
Abstract
Quantitation of BCR-ABL1 with the quantitative reverse transcriptase polymerase chain reaction (RT-PCR) is very important in monitoring chronic myeloid leukemia (CML), which relies on an RNA reference material. A genomic RNA reference material (RM) containing the BCR-ABL1 P210 fusion mutation was developed, and an absolute quantitative method based on one-step reverse transcription digital PCR (RT-dPCR) was established for characterizing the RM. The proposed dPCR method demonstrates high accuracy and excellent analytical sensitivity, as shown by the linear relationship (0.94 < slope < 1.04, R2≧0.99) between the measured and nominal values of b2a2, b3a2, and ABL1-ref within the dynamic range (104–101 copies/reaction). Homogeneity and stability assessment based on dPCR indicated that the RM was homogeneous and stable for 24 months at −80 °C. The RM was used to evaluate inter-laboratory reproducibility in eight different laboratories, demonstrating that participating laboratories could consistently produce copy concentrations of b3a2 and ABL1-ref, as well as the BCR-ABL1/ABL1 ratio (CV < 2.0%). This work suggests that the RM can be employed in establishing metrological traceability for detecting mutations in the BCR-ABL1 fusion gene, as well as in quality control for testing laboratories.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia chromosome (Ph) in patients with myeloproliferative neoplasms (MPNs) [1,2,3]. The proto-oncogene Abelson murine leukemia viral homolog 1 (ABL1) on chromosome 9 (9q34) fuses with the breakpoint cluster region (BCR) on chromosome 22 (22q11) [4]. This translocation results in the formation of the BCR-ABL1 fusion gene, leading to the expression of a protein with potent tyrosine kinase activity that disrupts multiple signaling pathways [5, 6]. In most cases, the ABL1 gene breakpoint occurs consistently between exons 1 and 2, while the BCR gene breakpoint varies, typically in exons 13 and 14, giving rise to two distinct products known as b2a2 (e13a2) and b3a2 (e14a2), both producing a 210 kDa BCR-ABL1 protein (P210) [7, 8]. Studies indicate that around two-thirds of patients with BCR-ABL1 P210 express b3a2, which may be related to a higher likelihood of deep molecular response and survival. In addition, 5–10% of all patients co-express both b2a2 and b3a2 [9, 10]. The BCR-ABL1 fusion gene serves as the molecular foundation of CML, functioning not only as a diagnostic and monitoring marker but also as a target for tyrosine kinase inhibitors (TKIs) in CML treatment. Therefore, monitoring the transcription level of the BCR-ABL1 fusion gene in CML patients is crucial for evaluation of treatment efficacy, early detection of drug resistance, and monitoring of disease progression to inform treatment decisions. Consequently, accurate quantitative detection of BCR-ABL1 is imperative for the management of CML and for conducting clinical studies of novel agents in CML.
The technical methods for the detection of leukemia-specific fusion genes are being gradually established and improved in China. The BCR-ABL1 fusion gene is currently detected by karyotyping, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR) techniques, and flow cytometric immune bead assay (CBA) [11]. Among the various techniques available, FISH cannot distinguish between fusion variants despite its highly standardized procedures, so FISH results are subjective. Real-time quantitative reverse transcription PCR (RT-qPCR) is widely used in laboratories to quantify BCR-ABL1 fusion gene transcripts, but significant differences are found among laboratories as a result of differences in testing kits due to a lack of International System of Units (SI)-traceable RNA reference material (RM) [12]. To ensure the comparability and consistency of detection results from different methods, platforms, and laboratories, it is necessary to establish a SI-traceable RNA RM for gene fusion measurement which can be used to implement the traceability required by ISO 17511 [13].
Many efforts have been made toward the standardization of BCR-ABL1 detection, including the establishment of primary and secondary standard reagents [14,15,16] and plasmid DNA reference materials [17]. However, no SI-traceable RNA RM is available so far to validate the RNA reverse transcription efficiency (RTE), due to the lack of a SI-traceable reference measurement procedure (RMP) for the RNA measurement. Reverse transcriptase digital polymerase chain reaction (RT-dPCR) is a unique approach to measurement of the absolute copy number of target DNA without using external standards, with a high degree of sensitivity and precision [18]. With regard to RNA quantification, it is essential to first evaluate RNA RTE [19]. Previous studies have demonstrated that different dPCR assays may exhibit varying RT efficiency based on RNA structure. Therefore, an RNA RMP with a defined RNA RTE is urgently needed to assign the reference value of BCR-ABL1 RNA RM.
Isotope dilution mass spectrometry (IDMS) is the primary method for quantifying small molecules in chemical analysis. It utilizes stable isotope-labeled analogues (i.e., 13C or 15N) to assess quantitative biases in sample preparation or detection by mass spectrometry (MS) [20, 21]. When combined with enzymatic digestion or chemical hydrolysis methods, IDMS can also be used for the quantification of DNA or RNA [22,23,24]. A previous study reported on the use of enzymatic digestion-IDMS as a method for measuring RNA concentration without the RT step, which was employed to evaluate the RTE of the RT-dPCR method for the detection of SARS-CoV-2 RNA.
