
Abstract
The National Institute of Standards and Technology (NIST) has prepared four seafood reference materials (RMs) for use in food safety and nutrition studies: wild-caught and aquacultured salmon (RM 8256 and RM 8257) and wild-caught and aquacultured shrimp (RM 8258 and RM 8259). These materials were characterized using genetic, metabolomic (1H-NMR, nuclear magnetic resonance and LC-HRMS/MS, liquid chromatography high-resolution tandem mass spectrometry), lipidomic, and proteomic methods to explore their use as matrix-matched, multi-omic differential materials for method development towards identifying product source and/or as quality control in untargeted omics studies. The results from experimental replicates were reproducible for each reference material and analytical method, with the most abundant features reported. Additionally, differences between the materials could be detected, where wild-caught and aquacultured seafood could be distinguished using untargeted metabolite, lipid, and protein analyses. Further processing of the fresh-frozen RMs by freeze-drying revealed the freeze-dried seafoods could still be reliably discerned. These results demonstrate the usefulness of these reference materials as tools for omics instrument validation and measurement harmonization in seafood-related studies. Furthermore, their use as differential quality control (QC) materials, regardless of preparation method, may also provide a tool for laboratories to demonstrate proficiency at discriminating between products based on source/species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
NIST is one of the National Metrology Institutes (NMIs) across the globe responsible for producing Reference Materials (RMs), materials sufficiently homogenous and stable with respect to specified properties that have been established to be fit for their intended purpose. NIST also produces Certified Reference Materials (CRMs), which are more rigorously characterized by metrologically valid procedures and include associated property values, uncertainty calculations, and a statement of metrological traceability [1]. CRMs are essential for providing higher-order calibration and validation materials to secondary standard producers. However, it would be unrealistic for NMIs to certify every material for every analyte due to the time and monetary investment required for this degree of characterization, which additionally is not appropriate for many research applications. Though RMs do not possess certified values, reference values are provided for specific analytes, rendering them useful for method development, between-method harmonization, and within-method precision as QC materials, an important component to help ensure labeling and regulatory requirements are met in the food industry [2].
The development of matrix-based RMs for use in food safety and nutrition studies has increased in recent years, and though a wide variety of RMs could be generated for these purposes, the cost of production, including processing, subsequent testing, storage, and shipping, must be taken into consideration, as well as the effect of production on the functionality of the material. Typically, the best practice is to use RMs that closely match the experimental samples with respect to matrix, species, preparation method, etc. However, other details such as availability, storage, and shipping costs/constraints must also be considered. Fresh-frozen (FF) materials, though more similar to food products, are costly to store and generally require dry ice for shipment, rendering these materials inaccessible to some customers due to shipping regulations. Freeze-dried (FD) materials are more biologically and chemically stable (provided that certified parameters are also stable at higher temperatures) and less expensive to store and ship. However, the drying process and method used have been shown to affect the metabolic composition of the material [3], whereby some metabolites increase while others decrease, and changes due to freeze-drying are inconsistent across different species [4]. Though processing method effects are not entirely understood, freeze-drying of biological samples is still a common method for many laboratories, including those conducting omics research [5].
Untargeted omics methods are useful for holistic screening and comparative studies, generating large amounts of data for wide-view comparative analyses. Putative compound identifications are made across a broad range of compound classes, which can then be investigated to answer more specific research questions. There are some disadvantages to these untargeted methods, however. The instrumentation and analysis can be costly and reference libraries used to make compound identifications can be incomplete, rendering a portion of the collected data unusable. Additionally, the field is in need of RMs to assess the suitability of the analytical method used, to benchmark measurements, and to evaluate instrument precision and harmonize instrument performance within studies and across laboratories [6]. Such RMs could exist as single, thoroughly characterized materials, or as suites comprised of multiple materials (e.g., differential materials) that each have distinct metabolic profiles, which would promote harmonization of measurements through the detection of differences between the materials. As such, there is added value in the characterization of RMs to provide information to the broader research community on overall material composition and compound profiles.
