
Abstract
The aims of this research were to systematically study how extraction, desorption and gas chromatography (GC) parameters affect the volatiles composition of mushrooms in headspace solid-phase microextraction (HS-SPME) analysis. The study was carried out both with reference compounds and with Cantharellus cibarius Fr. mushroom sample. The experiments were carried out with full-factorial multivariate designs. In the desorption studies, high fiber depth in the GC injector port instead of desorption time or temperature was the most important variable for maximizing peak areas. This could be a function related to the temperature gradient inside the injector as well as a faster transfer of volatiles to the column. Out of the tested fibers, divinylbenzene/carboxen/polydimethylsiloxane (DVB/Car/PDMS) was the most suitable coating for mushroom volatiles. Additionally, extraction time of 45 vs 30 min had a high effect, while doubling the sample volume had a minimal effect. 21 volatiles were identified in C. cibarius. Hexanal and 1-octen-3-ol were the most abundant volatile compounds. Overall, aldehydes and unsaturated C6–C10 alcohols and aldehydes were the most abundant compound groups. This study demonstrated that despite the wide adaptation and history of SPME, fiber injection depth, fiber coating and extraction time are important factors that should still be carefully tested as a part of method development for mushroom-type matrices. Finally, the results of this study show that it is important to look at the extracted gas chromatogram as a whole instead of focusing on a few peaks of interest.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The aroma of food is critical in appraising its sensory properties, and along with visual cues, orthonasal aroma is the first information a consumer receives from food. Aroma is composed of a set of volatile compounds. For them to have an impact on the overall aroma, they have to be present above their threshold concentrations for humans in the food headspace. Finally, the specific proportions of each aroma-contributing volatile compound in the sample headspace define the final impression perceived by chemical senses such as sense of smell [1].
Traditionally, the analysis of mushroom aroma compounds has focused on isolating the volatile compounds with different steam distillation [2] and solvent extraction [3] protocols, and their identification and quantitation with gas chromatography–mass spectrometric methods.
However, these solvent extracts do not fully represent the samples as consumers perceive food on a plate. Extracts contain compounds of highly differing volatility, which means that analyzing the extract gives an answer to a different research question regarding aroma than studying the headspace profile of a sample.
On the other hand, headspace measurements by themselves, while demonstrating the aroma profile as the consumer perceives it, have an inherent challenge: concentrating the sample is difficult. Thus, the sensitivity of headspace measurements is often too low for proper identification of sample analytes. Headspace solid-phase microextraction (HS-SPME) has become an established method over the 30 years since its conception [4], and circumvents many of these limitations. It unifies the sample preparation steps of extraction and concentration into one convenient process. However, it should be remembered that the extracted headspace profile with SPME is never fully equitable to the actual headspace profile and is more of an impression created by the used fiber coating [5]. Furthermore, SPME is an equilibrium method and as such, several variables affect the extracted profile. These include—aside from the selection of fiber coating—extraction time, headspace volume, and desorption conditions [6]. Thus, it is important to optimize the SPME method so that the resulting extract is as close to the actual headspace composition as possible.
Nordic forests are home to a wide selection of wild edible mushroom species, one of the most popular species among them being Cantharellus cibarius Fr. The last in-depth analysis on their volatile compound composition was done 40 years ago, and with solvent extraction-based methods [2]. Some of the wild mushroom species as well as the cultivated white button mushroom, Agaricus bisporus (J.E. Lange) Imbach, have been analyzed in later years using HS-SPME [7,8,9,10]. However, the species that have been analyzed in previous research and are found in Nordic forests are from locations that have very different climate and lighting conditions compared to Nordic forests, and it is reasonable to assume that this has an effect on the volatiles composition. Furthermore, previous HS-SPME studies on mushrooms left out some important parameters like the liner type and fiber desorption depth, and arrived in opposite conclusions especially regarding the choice of fiber [7, 8, 11]. Finally, the studies on interaction of different parameters has been minimal, as only one article on mushroom volatiles used full-factorial composite designs [11], and even recent publications using HS-SPME on other matrices have mainly utilized univariate study designs [12].