The objectives of this study are (1) to establish and validate an RMP for BCR-ABL1 transcript quantification at the RNA level by one-step reverse transcription dPCR (RT-dPCR) with a defined RTE determined by IDMS, (2) to develop a gRNA RM for BCR-ABL1 measurement, and (3) to assess inter-laboratory reproducibility by utilizing the gRNA RM.
Materials and methods
Study design
The study design is illustrated in Fig. 1. The scheme includes the following steps: (1) establishment and validation of the RT-dPCR using in vitro-transcribed (IVT) RNA and IDMS, (2) preparation of the genomic RNA (gRNA) candidate RM from the cell line, (3) characterization of the gRNA RM by the established RT-dPCR, and (4) evaluation of inter-laboratory reproducibility using the gRNA RM.

Study design. A Establishment of a one-step reverse transcription dPCR (RT-dPCR) method, including successful amplification of the wild-type (WT) and mutant assay in a single format, assessing the dynamic range, false-positive rate, quantitative consistency among different digital PCR platforms, and evaluating reverse transcription efficiency by isotope dilution mass spectrometry (IDMS). B Preparation of gRNA RM by culturing cell line, extraction and purification of gRNA, confirming the sequence, and vialing. C Characterizing the gRNA RM by assessing homogeneity and stability, and determining the reference value. D Evaluating inter-laboratory repeatability and reproducibility using the RM
Cell line, plasmid, and in vitro-transcribed RNA
Cell line K562 (containing BCR-ABL1 P210 transcript) was purchased from Meisen Chinese Tissue Culture Collections (Meisen CTCC). The culture medium comprised RPMI 1640, 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin (PS) (both from Gibco, USA) and was maintained by the National Institute of Metrology (NIM), China. The cell line was incubated in a 5% CO2 atmosphere at 37 °C, reaching a saturated cell density every 3 or 4 days, and was subcultured at a ratio of 1:3 or 1:4.
BCR-ABL1 P210 transcript (containing b2a2 and b3a2) and partial ABL1 (NM_005157.6, domain at the junction of exons 10 and 11) were synthesized and constructed into the same pBluescript II SK(+) vector by BGI (Beijing, China) to generate an IVT RNA molecule. The plasmid was linearized with Xba I (Takara, code no. 1093S) at the 3′ end and purified using a commercial DNA Clean-up Kit (CWBIO, Beijing, China) (Figure S1a). In vitro transcription reaction was performed using the MEGAscript™ T7 Transcription Kit (Thermo Fisher Scientific, USA), and Turbo DNase was added to remove the template DNA according to the instructions. The transcribed RNA was then purified with the MEGAclear™ kit (Thermo Fisher Science, USA). The quantity and quality of IVT RNA were assessed using the NanoDrop 2000 (Agilent, USA) and 2100 Bioanalyzer with the RNA 6000 Nano Kit (Agilent, USA), respectively. The successful transcription of RNA was confirmed using the RNA 6000 Nano Kit and analyzed on a 2100 Bioanalyzer (Agilent, USA) (Figure S1b).
Preparation of genomic RNA reference material
The cell line of total RNA was extracted by using an Ultrapure RNA kit (CWBIO, Beijing, China) according to the manufacturer’s instructions. The concentration and integrity number (RIN) of total RNA were estimated using the NanoDrop 2000 and analyzed on a 2100 Bioanalyzer Instrument with an RNA 6000 Nano Kit (both Agilent, USA), respectively. The purity and integrity of total RNA were confirmed by the value of A260/280 (1.8–2.0), A260/230 (\(>\) 2.0), and RIN \((\ge\) 9.3). The RNA stock solution was diluted to a concentration of 50 ng/μL using the RNA storage solution (Thermo Fisher Scientific, USA) with 5 ng/μL of yeast total RNA (Sigma, Code No. 63231630, USA), packaged in 30 μL per vial, stored at −80 °C and used as the candidate reference material (RM).
Sanger sequencing
The candidate RM was converted into complementary DNA (cDNA) by using a reverse transcription kit (Takara, code no. 6215A). Subsequently, the cDNA was sequenced by Sanger sequencing (ABI 3730XL, USA) to confirm the presence of BCR-ABL1 transcript by Sangon Biotech Co., Ltd. (Shanghai, China). The sequence data were then analyzed using SeqMan software.
Reverse transcription digital PCR (RT-dPCR)
The primer and probe sequences used for detecting the targets of BCR-ABL1 (b2a2 and b3a2) and the ABL1 reference gene (ABL1-ref) are listed in Table S1 [2, 16, 18]. The optimization of primer and probe concentrations is shown in Figure S2a–c. The RT-dPCR analysis was performed on a QX200 Droplet Digital PCR system (Bio-Rad, USA). A 20-μL reaction mixture was prepared, consisting of 5 µL of supermix (Bio-Rad, USA), 2 μL of reverse transcriptase, 1 μL of 300 mM DL-Dithiothreitol (DTT), primer and probe, and 2 μL of template and RNase-free water top-up. The final concentrations of primers and probes were 500 nM and 400 nM for b2a2, 400 nM and 300 nM for b3a2, and 600 nM and 300 nM for ABL1-ref, respectively.
