAD and FMN. We have reported the applicability of this methodology to real samples of milk and milk products also supporting its suitability for routine application. Furthermore, our study revealed that this method is very effective for studying the degradation of RF into LF and LC in light-exposed milk and the related appearance of the sunlight off-flavor. Our data, although at preliminary level, indicated that the type of packaging has a deeper impact on RF preservation in milk than the heat-treatment itself. This aspect is of increasing interest for both the producers and consumers due to its impact on the shelf storage stability of consumption milk.
References
Ball GFM (2004) Flavins: riboflavin, FMN and FAD (vitamin B2). In: Ball GFM (ed) Vitamins: their role in the human body. Blackwell Publishing Ltd, Oxford
Sheraz MA, Kazi SH, Ahmed S, Anwar Z, Ahmad I (2014) Photo, thermal and chemical degradation of riboflavin. Beilstein J Org Chem 10:1999–2012
Gliszczynska-Swigło A, Koziołowa A (2000) Chromatographic determination of riboflavin and its derivatives in food. J Chromatogr A 881:285–297
Monteiro MC, Perrone D (2013) Chemistry and biochemistry of riboflavin and related compounds. In: Preedy VR (ed) B vitamins and folate chemistry, analysis, function and effects. RCS Publishing, Cambridge
Depeint F, Bruce WR, Shangari N, Mehta R, O’Brien PJ (2006) Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem Biol Interact 163:94–112
Nollet LML, Toldrá F (2013) Water soluble vitamins. In: Nollet LML (ed) Food analysis by HPLC. Taylor & Francis Group, New York
Golbach JL, Ricke SC, O’Bryan CA, Crandall PG (2014) Riboflavin in nutrition, food processing and analysis—a review. J Food Res 3:23–35
Bitsch R, Bitsch I (2011) HPLC determination of riboflavin in fortified foods. In: Rychlik M (ed) Fortified foods with vitamins: analytical concepts to assure better and safer products. Wiley, New York
Choe E, Huang R, Min DB (2005) Chemical reactions and stability of riboflavin in foods. J Food Sci 70:R28-R36
Cardoso DR, Libardi SH, Skibsted LH (2012) Riboflavin as a photosensitizer. Effect on human health and food quality. Food Funct 3:487–502
Jung MY, Yooh SH, Lee HO, Min DB (1998) Single oxygen and ascorbic acid effects on dimethyl sulfide and off-flavour in skim milk exposed to light. J Food Sci 63:408–412
Lee JH, Min DB (2009) Changes of headspace volatiles in milk with riboflavin photosensitization. J Food Sci 74:C563-C568
Shuping W, Zhiqin J, Heting L, Li Y, Daixun Z (2001) Sensitized photooxygenation of cholesterol and pseudocholesterol derivatives via singlet oxygen. Molecules 6:52–60
Solah VA, Staines V, Honda S, Limley HA (2007) Measurement of milk color and composition: effect of dietary intervention on western Australian holstein-friesian cow’s milk quality. J Food Sci 72:S560-S566
Albalá-Hurtado S, Veciana-Nogués MT, Izquierdo-Pulido M, Mariné-Font A (1997) Determination of water-soluble vitamins in infant milk by high-performance liquid chromatography. J Chromatogr A 778:247–253
Hall NK, Chapman TM, Jung Kim H, Min DB (2010) Antioxidant mechanisms of Trolox and ascorbic acid on the oxidation of riboflavin in milk under light. Food Chem 118:534–539
Gatti R, Gioia MG (2005) Liquid chromatographic determination with fluorescence detection of B6 vitamers and riboflavin in milk and pharmaceuticals. Anal Chim Acta 538:135–141
Severo Silva L Jr, Trevisan MG, Rath S, Poppi RJ, Feyes FGR (2005) Chromatographic determination of riboflavin in the presence of tetracyclines in skimmed and full cream milk using fluorescence detection. J Braz Chem Soc 16:1174–1178
Viñas P, Balsalobre N, López-Erroz C, Hernández-Córdoba M (2004) Liquid chromatographic analysis of riboflavin vitamers in foods using fluorescence detection. J Agric Food Chem 52:1789–1794
Russell LF, Vanderslice JT (1992) Comments on the standard fluorometric determination of riboflavin in foods and biological tissues. Food Chem 43:79–82
Gentili A, Caretti F, D’Ascenzo G, Marchese S, Perret D, Di Corcia D, Mainero Rocca L (2008) Simultaneous determination of water-soluble vitamins in selected food matrices by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom 22:2029–2043
Phillips MM (2015) Liquid chromatography with isotope-dilution mass spectrometry for determination of water-soluble vitamins in foods. Anal Bioanal Chem 407:2965–2974
Cellar AN, McClure SC, Salvati ML, Reddy TM (2016) A new sample preparation and separation combination for precise, accurate, rapid, and simultaneous determination of vitamins B1, B2, B3, B5, B6, B7, and B9 in infant formula and related nutritionals by LC–MS/MS. Anal Chim Acta 934:180–185