Thus, there was a need to explore which parameters would yield the optimum signal for Finnish mushroom species. The aims of this research were to systematically study how extraction, desorption and gas chromatography parameters affect the volatiles composition of mushrooms in headspace solid-phase microextraction (HS-SPME) analysis. This was done to assess (1) which desorption parameters have a systematic effect on peak shapes, (2) if there are major interactions between mushroom volatile compounds and SPME fiber coatings, and (3) whether the column stationary phase has a stronger impact on resolution than pre-columns or focusing effects.
Materials and methods
Research workflow
The research was carried out in four stages. The rationale behind this workflow was to split the analysis in independent stages so that interacting variables and thus the number of samples would be minimized. First, desorption conditions were optimized. Then, the resolving power of two similar columns differing mainly in their film thickness, with or without a preceding retention gap was investigated. After deciding the appropriate column system, the oven program was further refined. Finally, the effects of different variables in the volatiles extraction were studied.
Samples
Standard mixture for desorption studies
Desorption parameters were studied with a reference compound mixture containing seven compounds. 1-octen-3-ol, eugenol and nonanal were bought from Aldrich (St. Louis, MO). 1-hexanol, methional and (R)-(+)-limonene were bought from Sigma (St. Louis, MO). P-xylene was bought from Fluka Chemie AG (Buchs, Switzerland). Benzaldehyde was brought from BDH Chemicals (Poole, UK). All reagents were of ≥ 95% purity. The compounds were selected by consulting earlier literature of relevant mushroom volatiles studies [2, 8, 9, 13], as well as preliminary analyses of mushroom samples. Selected compounds also spanned the total chromatographic retention index range on the selected column and represented different compound groups (alcohols, aldehydes, terpenoids, and aromatic hydrocarbons). A master solution of 600 µg/ml concentration for each compound in hexane was made and stored in − 20 °C. A fresh 18 µg/ml dilution was prepared at the start of each day from the master solution. This concentration resulted in comparable peak areas with mushroom samples.
For analysis, 3.0 µl of the desorption solution was pipetted in a 5-ml glass vial equipped with a septum cap. The headspace was equilibrated for 10 min in ambient temperature and pressure, after which a DVB/CAR/PDMS fiber was placed in the middle of the vial, exposed to the headspace and the compounds were extracted for 15 min. Finally, the fiber was exposed in the injection port with the desorption conditions dictated by the experimental design.
Alkane standard
The C5–C20 alkane standard (ASTM-P-0050) from Accustandard (New Haven, CT) was used for building the linear retention index. HPLC-grade hexane was used as the solvent. A 50 µg/ml solution of the alkane standard was created, and the sample was extracted as described above for the desorption mixture.
Additional reference compounds
Pentanal, 1-octene, (E)-2-heptenal, 3-octanone, 2-pentylfuran, (E)-2-nonenal, decanal and (E,E)-2,4-decadienal were bought from Sigma Aldrich (St. Louis, MO) and used for identifications in the mushroom sample.
Mushroom sample
Chanterelle (C. cibarius) was used during the selection of column, optimization of oven programs as well as during assessment of SPME extraction parameters. The chanterelle sample was collected in Pargas, Finland (60.30°N, 22.30°E), in September 2015. After arriving to the laboratory within 24 h after collection, each mushroom was cut to 3–5 pieces of similar size, and blanched in boiling water for 2 min. After cooling down in ambient temperature, the mushrooms were frozen at − 20 °C.
Before the main analysis, the sample lot was cut, pooled and divided into aliquots. In short, 500 g of frozen sample material was cut into 2–7 mm cubes. All materials were combined, mixed, spread over a tray and divided into either 10.0 g or 20.0 g sections using the cone quartering method. The individual samples were packaged in re-sealable plastic bags. After this, the samples were stored at − 20 °C for 3 months, and analyzed in 2 weeks.