Droplets of each reaction mixture were generated on a QX200 droplet generator (Bio-Rad, USA) and transferred to a conventional PCR thermal cycler (VeritiPro, Applied Biosystems) for heating and amplification. The optimization of the annealing temperature is shown in Figure S2d–f. The final PCR conditions included 60 min reverse transcription at 45 °C; initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s and annealing at 60 °C for 30 s; and final extension at 98 °C for 10 min. The plate was then transferred to the QX200 reader (Bio-Rad, USA) and analyzed with QuantaSoft v.1.7.4.0917 software (Bio-Rad, USA). The workflow and data analysis were described in our previous report [25].
Limit of blank (LoB), detection (LoD), and quantification (LoQ)
To determine the dynamic range of the dPCR assays for the two transcripts of BCR-ABL1 P210 and ABL1-ref target, serial dilutions of the above IVT RNA were prepared by gravimetry. Each dilution was measured in three to six replicates using the established three singleplex dPCR assays. 60 measurements were performed on blank samples to determine the LoB according to EP17-A [26]. To establish the LoD, 60 measurements from five samples with low concentration were used to determine the LoD according to the guideline of EP17-A [26]. The coefficient of variation (CV) was calculated by the several corresponding replicates at each concentration to determine LoQ based on CV < 25%.
Reverse transcription efficiency assessment
To evaluate the reverse transcription efficiency (RTE) of RT-dPCR, isotope dilution mass spectrometry (IDMS), an RNA quantification method without reverse transcription, was employed [19]. An IVT RNA including BCR-ABL1 P210 transcript (containing b2a2 and b3a2) and a partial ABL1 gene (NM_005157.6, domain at the junction of exons 10 and 11) was used for the evaluation. Following verification of the purity of the IVT RNA using the Agilent 2100 Bioanalyzer with the RNA 6000 Nano Kit, it was digested into nucleotide monophosphates (NMPs) with snake venom phosphodiesterase (SVP) and then analyzed using IDMS.
Four certified reference materials (CRMs) of pure NMPs (adenosine 5′-monophosphate [AMP, GBW(E)100,154], guanosine 5′-monophosphate disodium salt [GMP, GBW(E)100,068], cytidine 5′-monophosphate [CMP, GBW(E)100,067], and uridine 5′-monophosphate disodium salt [UMP, GBW(E)100,069]) (NIM, China) were diluted with TE0.1 buffer (10 mol/L pH 8.0Tris-HCl, 0.1 mmol/L pH 8.0 ethylenediaminetetraacetic acid [EDTA]) to prepare the NMP mix solution. An internal standard mix of four isotope-labeled (13C, 15N) NMPs (LNMPs) (Silantes, Germany) was prepared with SVP buffer (100 mM pH 8.8 Tris–HCl, 10 mM ammonium acetate, and 100 mM magnesium acetate). The concentrations of the four NMPs and LNMPs in the mix solution were determined based on the estimated concentrations of native NMP in the IVT RNA sample, ensuring a molar ratio of NMP to LNMP of approximately 1:1 in the sample. Five concentrations of standard solutions were prepared, with mass ratios of NMP/LNMP of 0.4, 0.8, 1.2, 1.6, and 2.0. The procedure for optimization of IDMS was described in detail in our previous report [19]. Two tubes of RNA samples were used, with each undergoing digestion twice in each experiment, and each digestion analyzed with three replicates. Consequently, a total of 12 sets of data were collected. The overall average of the four NMP measurements was calculated as the concentration of IVT RNA.
Inter-laboratory assessment
The repeatability and reproducibility of the established RT-dPCR were evaluated by analyzing the RM in eight different laboratories using the QX200 (Bio-Rad, CA, USA), DQ24 (Sniper, Suzhou, China), or Microdrop-100 ddPCR (FOREVERGEN, Guangdong, China) platform according to ISO 5725-1 [27]. Details of information on laboratories are listed in Table S2. The Grubbs test was used to check for outliers and Mandel’s h and k statistics were used to assess the consistency between and within laboratories according to ISO 5725-2 [28].
Results
Validation of RT-dPCR
Three singleplex dPCR assays for b2a2, b3a2, and ABL1-ref were optimized and established. Good separation between positive and negative fluorescence amplitude was observed for all targets. The two-dimensional scatterplot is shown in Figure S3.
Cross-reactions between b2a2, b3a2, and ABL1-ref were assessed by a combination of the targeted assays with WT RNA template extracted from 293 T and IVT RNAs. Both b2a2 and b3a2 assays demonstrated good specificity, with only positive amplification observed for samples containing their specific targets (Figure S4a and b). Additionally, the ABL1-ref assay showed positive amplification only in WT RNA when amplifying IVT RNAs of the mutation (MU, containing b2a2 and b3a2 variants) and WT RNA (Figure S4c). This indicates that there was no cross-reaction between the BCR-ABL1 variants and the reference gene.