To this end, NIST utilized four new seafood RMs to explore the metabolomic, lipidomic, and proteomic profiles of salmon and shrimp originating from either wild-caught (WC) or aquacultured (AQ) sources. The species of the materials were characterized using Sanger sequencing. The materials were then probed via differential analysis using untargeted omics (NMR and LC-HRMS/MS) to identify highly abundant components and provide initial characterization of each material for use as possible control materials. Additionally, NIST partnered with the Joint Research Centre (JRC) of the European Commission to evaluate differences in biochemical constituents in response to different production methods, whereby the FF RMs were further processed by freeze-drying. The FD materials were included in comparisons of the FF materials using the same omics methodologies. The successful use of RMs in omics-based studies can provide a powerful means to harmonize measurements made across laboratories and platforms, thereby increasing confidence and consistency in measurements, including those pertaining to seafood-related studies.
Materials and methods
The following NIST RMs were used for multi-omics analysis: RM 8256 Wild-caught Coho Salmon, RM 8257 Aquacultured Coho Salmon, RM 8258 Wild-caught Shrimp, and RM 8259 Aquacultured Shrimp. Both salmon materials were prepared from the edible parts (muscle and skin only) of 12 WC fish and approximately 30 AQ fish. The AQ fish were smaller than the WC, so more individuals were required for RM production. All fish were frozen following catch/harvest and maintained frozen until processing, when fish were partially thawed and filleted, and meat was chopped into smaller pieces before refreezing. The shrimp materials were prepared from edible shrimp meat (heads and shells removed) and RM 8258 was prepared from a mixture of wild-caught brown shrimp and Atlantic white shrimp [7]. As with the salmon, shrimp were frozen following catch/harvest, partially thawed to remove the shells and tails, and refrozen. All materials were cryohomogenized and resultant homogenate was stored frozen (≤ − 80 °C) prior to any further processing/analysis.
During production and before bottling, a portion of each FF homogenate was set aside for freeze-drying in an Epsilon 1-6D freeze-dryer (Martin Christ, Osterode, DE) at the JRC in Geel, Belgium. All tools and equipment contacting the different matrices were cleaned using DNA-erase (MP Biomedicals, Solon, OH, USA) to preclude possible carry-over of material between different RMs. Freeze-drying shelves were cooled to − 40 °C and held for ≈ 7 h. The chamber pressure was then reduced to 10 Pa and held for ≈ 35 h. During this hold time, the shelf temperature was gradually increased in ten-degree increments approximately every 5 h. The pressure was then reduced to 1 Pa in the secondary drying step and held for an additional ≈ 20 h, while the shelves gradually increased to room temperature. The total run time, including ramps, was 70 h. This resulted in materials with approximately 3% residual water content (mass fraction) as determined using the Karl Fischer titration method. Between each material, the trays used in the freeze dryer were thoroughly cleaned to prevent carry-over as detailed above. Following the freeze-drying process, each material was milled using three separate mill chambers, two times for 3 min each at 13 cps (cycles per second) in a SPEX 6870 Freezer/Mill (Metuchen, NJ, USA). Material from the containers was then pooled and blended for 30 min using a small T2 Turbula mixer (WAB, Basel, CH) prior to being shipped back to NIST in Charleston, SC, for analysis.
Genetic analysis
For each RM, suppliers provided information on species identity based on morphology, and species analysis for all FF and FD materials was conducted using Sanger sequencing. Of note, in the US market, domestic WC shrimp species (here Penaeus aztecus; syn. Farfantepenaeus aztecus) are not known to be commercially available from AQ sources. Conversely, the primary AQ species (Penaeus vannamei; syn. Litopenaeus vannamei) is not documented in the domestic WC fishery. Therefore, the WC and AQ shrimp were expected to represent different species.
Briefly, DNA was extracted from the tissues with a Qiagen BioSprint 15 robotic workstation (Qiagen, Hilden, Germany) and a portion of the mitochondrial DNA (mtDNA) cytochrome c oxidase subunit III and NADH dehydrogenase 3 (COIII/ND3) region was amplified from the salmon samples, while a portion of the mtDNA 16S region was amplified from the shrimp samples. These targets are considered the source of “comparability of identity” for the respective seafood types. The PCR products were sequenced and the resulting sequences were evaluated for quality, edited, and aligned using Sequencher™ v5.1 (Gene Codes Corporation, Ann Arbor, MI, USA). Using MEGA X [8], the sequences were then aligned with the appropriate National Oceanic and Atmospheric Administration (NOAA) Northwest Fisheries Sciences Center databases and identifications were made based on sequence similarity and topography of phylograms. Mitochondrial DNA sequences from known species references were used to establish inclusivity, and mtDNA sequences from close relatives were used to establish exclusivity. Extraction and PCR blanks and known voucher specimens were used as negative and positive controls, respectively.