SPME fibers
Three types of 24 gauge, 1 cm SPME fibers from Supelco (Bellefonte, PA) were used: 50/30 µm divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS), 75 μm carboxen/polydimethylsiloxane (CAR/PDMS), and 65 μm polydimethylsiloxane/divinylbenzene (PDMS/DVB) coatings. The selected fibers were typical alternatives used in previous SPME studies on fungal volatiles [7,8,9, 11, 14] as well as recent studies on other matrices using SPME [12, 15, 16]. Each fiber was thermally conditioned in accordance to the manufacturer’s specifications before use.
Instrumentation
The study was conducted with two gas chromatography instruments, both equipped with flame ionization detectors. The first and second experiment on desorption parameters were done with a Shimadzu GC-2010 Plus gas chromatograph (Shimadzu Corporation, Kyoto, Japan). Other studies were done with an HP-6890 series gas chromatograph (Agilent Technologies, Santa Clara, CA). Both the GC-2010 and HP-6890 had split/splitless injectors, flame ionization detectors and were equipped with 0.75 mm ID SPME liners from Shimadzu and Restek Corporation (Bellefonte, PA), respectively.
The analytical column for the desorption parameters was a 30 m × 0.25 mm × 0.50 µm DB-5 capillary column (Agilent J&W, Santa Clara, CA). In the column selection stage, a 30 m × 0.25 mm × 1.00 µm RTX-5 Sil MS column by Restek Corporation (Bellefonte, PA) was added to comparison. Additionally, the effect of adding a 3 m × 0.25 mm uncoated fused silica retention gap from Supelco (Bellefonte, PA) was studied. The retention gap was installed in front of the analytical column with a capillary column butt connector from Supelco (Bellefonte, PA).
The oven temperature was optimized in multiple stages following previous publications [3, 9], and will be outlined in the relevant sections. The carrier gas was helium, with a linear flow of 40 cm/s for desorption studies and 33 cm/s for the rest of the experiments. The linear flow velocity was verified with the retention time of injected propane gas.
Experiments
Desorption studies
Different levels of desorption parameters (desorption time, injector temperature and fiber depth in the injector) were compared in three sample sets (Table 1), each using the same freshly diluted reference standard mixture as a sample. The experimental design for each experiment was a full-factorial design run in randomized duplicate. The first experiment contained also three center points for standard deviation estimation.
During desorption studies, the following oven program was used: 40 °C held for 5 min, increased at 6 °C/min to 170 °C, and held at final temperature for 5 min, resulting in an approximately 27 min analysis time. Desorption time corresponded to the length of the splitless time. After this, the fiber was removed from the injector and a 20:1 split ratio was used.
Column selection and oven program
The column comparison and oven program development aimed to make peaks narrower, to improve peak symmetry and to improve resolution especially in the crowded retention index range 950–1050. Thus, both columns with or without the retention gap (thus four combinations) were tested with three progressively complex oven programs with the chanterelle sample.
Both tested columns have phenyl substituted polysiloxane stationary phases and thus are comparable in terms of polarity and other resolving characteristics. The used phase has been typical in mushroom volatiles analysis [3, 7, 8, 13]. The columns are also of identical length and inner diameter. The hypothesis was as discussed in the literature [6] that using a thick-film stationary phase column as well as a retention gap would enhance sample focusing.
After the column selection, the oven program was further refined with the mushroom sample. The final oven program was the following: 35 °C held for 5 min, increased at 5 °C/min to 90 °C, then at 2 °C/min to 100 °C, at 7 °C/min to 260 °C and held at this final temperature for 2 min.
Extraction parameters
Three extraction parameters were optimized: fiber coating (three types, stated above), sample mass (two levels, 10 g and 20 g) and extraction time (2 levels, 30 min and 45 min). The sample mass levels were based on pilot studies as well as published literature [8, 9] and extrapolated to the used extraction vial volume. Extraction time points were likewise based on published literature [11]. The experimental design was a full-factorial model run in duplicate, resulting in 24 samples. The run order was randomized both for the parameter combinations and the used sample aliquots.