The variability of BCR-ABL1 RMP across different dPCR platforms was evaluated to identify potential biases impacting the accuracy of the results. Five commercial dPCR platforms were selected to detect the template panel combinations for cross-comparison as shown in Fig. 2. This template panel consists of BCR-ABL1 P210 expressing b2a2 and b3a2 variants at a concentration that simulates the concentration of clinical samples (approximately 104 copies/reaction), with or without high background concentrations (approximately 104 copies/reaction) of the WT ABL1.

Comparison of digital PCR platform. a Quantification of samples containing BCR-ABL1 b2a2 or b3a2 molecules at approximately 104 copies/reaction with or without a background of 104 copies/reaction reference gene (ABL1-ref) molecules per reaction using five dPCR platforms (n = 3). b False-positive rate (FPR) for b2a2, b3a2, and ABL1-ref assays
Quantification results of IVT RNA samples of each target fusion variant with or without WT were relatively consistent across the five dPCR platforms (Fig. 2a1 and 2a2). This indicates that the quantitative result of the mutant type was not affected by the presence of WT. The differences among the other four dPCR platforms ranged from 0.93-fold to 1.15-fold compared with QX200 (Tables S3 and S4), indicating no significant variation in the accuracy of the RMP among these dPCR platforms. Additionally, the false-positive rates (FPR) of MU (b2a2 or b3a2) and WT (ABL1-ref) assays were assessed using templates for the WT and MU, respectively, as shown in Fig. 2b1 and b2. The assay specificity results measured by FPR were close to 0 for QX200, while they were relatively higher on the other three platforms. Notably, the QIAcuity One platform exhibited the highest FPR for b2a2 and b3a2, corresponding to higher concentrations of the target gene, suggesting potential contamination during the automatic operation of this dPCR platform. The precision and repeatability of the assays were evaluated based on the CV of this template panel on each platform. All platforms demonstrated higher precision and repeatability with CV < 5.5% for fusion variant quantification of approximately 104 copy number samples (Tables S3 and S4). The concentrations of these samples are consistent with clinically detectable levels, indicating that all five dPCR instruments could serve as reliable platforms for quantifying the BCR-ABL1 fusion gene.
Dynamic range of the RMP
Due to the precision of dPCR, relying on an absolute measurement of the number of partitions, referred to as lambda (λ), the linear range was tested by varying the λ [29]. The dynamic range of the RMP was then evaluated using a dilution series of the IVT RNA containing b2a2, b3a2, and ABL1-ref from λ approximately 8.3 to 0.1, corresponding to the upper limit of the dynamic range, which was 1.2 × 105 copies/partition (Fig. 3a). Three dPCR assays displayed good linearity between the measured value and the prepared value in the tested interval within the range of approximately 104 to 101 copies/reaction (R2 \(\ge\) 0.99) (Fig. 3b). The slope (0.94 < slope < 1.04) was close to 1, as the measured concentration was very close to its corresponding nominal concentration by gravimetric dilution, indicating that RT-dPCR is highly accurate in detecting BCR-ABL1 transcript and the reference gene at concentrations of higher than 10 copies/reaction. Since the distribution of the 60 blank measurements was non-normally distributed, the LoB was estimated using the nonparametric method as the 95th percentile of the measurements, which corresponds to 57.5 ordinal observations (60*[95/60 + 0.5]). As the 60 blank measurements were predominantly 0, only the 15 highest blank values of the LoB are shown in Table S8. The LoB values for BCR-ABL1 b2a2, b3a2, and ABL1-ref were determined to be 0.95 copies/reaction, 1 copies/reaction, and 0.20 copies/reaction. The distribution of 60 measurement results for five low-concentration samples does not conform to a Gaussian distribution (Figure S5). Thus, nonparametric statistics were used. Consequently, the LoD values for BCR-ABL1 b2a2, b3a2, and ABL1-ref were determined to be 1.74 copies/reaction, 2.52 copies/reaction, and 1.30 copies/reaction [26]. The results indicate that the LoD of b2a2, b3a2, and ABL1-ref was two copies/reaction based on the criterion that more than 19 of the 20 measurements were positive. In accordance with the criteria for a CV lower than 25%, the LOQ of BCR-ABL1 b2a2, b3a3, and ABL1-ref was determined to be 31, 30, and 31 copies/reaction, respectively (Table S5–S7).