Hampel D, Allen LH (2016) Analyzing B-vitamins in human milk: methodological approaches. Crit Rev Food Sci Nutr 56:494–511
Redeuil K, Bénet S, Affolter M, Thakkar SK, Campos-Giménez E (2017) A novel methodology for the quantification of B-vitamers in breast milk. J Anal Bioanal Tech 8:1000352
Cataldi TRI, Nardiello D, De Benedetto GE, Bufo SA (2002) Optimizing separation conditions for riboflavin, flavin mononucleotide and flavin adenine dinucleotide on capillary zone electrophoresis with laser-induced fluorescence detection. J Chromatogr A 968:229–239
Webster JB, Duncan SE, Marcy JE, O’Keefe SF (2009) Controlling light oxidation flavor in milk by blocking riboflavin excitation wavelengths by interference. J Food Sci 74:S390-S398
Osório MV, Marques SS, Oliveira HM, Barreiros L, Segundo MA (2016) Fluorometric method based on molecular recognition solid-phase extraction for determination of riboflavin in milk and infant formula. J Food Compos Anal 45:141–146
Wold JP, Skaret J, Dalsgaard TK (2015) Assessment of the action spectrum for photoxidation in full fat bovine milk. Food Chem 179:68–75
Zandomeneghi M, Carbonaro L, Zandomeneghi G (2007) Biochemical fluorimetric method for the determination of riboflavin in milk. J Agric Food Chem 55:5990–5994
Drössler P, Holzer W, Penzkofer A, Hegemann P (2002) pH dependence of the absorption and emission behavior of riboflavin in aqueous solution. Chem Phys 282:429–439
Magnusson B, Örnemark U (2014) Eurachem guide: the fitness for purpose of analytical methods—a laboratory guide to method validation and related topics. ISBN 978-91-87461-59-0http://www.eurachem.org. Accessed 21 Jan 2018
Andrés-Lacueva C, Mattivi F, Tonon D (1998) Determination of riboflavin, flavin mononucleotide and flavin adenine dinucleotide in wine and other beverages by high-performance liquid chromatography with fluorescence detection. J Chromatogr A 823:355–363
Islam MS, Honma M, Nakabayashi T, Kinjo M, Ohta N (2013) pH dependence of the fluorescence lifetime of FAD in solution and in cells. Int J Mol Sci 14:1952–1963
Capo-chichi CD, Guéant JL, Feillet F, Namour F, Vidailhet M (2000) Analysis of riboflavin and riboflavin cofactor levels in plasma by high-performance liquid chromatography. J Chromatogr B 739:219–224
Koop J, Monschein S, Macheroux EP, Knaus T, Macheroux P (2014) Determination of free and bound riboflavin in cow’s milk using a novel flavin-binding protein. Food Chem 146:94–97
Kanno C, Kanehara N, Shirafuji K, Tanji R, Imai T (1991) Binding form of vitamin B2 in bovine milk: its concentration, distribution and binding linkage. J Nutr Sci Vitaminol 37:15–27
Gliszczynska-Swigło A, Rybicka I (2015) Simultaneous determination of caffeine and water-soluble vitamins in energy drinks by HPLC with photodiode array and fluorescence detection. Food Anal Method 8:139–146
Cattaneo S, Masotti F, Pellegrino L (2009) Liquid infant formulas: technological tools for limiting heat damage. J Agric Food Chem 57:10689–10694
Schmidt A, Schreiner MG, Mayer HK (2017) Rapid determination of the various native forms of vitamin B6 and B2 in cow’s milk using ultra-high performance liquid chromatography. J Chromatogr A 1500:89–95
Sunaric S, Denic M, Kocic G (2012) Evaluation of riboflavin content in dairy products and non-dairy substitutes. Ital J Food Sci 24:352–357
Huang R, Kim HJ, Min DB (2006) Photosensitizing effect of riboflavin, lumiflavin, and lumichrome on the generation of volatiles in soy milk. J Agric Food Chem 54:2359–2364
LeBlanc JG, Laiño JE, del Valle MJ, Vannini V, van Sinderen D, Taranto MP, de Valdez GF, de Giori GS, Sesma F (2011) B-Group vitamin production by lactic acid bacteria—current knowledge and potential applications. J Appl Microbiol 111:1297–1309
Fracassetti D, Gabrielli M, Encinas J, Manara M, Pellegrino L, Tirelli A (2017) Approaches to prevent light-struck taste in white wine. Aust J Grape Wine Res 23:329–333
Acknowledgements
We are grateful to Dr. Claudio Gardana from our Department for performing the UPLC MS/MS analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest to this work.
Ethics requirements
This article does not contain any studies with human or animal subjects.
Rights and permissions
About this article

Cite this article
Fracassetti, D., Limbo, S., D’Incecco, P. et al. Development of a HPLC method for the simultaneous analysis of riboflavin and other flavin compounds in liquid milk and milk products. Eur Food Res Technol 244, 1545–1554 (2018). https://doi.org/10.1007/s00217-018-3068-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-018-3068-6