Each sample aliquot was weighed straight from − 20 °C storage to a 90-ml glass vial. Equilibrium time was 30 min. Equilibrium and extraction temperature was each time 30 °C and controlled by a water bath surrounding the extraction flask. After SPME extraction, the volatiles were desorbed from the fiber using the best parameters found during desorption studies.
Peak characteristics criteria
Throughout the study, the following four peak criteria were set as desirable: (1) the highest possible areas; (2) highest possible signal-to-noise ratios; (3) lowest possible full widths at half height, and (4) as symmetrical peaks as possible, i.e., tailing factors as close to 1 as possible.
Furthermore, the fiber that achieved these with the widest representation of compounds over the whole retention index range was selected as ideal.
Identification
The compounds were identified with reference compounds, by comparing the retention indices to literature data on the NIST Chemistry WebBook [17], and by gas chromatography–mass spectrometry analysis. For GC–MS analysis, a GC of the same HP 6890 model as in the main studies was attached to the HP 5973 mass sensitive detector. All mass spectra were acquired in the electron impact (EI) mode. The mass ranged from 30 to 700 m/z, with a scan rate of 2.23 scans/s. The interface temperature was set to 150 °C, and other GC analytical conditions were as described above for the extraction parameter study. The MS peaks were identified with the Wiley 275 mass spectral library as well as with the in-house mass library created from reference compounds.
Statistical analysis
Gas chromatography (GC) chromatograms were integrated and the following key parameters were extracted from the data: peak areas, signal-to-noise ratios, full widths at half height, and tailing factors.
Initial evaluation of the effects of desorption parameters on peak areas of example compounds 1-octen-3-ol and nonanal were done with linear regression. The effect of extraction parameters on the total area of peaks was examined with two-way analysis of variance (ANOVA). These analyses were done using SPSS version 23 (SPSS Inc., Chicago, IL). Limit for statistical significance was 0.05.
As unsupervised data analysis, principal component analysis (PCA) was done for desorption studies and extraction parameter datasets with autoscaled data. After discarding variables with minimal variation and strongly collinear peak parameters, the PCA was refined. After this, partial least squares regression discriminant analysis (PLS-DA) was done with either desorption time, temperature and fiber depth (desorption studies), or fiber type, sample mass and extraction time (extraction studies) as Y-variables and peak parameters for each compound as X-variables. All data was autoscaled. PCA and PLS-DA models were created with The Unscrambler X version 10.4 (CAMO Software AS, Oslo, Norway).
Results and discussion
Desorption conditions
First sample set
Surprisingly, in the linear regression of 1-octen-3-ol and nonanal, fiber depth alone explained 70 and 80% of the variance of the peak areas, respectively. Furthermore, temperature and desorption time were not statistically significant regression predictors for 1-octen-3-ol area, and even for nonanal, fiber depth had an order of magnitude higher coefficient in the regression model than the other two parameters. This is also visualized in the PCA model (Supplementary information, Figure S1): all peak areas as well as fiber depth have a high negative loading on the first principal component.
The effect is further elucidated on the PLS model (Fig. 1), in which first three factors explain 83% of Y-variable variation and 84% of X-variable variation. Desorption parameters had a minimal effect on peak widths as the relative standard deviation on the first dataset was < 3% for all compounds, so peak widths were left out of the displayed model. The peak areas are closely associated with fiber depth, pointing out that increased fiber depth is optimal. Interestingly, longer desorption time seems to correlate with lesser peak tailing. Likewise, higher temperatures correlated with tailing of some compounds. However, looking at data in which Y-variable categories have been split (data not shown), it can be seen that it is mostly the lowest 2-min desorption time which is associated with tailing peaks.

PLS plot of factors 1 and 2 (A) and 1 and 3 (B) the first desorption study. This model includes peak areas and tailing factors which were found to account for the largest data variance in the PCA model
Second sample set
In the PLS model (Fig. 2) the first two factors explain over 90% of variation in the desorption variables and 65% of variation in peak parameters. Most peak areas are associated with higher temperatures and correlate inversely with peak tailing. Additionally, the widths of four of the peaks are associated with longer desorption times. This points to the 240 °C injector temperature and a 3-min desorption time being optimal values.