Validated range of the candidate primary reference measurement procedure. Evaluation of linearity of RNA samples containing BCR-ABL1 b2a2, b3a2, and ABL1-ref over the (a) extended λ range (0.1 < λ < 8.3) and (b) low λ range (λ < 0.2)
RTE evaluation by IDMS
The purity of the IVT was determined to be 99.2% using the Bioanalyzer, as shown in Figure S1, which meets the requirement for using IDMS to evaluate RTE. The concentration of NMPs in the fully digested mixture was determined by establishing standard curves. The standard curves for AMP/LAMP, CMP/LCMP, UMP/LUMP, and GMP/LGMP exhibited strong linear correlations with R2 values of 0.9990, 0.9993, 0.9991, and 0.9991, respectively (Figure S6). The concentration of intact RNA was calculated based on the mass concentration of each NMP detected by IDMS. The RNA mass fractions calculated from the measured concentrations of AMP, GMP, CMP, and UMP were 0.88 μg/g, 0.95 μg/g, 0.91 μg/g, and 1.00 μg/g, respectively (Fig. 4a).

The quantification results of IVT RNA by IDMS and RT-dPCR. a A comparison of the RNA mass concentrations calculated from four NMPs by IDMS. The error bar represents expanded uncertainty (k = 2), and the horizontal axis represents the mean of the results. b A comparison of the copy number concentration result of in vitro-transcribed RNA by RT-dPCR and IDMS
The IVT mass fraction measured by IDMS was converted into copy number concentration based on the Avogadro constant and the molecular weight of RNA, representing the absolute copy number concentration of the RNA molecule (denoted as \({C}_{RNA}\)). The cDNA copy number concentration measured by RT-dPCR was recorded as \({C}_{cDNA}\), and the RTE was calculated according to the formula RTE = \({C}_{cDNA}/{C}_{RNA}\). The uncertainty evaluation of RTE is provided in Table S9.
As the droplet volume is a critical parameter for the accuracy of the dPCR result, we measured the droplet volume based on the sizes of 2130 droplets from eight wells determined by microscopy (Olympus, BX51). The average droplet volume for this one-step reverse transcription PCR master mix was determined to be (0.769 ± 0.012) nL, which agreed well with a previous report (0.763 ± 0.016) nL by the National Measurement Institute of Australia [30]. The average copy numbers of b2a2, b3a2, and ABL1-ref genes corrected by droplet volume were 1.17 \(\times\) 109, 1.21 \(\times\) 109, and 1.17 \(\times\) 109 copies/μL, respectively. The average copy number of the IVT RNA measured by IDMS was 1.09 \(\times\) 109 copies/μL (Fig. 4b). The RTE values for b2a2, b3a2, and ABL1-ref gene were 1.07, 1.11, and 1.07, respectively, which were close to 1. This further confirmed the high accuracy of the established RT-dPCR.
Confirmation of BCR-ABL1 fusion variant in the candidate RMs
The fusion variant of BCR-ABL1 in the candidate RM was confirmed by Sanger sequencing and is shown in Figure S7. The result demonstrated that the candidate RM, derived from K562 cell line gRNA, carries the fusion mutation of BCR-ABL1 P210, formed by exon 14 of the BCR gene with exon 2 of the ABL1 gene, denoted as b3a2.
Homogeneity and stability of the candidate RMs
The homogeneity and stability of the candidate RMs were evaluated according to ISO 17034 and ISO guide 35 [31, 32], and detailed information is provided in the supplemental material (Tables S10–S11 and Figure S8). The F values for the homogeneity test of b3a2 and ABL1-ref copy numbers and BCR-ABL1/ABL1 ratio were 3.8, 1.9, and 0.8, respectively. Except for b3a2, all calculated F values were lower than the F critical value [33]. However, the CVs for all targets were within 1.4%, as shown in Table S10, indicating that the candidate RM was homogeneous. Regarding long-term stability, no significant differences were observed in the copy number of b3a2 and ABL1-ref and the ratio of BCR-ABL1/ABL1 at −80 °C over 12 months (P > 0.05, Figure S8 and Table S11). Additionally, based on our previous data [34], we have similar RNA reference material that remained stable for 2 years at −80 °C. Thus, we anticipate that the total RNA RM for BCR-ABL1 developed in this study can also remain stable for at least 2 years at −80 °C. The measurement uncertainty and the reference value are provided in Table 1.
Inter-laboratory repeatability and reproducibility
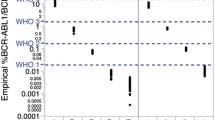
With the participation of eight different laboratories, the performance of RMP and its associated measurement uncertainty were further evaluated using candidate RM under reproducible conditions. The results across all eight laboratories and three platforms of b3a2 and ABL1-ref concentrations, as well as BCR-ABL1/ABL1 ratio, were in good agreement with the reference values within the extended uncertainty range (Figure S9). All three types of platforms are droplet-based digital polymerase chain reactions. This allows for more digital PCR-based platforms to further evaluate the performance criteria of candidate RMP [35, 36]. Thus, the overall reproducibility of the candidate RMP was calculated as less than 2% for the inter-laboratory CV of b3a2, ABL1-ref, and %BCR-ABL1/ABL1 between all laboratories, indicating good repeatability and reproducibility (Table S12).