PLS model of the second desorption study. This model includes peak areas, full widths at half height and tailing factors
Third sample set
In the PLS model (Fig. 3), the first two factors explain 75% of Y-variable and 69% of peak parameter (X-variable) variation. Factor 1 is still associated with all peak areas, with both fiber depth and peak areas having high positive loadings. This reinforces the fact that for fast desorption of analytes, maximal fiber depth is ideal. Factor 2 is associated with desorption time. However, there is no systematic link to peak characteristics; for four compounds, longer desorption time associates with wider peaks, while it is the opposite for the other three compounds. Interestingly, longer desorption time seems to be linked to less tailing peaks on most compounds.

PLS correlations loadings plot of the third desorption study. This model includes peak areas, full widths at half height and tailing factors
The three sample sets thus show that out of the tested variables, optimal desorption parameters were maximal (60 mm) fiber depth in the holder, 240 °C injector temperature, and 3 min desorption time.
Out of these variables, using a higher desorption temperature was as recommended in the literature [6], but the strong effect of fiber depth was surprising. The peak area increase could be caused by faster access of the volatiles to the beginning of the column. As there is no need to vaporize any solvents, the main role of the liner is to ensure a high linear velocity and thus fast desorption of the analytes to the column [18]. The benefits of narrow-bore glass inserts as used in this study have been demonstrated well [19], with still achieving narrower peaks by using increased flow rates during the desorption. In future studies, this phenomenon could be utilized by using the pulsed splitless injection mode for the desorption period. It could be postulated that setting the fiber closer to the column helps with peak focusing: there is less time that the desorbed compounds spend in the liner, and thus they will be focused rapidly at the beginning of a cool column. This kind of effect, paired with the use of a thick-film column, was also demonstrated by Langenfeld et al. [18]. Ouyang et al. [20] have reported an analogous phenomenon with aromatic hydrocarbons: sample transfer is more efficient when the GC column penetrates deeper in the liner and thus is closer to the SPME fiber during desorption.
In addition to the mechanism postulated above, SPME guidebooks recommend setting the desorption depth to correspond with the hottest part of the injector [6], which typically means the center part of the liner. The 95-mm liner in the Shimadzu instrument causes the SPME fiber to reside in the upper third of the liner, even with the highest depth setting. However, in the shorter HP/Agilent 78.5 mm liner, the fiber is almost at the center point with the maximum setting.
Finally, it should be mentioned that the effect size is relatively small. Changing the fiber depth only caused a ~ 10% difference in peak areas compared to the general average. As will be pointed out, extraction parameters had a much higher effect on peak parameters.
Column and oven program
The thicker 1.0-µm film in the RTX-5 Sil MS column was clearly beneficial. This is also recommended in literature [6]. This column resolved several convoluted peaks compared to the DB-5 and in general the peaks were also slightly more symmetrical (less fronting and tailing).
In comparison, the retention gap had a minor effect. As a benefit, it did allow a partial, but not baseline-separated resolution of extra peaks at retention time < 5 min when combined with the RTX-5 Sil MS column. Additionally, the early eluting compounds had up to 10% narrower peak widths at half height; however, this trend was reversed at the end of the chromatogram. There was neither a systematic effect to signal-to-noise ratios or peak symmetry. Thus, the retention gap had no marked benefits to the analysis. Selection of an appropriate column and optimization of the oven program yielded larger improvements in resolution. Finally, as creating a column-to-column connection can cause dead volume and leaks in the analysis system, it was concluded that a retention gap with the tested specifications is not needed. In fact, the effect of the 3-m retention gap was comparable to lowering the linear velocity from 40 cm/s to 33 cm/s. With this change, early eluting compounds had up to 20% higher resolution. Peak areas stayed the same, but early eluting compounds had ~ 10% lower S/N and late-eluting compounds up to 20% higher S/N.