The Mandel’s h and k statistics were plotted to assess consistency between and within laboratories (Fig. 5). All testing laboratories showed good agreement in the copy number concentration of b3a2 and ABL1-ref and the ratio of BCR-ABL1/ABL1 as indicated by the Mandel’s h plot at a significance level of 1% (Fig. 5a), demonstrating good reproducibility of the RMP. Mandel’s k plot revealed that Laboratory 3 had poor repeatability in copy number concentration of b3a2 at a significance level of 1%, followed by poor repeatability of Laboratory 4 at a significance level of 5% (Fig. 5b). Overall, at least seven participating laboratories had good repeatability and consistency.

Consistency assessment by Mandel’s between-laboratory statistic, h (a) and within-laboratory statistic, k (b), for the average copy number concentration of b3a2 and ABL1-ref and %BCR-ABL1/ABL1 of the reference material. The solid and dashed lines indicate the 1% and 5% significance levels, respectively
Discussion
Digital PCR, as a third-generation PCR technique, is a method based on a microfluidic or water emulsion droplet technique in which a sample is divided into several thousand to several hundred thousand partitions. This is followed by PCR amplification of the template in each partition, resulting in high sensitivity, precision, reliable quantitation, and exquisite reproducibility without the need for calibration curves [37]. Both the European Leukemia Net (ELN) and the National Comprehensive Cancer Network (NCCN) recommend RT-qPCR for monitoring the prediction, relapse, or molecular remission of CML. However, RT-qPCR is not as sensitive and accurate as RT-dPCR for low-copy rare targets in CML patients with complete molecular response (CMR, corresponding to MR4.0 and MR4.5) status prior to discontinuation of the drug [38,39,40]. Therefore, RT-dPCR can be an ideal alternative for the detection of very low levels of disease [37].
For DNA molecules, digital PCR has advanced as an absolute quantification method that does not rely on an external calibrator. It can determine copy number concentration and fractional abundance values for HER2, BRAF V600E DNA, and EGFR reference materials [35, 36, 41]. When dealing with RNA molecules, cDNA must first be obtained through reverse transcription (RT) before PCR amplification. However, the RNA-to-cDNA conversion step is often seen as inefficient and variable. Previous studies have shown that the efficiency of this conversion process varies significantly based on the type of reverse transcriptase used and the RT reaction conditions, leading to substantial differences in the quantification outcomes of RNA molecules [25, 42]. To ensure accurate quantification of RNA molecules, it is essential to develop an optimized workflow that identifies the most suitable RT type and reaction conditions for the target gene. Furthermore, a method was established for in vitro quantification of transcribed RNA by isotope dilution mass spectrometry (IDMS) without RT reaction, which is not specific to RNA sequences and is independent of the RNA reverse transcription process, and is thus ideal for accurate quantification in the development of higher-order reference materials and RT reagents [19]. Therefore, this study validated the accuracy of the established RT-dPCR method for BCR-ABL1 fusion genes by quantifying IVT using different commercial digital PCR platforms and IDMS. The results were highly consistent between the two methods based on different principles, which bolstered confidence in the reliability and supporting evidence for the applicability of the established RT-dPCR method as a primary RMP for quantifying the BCR-ABL1 fusion gene.
The main goal of an inter-laboratory study is to assess the established RMP’s reproducibility performance using the same RM. Reproducibility refers to the degree of consistency when test results are obtained for the same item in different laboratories, with different operators using different equipment, and using the same method by ISO 5725-1 [27]. To ensure statistical validity, a sufficient number of laboratories should participate in the inter-laboratory study. Our study has shown consistent reported values among eight different laboratories, demonstrating good performance of the proposed RMP and ensuring its high quality and wide usability.
In conclusion, a reference measurement procedure for the quantitative detection of BCR-ABL1 P210 express b2a2, b3a2, and the reference gene (ABL1-ref) in CML based on one-step reverse transcription dPCR (RT-dPCR) was established and validated by IDMS. The results indicate that our proposed RT-dPCR method has high accuracy and good analytical sensitivity, making it suitable for characterizing RNA reference material containing the BCR-ABL1 P210 (b2a2, b3a2) fusion. Most importantly, a homogeneous BCR-ABL1 fusion gene (b3a2) reference material was established, which can be used to establish the metrological traceability of BCR-ABL1 P210 measurement to ensure the comparability and reliability of clinical diagnostic results.
References
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–56.
Ayatollahi H, Keramati MR, Shirdel A, Kooshyar MM, Raiszadeh M, Shakeri S, et al. BCR-ABL fusion genes and laboratory findings in patients with chronic myeloid leukemia in northeast Iran. Caspian J Intern Med. 2018;9:65–70. https://doi.org/10.22088/cjim.9.1.65.
Karow A, Gohring G, Sembill S, Lutterloh F, Neuhaus F, Callies S, et al. The cytogenetic landscape of pediatric chronic myeloid leukemia diagnosed in chronic phase. Cancers (Basel). 2022;14. https://doi.org/10.3390/cancers14071712.
Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3. https://doi.org/10.1038/243290a0.
Heim D, Ebnöther M, Favre G. Chronic myeloid leukemia - update 2020. Ther Umsch. 2019;76:503–9. https://doi.org/10.1024/0040-5930/a001124.
Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619–30. https://doi.org/10.1182/blood-2008-03-144790.
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–84.
Vinhas R, Cordeiro M, Pedrosa P, Fernandes AR, Baptista PV. Current trends in molecular diagnostics of chronic myeloid leukemia. Leuk Lymphoma. 2017;58:1791–804. https://doi.org/10.1080/10428194.2016.1265116.
Jain P, Kantarjian H, Patel KP, Gonzalez GN, Luthra R, Kanagal Shamanna R, et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood. 2016;127:1269–75. https://doi.org/10.1182/blood-2015-10-674242.
Baccarani M, Castagnetti F, Gugliotta G, Rosti G, Soverini S, Albeer A, et al. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. An international overview. Leukemia. 2019;33:1173–83. https://doi.org/10.1038/s41375-018-0341-4.
Yu S, Cui M, He X, Jing R, Wang H. A review of the challenge in measuring and standardizing BCR-ABL1. Clin Chem Lab Med. 2017;55:1465–73. https://doi.org/10.1515/cclm-2016-0927.
Zhen C, Wang YL. Molecular monitoring of chronic myeloid leukemia: international standardization of BCR-ABL1 quantitation. J Mol Diagn. 2013;15:556–64. https://doi.org/10.1016/j.jmoldx.2013.05.010.
ISO 17511:2003. In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values assigned to calibrators and control materials. Geneva, Switzerland: ISO; 2003.
Fu Y, Zhang R, Wu Q, Zhang J, Bao L, Li J. External quality assessment of p210 BCR-ABL1 transcript quantification by RT-qPCR: Findings and recommendations. Int J Lab Hematol. 2018;41:46–54. https://doi.org/10.1111/ijlh.12919.
Labourier E, Cross NCP, Radich J, Pane F, Pagnano KB, Müller MC, et al. Establishment and validation of analytical reference panels for the standardization of quantitative BCR-ABL1 measurements on the international scale. Clin Chem. 2013;59:938–48. https://doi.org/10.1373/clinchem.2012.196477.
White HE, Matejtschuk P, Rigsby P, Gabert J, Lin F, Lynn Wang Y, et al. Establishment of the first World Health Organization International Genetic Reference Panel for quantitation of BCR-ABL mRNA. Blood. 2010;116:e111–7. https://doi.org/10.1182/blood-2010-06-291641.
Zhang J-W, Fu Y, Wu Q-S, Bao L-H, Peng R-X, Zhang R, et al. Standardization of BCR-ABL1 quantification on the international scale in China using locally developed secondary reference panels. Exp Hematol. 2020;81:42-9.e3. https://doi.org/10.1016/j.exphem.2019.12.002.
Maier J, Lange T, Cross M, Wildenberger K, Niederwieser D, Franke GN. Optimized digital droplet PCR for BCR-ABL. J Mol Diagn. 2019;21:27–37. https://doi.org/10.1016/j.jmoldx.2018.08.012.
Niu C, Wang X, Gao Y, Qiao X, Xie J, Zhang Y, et al. Accurate quantification of SARS-CoV-2 RNA by isotope dilution mass spectrometry and providing a correction of reverse transcription efficiency in droplet digital PCR. Anal Bioanal Chem. 2022;414:6771–7. https://doi.org/10.1007/s00216-022-04238-6.
Burke DG, Griffiths K, Kassir Z, Emslie K. Accurate measurement of DNA methylation that is traceable to the international system of units. Anal Chem. 2009;81:7294–301. https://doi.org/10.1021/ac901116f.
Lowenthal MS, Quittman E, Phinney KW. Absolute quantification of RNA or DNA using acid hydrolysis and mass spectrometry. Anal Chem. 2019;91:14569–76. https://doi.org/10.1021/acs.analchem.9b03625.
Heiss M, Reichle VF, Kellner S. Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS. RNA Biol. 2017;14:1260–8. https://doi.org/10.1080/15476286.2017.1325063.
Sarin LP, Kienast SD, Leufken J, Ross RL, Dziergowska A, Debiec K, et al. Nano LC-MS using capillary columns enables accurate quantification of modified ribonucleosides at low femtomol levels. RNA. 2018;24:1403–17. https://doi.org/10.1261/rna.065482.117.
Dong L, Zang C, Wang J, Li L, Gao Y, Wu L, et al. Lambda genomic DNA quantification using ultrasonic treatment followed by liquid chromatography-isotope dilution mass spectrometry. Anal Bioanal Chem. 2012;402:2079–88. https://doi.org/10.1007/s00216-011-5644-5.
Niu C, Dong L, Gao Y, Zhang Y, Wang X, Wang J. Quantitative analysis of RNA by HPLC and evaluation of RT-dPCR for coronavirus RNA quantification. Talanta. 2021;228:122227. https://doi.org/10.1016/j.talanta.2021.122227.
CLSI. EP17-A Protocols for Determination of Limits of Detection and Limits of Quantitation; Approved Guideline, Clinical and Laboratory Standards Institute; 2004.