In the oven program, the largest gains in resolution could be attained by slowing down heating in the 80–120 °C temperature range as much as possible. The final program is a good compromise between maintaining small peak widths and resolving peaks over the whole retention index range. The largest challenge in using polysiloxane columns for mushroom volatiles analysis seems to be that the area surrounding 1-octen-3-ol is very challenging to make fully baseline separated for all compounds.
Extraction parameters
21 identified peaks were integrated (Table 2). The retention times were stable across all identified compounds, with standard deviations on peak retention times being 0.3–0.7 s depending on the compound.
As previously reported [7, 8, 11], different SPME fibers had very different extraction profiles (Figures S2–3). The least suited coating for an overall mushroom profile was carboxen/PDMS. This fiber had very high adsorption for peaks until hexanal, in other words in the retention index range 300–800, but progressively diminishing adsorption for further compounds. Furthermore, these peaks were 10–30% wider compared to the average width at half height of 2.9 s on the other two fibers. This held true even for peak areas that were smaller compared to other fibers, and they were almost systematically tailing. In fact, the separation for most peaks was too poor to even calculate tailing factors. Therefore, carboxen/PDMS fiber was left out from further comparison. In general, DVB/Car/PDMS seemed to contain benefits from both fiber coatings and followed the peak average most closely.
Two-way ANOVA on the total area of peaks shows that only fiber type and extraction time are statistically significant variables (p < 0.01). Neither sample volume nor any of the interaction terms were statistically significant. Furthermore, a model with these two main effects already explained 80% of the variation in total peak areas. Demonstrating univariate differences, two-way ANOVA was done also for hexanal and 1-octen-3-ol as an example. As with the sum of peak areas, only fiber type and extraction time are statistically significant (p < 0.05) variables. DVB/Car/PDMS fiber had 24% and 18% bigger peaks, increasing the extraction time from 30 to 45 min increased peak areas by 28 and 36%, but doubling the sample mass actually resulted in lower signal areas. In fact, the extraction time was the only extraction variable where one factor level resulted in consistently higher peak areas. This kind of univariate comparison, however, leaves out essential information on compound-level variations.
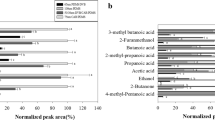
There is also clear separation between the DVB/PDMS and the DVB/Car/PDMS fibers in multivariate models. In Fig. 4, it is shown that factor 1 is mostly associated with extraction time, while factor 2 associates mainly with fiber type. Factor 3, on the other hand, associates with sample volume. The first three factors explain 88% of the variation in peak areas and 78% of variation in extraction parameters.

PLS correlation loadings plots of the variables in the extraction parameter experiment using peak areas. a Factors 1 and 2, b factors 1 and 3
While peak area was more closely correlated with DVB/Car/PDMS fibers, samples run with that fiber had slightly wider peaks (data not included in the model). However, the relative standard deviation for peak widths in the whole dataset was ≤ 10%, meaning that on an average peak width at half height being less than 3 s, this compromise is of minimal importance.
However, it should be remembered that comparison of absolute peak areas and thus concentrations of volatile compounds is not alone a sufficient base for studying the aroma profile of mushrooms. Different volatile compounds have major differences in their odor thresholds. Comparing literature values [21] to compounds identified in the C. cibarius samples, extreme cases would be benzaldehyde and (E,E)-2,4-decadienal, where the former has an over three magnitudes higher detection threshold than the latter. However, the peak of benzaldehyde is only four times that of (E,E)-2,4-decadienal. Thus, optimizing the extraction efficiency of a seemingly large peak might not influence the actual aroma profile at all, while even minuscule gains to small peaks with potent odor intensity are important. In this study, the carboxen/divinylbenzene/polydimethylsiloxane and the longer, 45 min extraction time were associated with higher peak areas of almost all identified compounds, meaning that for unsupervised analysis of odor-contributing volatile compounds, these parameters are the best option.
Based on these results, selecting the carboxen/divinylbenzene/polydimethylsiloxane fiber is appropriate for mushrooms. Furthermore, major gains could be attained by using the longer 45 min extraction time. Additionally, keeping the sample amount at 10 g does not cause a significant loss in sensitivity. This observation is similar to what was noticed on HS-SPME on virgin olive oils [12].
Volatile compounds in chanterelle
The relative content of identified volatile compounds in chanterelles is displayed in Table 2. Hexanal and 1-octen-3-ol were the most abundant compounds and other compounds were present at an order of magnitude smaller concentrations. Overall, aldehydes and unsaturated C6–C10 alcohols and aldehydes were the most abundant compound groups. Dunkel et al. [1] reviewed the precursors of 226 key food odorants, and according to the review, the precursors of these C6–C10 compounds are fatty acids. Politowicz et al. [10] recently studied the effect of drying conditions on the volatiles in C. cibarius and found that 1-octen-3-ol, 1-hexanol and hexanal were the most abundant volatile compounds in freeze-dried chanterelles. De Pinho et al. [8] reported earlier that main compounds in their chanterelle samples were 1-octen-3-ol, 1-octen-3-one and 1-hexanol. Comparing these published results to ours shows that the samples in this study had a significantly higher content of hexanal. It should be noted that the sample pretreatment was very different—our samples were cooked and frozen while samples in other studies were not cooked at all and were preserved by different drying processes. It is possible that the cooking step in our sample preparation has resulted in lipid oxidation and thus increased formation of hexanal from other volatile compounds. Furthermore, as Politowicz et al. [10] discuss growth conditions were different between these studies which could explain the difference.
Conclusions
This study points out that fiber type and extraction time are important factors that should be carefully tested as a part of SPME analysis development for mushroom-type matrices. Interestingly, there was no interaction between extraction variables, which could simplify experimental designs. Desorption parameters indeed affected the chromatographic profile, but interestingly fiber depth had the most systematic effect and it should be checked as part of optimization. This was surprising as fiber positioning in the injector is just quickly glanced over in textbooks on the subject [6]. This study also demonstrated that with modern analysis methodology, a thick-film column indeed suffices for focusing volatiles present in mushrooms. The results likewise show the importance of looking at the extracted profile as a whole so that the researcher in charge can make informed decisions on the compromises related to each selected analysis factor. Finally, the identified compounds indicate that fatty acids are the precursors of the majority of volatile compounds in C. cibarius.
References
Dunkel A, Steinhaus M, Kotthoff M et al (2014) Nature’s chemical signatures in human olfaction: a foodborne perspective for future biotechnology. Angew Chem Int Ed Engl 53:7124–7143. https://doi.org/10.1002/anie.201309508
Pyysalo H, Berg A, Lund H et al (1976) Identification of volatile compounds in seven edible fresh mushrooms. Acta Chem Scand 30b:235–244. https://doi.org/10.3891/acta.chem.scand.30b-0235
Grosshauser S, Schieberle P (2013) Characterization of the key odorants in pan-fried white mushrooms (Agaricus bisporus L.) by means of molecular sensory science: comparison with the raw mushroom tissue. J Agric Food Chem 61:3804–3813. https://doi.org/10.1021/jf4006752
Pawliszyn J (1997) Solid phase microextraction: theory and practice. Wiley, New York
Järvenpää E, Nurmela K (2010) Applicability of SPME techniques for analysis of volatile compounds in complex matrices. In: Jestoi M, Järvenpää E, Peltonen K (eds) First dice your dill (Anethum graveolens L.)—new methods and techniques in sample handling. University of Turku, Finland
Kudlejova L, Risticevic S, Vuckovic D (2012) Solid-phase microextraction method development. In: Pawliszyn J (ed) Handbook solid phase microextraction. Elsevier, Toronto, pp 201–249. https://doi.org/10.1016/B978-0-12-416017-0.00007-3
Costa R, Tedone L, De Grazia S et al (2013) Multiple headspace-solid-phase microextraction: an application to quantification of mushroom volatiles. Anal Chim Acta 770:1–6. https://doi.org/10.1016/j.aca.2013.01.041