ISO 5725–1:1994, Accuracy (Trueness and Precision) of Measurement Methods and Results – Part 1: General Principles and Definitions, Geneva, Switzerland, ISO; 1994.
ISO 5725–2:1994, Accuracy (Trueness and Precision) of Measurement Methods and Results – Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method, Geneva, Switzerland, ISO; 1994.
Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, et al. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods. 2010;50:271–6. https://doi.org/10.1016/j.ymeth.2010.01.003.
Burke D. Title of document. In: Quantitative sars cov 2 certified reference material, SARS-CoV-2 Standard NMIA NA050 to NA055. Report ID: 210329, https://www.industry.gov.au/sites/default/files/2020-12/sars-cov-2_c_of_a_b200921_feb_2021.pdf. Accessed 29 Mar 2021.
ISO 17034. General requirements for the competence of reference material producers, Geneva, Switzerland: ISO; 2016.
ISO GUIDE 35:2017. Reference materials — Guidance for characterization and assessment of homogeneity and stability. Switzerland: ISO; 2017.
Van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13:1901–28. https://doi.org/10.1038/sj.leu.2401592.
Yu Y, Hou W, Liu Y, Wang H, Dong L, Mai Y, et al. Quartet RNA reference materials improve the quality of transcriptomic data through ratio-based profiling. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-023-01867-9.
Wang X, Zhang Y, Niu C, Wang S, Li L, Guo Y, et al. Establishment of primary reference measurement procedures and reference materials for EGFR variant detection in non-small cell lung cancer. Anal Methods. 2021;13:2114–23. https://doi.org/10.1039/d1ay00328c.
Dong L, Wang X, Wang S, Du M, Niu C, Yang J, et al. Interlaboratory assessment of droplet digital PCR for quantification of BRAF V600E mutation using a novel DNA reference material. Talanta. 2020;207:120293. https://doi.org/10.1016/j.talanta.2019.120293.
Colafigli G, Scalzulli E, Di Prima A, Pepe S, Loglisci MG, Diverio D, et al. Digital droplet PCR as a predictive tool for successful discontinuation outcome in chronic myeloid leukemia: Is it time to introduce it in the clinical practice? Crit Rev Oncol Hematol. 2021;157:103163. https://doi.org/10.1016/j.critrevonc.2020.103163.
Cross NCP, Ernst T, Branford S, Cayuela JM, Deininger M, Fabarius A, et al. European LeukemiaNet laboratory recommendations for the diagnosis and management of chronic myeloid leukemia. Leukemia. 2023;37:2150–67. https://doi.org/10.1038/s41375-023-02048-y.
Franke GN, Maier J, Wildenberger K, Cross M, Giles FJ, Müller MC, et al. Comparison of real-time quantitative PCR and Digital Droplet PCR for BCR-ABL1 Monitoring in Patients with Chronic Myeloid Leukemia. J Mol Diagn. 2020;22:81–9. https://doi.org/10.1016/j.jmoldx.2019.08.007.
Wang WJ, Zheng CF, Liu Z, Tan YH, Chen XH, Zhao BL, et al. Droplet digital PCR for BCR/ABL(P210) detection of chronic myeloid leukemia: A high sensitive method of the minimal residual disease and disease progression. Eur J Haematol. 2018;101:291–6. https://doi.org/10.1111/ejh.13084.
Wang X, Xing D, Liu Z, Zhang Y, Cheng B, Sun S, et al. Establishment and evaluation of digital PCR methods for HER2 copy number variation in breast cancer. Anal Bioanal Chem. 2023;415:725–33. https://doi.org/10.1007/s00216-022-04466-w.
Schwaber J, Andersen S, Nielsen L. Shedding light: The importance of reverse transcription efficiency standards in data interpretation. Biomol Detect Quantif. 2019;17:100077. https://doi.org/10.1016/j.bdq.2018.12.002.
Acknowledgements
We appreciate the Institute of Biotechnology, Chinese Academy of Agricultural Sciences, FOREVERGEN Co., Ltd., and Sniper Medical Technology Co., Ltd. for their participation in the inter-laboratory assessment.
Funding
This work was supported by the National Key Research and Development Program of China (2023YFF0613300).
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology: Lianhua Dong. Methodology: Lianhua Dong, Yi Yang, Chunyan Niu, Xia Wang. Investigation and data curation: Yi Yang, Shujun Zhou, Huafang Gao, Xiaohua Jin, Shangjun Wang, Meihong Du, Xiaoyan Cheng, Lingxiang Zhu. Writing—original draft preparation: Lianhua Dong, Yi Yang. Supervision: Lianhua Dong. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
Corresponding author
Ethics declarations
Ethical approval
This study does not involve any ethical concerns.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, Y., Wang, X., Niu, C. et al. Establishment of genomic RNA reference materials for BCR-ABL1 P210 measurement. Anal Bioanal Chem (2024). https://doi.org/10.1007/s00216-024-05492-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00216-024-05492-6