de Pinho GP, Ribeiro B, Gonçalves RF et al (2008) Correlation between the pattern volatiles and the overall aroma of wild edible mushrooms. J Agric Food Chem 56:1704–1712. https://doi.org/10.1021/jf073181y
Aprea E, Romano A, Betta E et al (2015) Volatile compound changes during shelf life of dried Boletus edulis: comparison between SPME-GC-MS and PTR-ToF-MS analysis. J Mass Spectrom 50:56–64. https://doi.org/10.1002/jms.3469
Politowicz J, Lech K, Sánchez-Rodríguez L et al (2017) Volatile composition and sensory profile of Cantharellus cibarius Fr. as affected by drying method: aroma profile of fresh and dried Cantharellus cibarius. J Sci Food Agric. https://doi.org/10.1002/jsfa.8406
San Román I, Alonso ML, Bartolomé L et al (2014) Analytical strategies based on multiple headspace extraction for the quantitative analysis of aroma components in mushrooms. Talanta 123:207–217. https://doi.org/10.1016/j.talanta.2014.01.021
Perestrelo R, Silva C, Silva P, Câmara JS (2017) Global volatile profile of virgin olive oils flavoured by aromatic/medicinal plants. Food Chem 227:111–121. https://doi.org/10.1016/j.foodchem.2017.01.090
Cho IH, Lee SM, Kim SY et al (2007) Differentiation of aroma characteristics of pine-mushrooms (Tricholoma matsutake Sing.) of different grades using gas chromatography—olfactometry and sensory analysis. J Agric Food Chem 55:2323–2328. https://doi.org/10.1021/jf062702z
Jeleń HH (2003) Use of solid phase microextraction (SPME) for profiling fungal volatile metabolites. Lett Appl Microbiol 36:263–267. https://doi.org/10.1046/j.1472-765X.2003.01305.x
D’Agostino MF, Sanz J, Sanz ML et al (2015) Optimization of a solid-phase microextraction method for the gas chromatography–mass spectrometry analysis of blackberry (Rubus ulmifolius Schott) fruit volatiles. Food Chem 178:10–17. https://doi.org/10.1016/j.foodchem.2015.01.010
Soares RD, Welke JE, Nicolli KP et al (2015) Monitoring the evolution of volatile compounds using gas chromatography during the stages of production of Moscatel sparkling wine. Food Chem 183:291–304. https://doi.org/10.1016/j.foodchem.2015.03.013
Stein SE (1997) Retention indices. NIST chemistry webbook NIST standard reference, database 69
Langenfeld JJ, Hawthorne SB, Miller DJ (1996) Optimizing split/splitless injection port parameters for solid-phase microextraction. J Chromatogr A 740:139–145. https://doi.org/10.1016/0021-9673(96)00140-9
Ilias Y, Bieri S, Christen P, Veuthey J-L (2006) Evaluation of solid-phase microextraction desorption parameters for fast GC analysis of cocaine in coca leaves. J Chromatogr Sci 44:394–398
Ouyang G, Chen Y, Setkova L, Pawliszyn J (2005) Calibration of solid-phase microextraction for quantitative analysis by gas chromatography. J Chromatogr A 1097:9–16. https://doi.org/10.1016/j.chroma.2005.08.017
Odor Detection Thresholds & References. http://www.leffingwell.com/odorthre.htm. Accessed 10 Aug 2017
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the University of Turku Graduate School (UTUGS) and its Doctoral Program in Molecular Life Sciences (DPMLS), Niemi foundation research grant (2015), by Academy of Finland (MS252005) and the project “Innovative Technologies and Concepts for Business Growth Based on Finnish Mushrooms” (3135/31/2015), funded by Tekes (the Finnish Funding Agency for Innovation).
Conflict of interest
The authors declare that they have no conflict of interest.
Human or animal participants statements
This article does not contain any studies with human or animal subjects.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Aisala, H., Linderborg, K.M. & Sandell, M. Fiber depth, column coating and extraction time are major contributors in the headspace solid-phase microextraction–gas chromatography analysis of Nordic wild mushrooms. Eur Food Res Technol 244, 841–850 (2018). https://doi.org/10.1007/s00217-017-3005-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-017-3005-0