Phylograms were constructed and phylogenetic relationships of the sequences were inferred using the Neighbor-Joining method [9] with 500 bootstrap replicates, p-distances, and pairwise deletion. All codon positions were included. The salmon alignment was 326 bp long and the shrimp alignment was 581 bp long. Evolutionary analyses were conducted in MEGA X and optimal trees are shown in Figures S1–S4.
Metabolomic/lipidomic sample preparation and analysis
Four aliquots of each FF RM were randomly selected across the production batches for analysis. The water fraction of the shrimp and salmon materials was assumed to be 78% and 71%, respectively, based on previous studies with these organisms [10, 11]. Approximately 200 mg ± 8.5 mg (standard deviation, SD) of each FF sample was subsampled into previously cooled glass tubes and kept frozen until extraction. Additionally, 45 mg ± 1.1 mg SD of FD shrimp and 58 ± 0.6 mg SD of FD salmon were subsampled and kept at room temperature until extraction. Material from four jars (one from each RM) was combined to create a pooled sample for QC for each preparation type (e.g., Pooled CM-Wet and Pooled CM-Dry). These materials were extracted in quadruplicate along with the experimental samples and quality control results are shared in the Supplementary Information (Figures S5–S7).
All extraction solvents and samples were kept on ice during the procedure and experimental samples were extracted in batches across two different investigators over a two-day period. A 2:2:1.8 solvent system of methanol:chloroform:water based on Wu et al. [11] was added to each sample and extracted according to Schock et al. [10]. The volumes were added in a two-step method. First, 4.0 mL/g wet mass of cold methanol was added to each sample followed by 0.82 mL/g wet mass of cold water for shrimp or 0.89 mL/g wet mass of cold water for salmon and the samples were vortexed for 1 min each. Next, 4.0 mL/g wet mass of cold chloroform was added to each sample followed by 2.0 mL/g wet mass of cold water. The samples were vortexed for 1 min each then allowed to sit on ice for 10 min. Samples were centrifuged (Eppendorf 5810R centrifuge, Eppendorf AG, Hamburg, DE) at 2000 × g for 5 min at 4 °C for phase separation.
The aqueous polar layer was removed using a glass pipet, taking care not to disturb the protein layer and phase transition, and placed in a pre-weighed microcentrifuge tube for 1H-NMR analysis (sample set A). An aliquot of 500 µL was removed from each microcentrifuge tube and placed in an additional pre-weighed microcentrifuge tube for MS analysis (sample set B). Samples were dried and rehydrated as previously described [10]. Samples from set A (550 µL) were transferred into 5-mm NMR tubes for NMR spectroscopy analysis. Sample set B was stored at − 80 °C prior to LC–MS/MS analysis, when the samples were resuspended in 2% methanol in water. The nonpolar fraction was removed using a glass pipet, placed in pre-weighed autosampler vials, dried in the vacufuge, weighed, and stored at − 80 °C until analysis. Nonpolar samples were resuspended in 1 mL of 60% acetonitrile in water, placed in clean microcentrifuge tubes, and spun at 4 °C for 5 min at 12,000 × g. Approximately 50 µL was transferred to each of two clean autosampler vials with inserts for positive ionization and negative ionization data-dependent MS acquisition.
NMR spectroscopy
Both one-dimensional (1D) 1H NMR spectra and two-dimensional (2D) 1H-13C heteronuclear single quantum correlation (HSQC) NMR spectra were acquired on a Bruker Avance II 700 MHz NMR spectrometer equipped with a TCI cryoprobe and Samplejet (Bruker Biospin, Inc., Billerica, MA). Full NMR method details and instrument settings can be found in the Supplementary Information. All raw data files are available on the Mass Spectrometry Interactive Virtual Environment (MassIVE) [12], dataset MSV000092716 and representative 1D NMR spectra are provided in Figure S8.
Following data acquisition, the top ten abundant metabolites for each RM were determined using the automatic peak picking feature in TopSpin3.5 software (Bruker Biospin, Billerica, MA) and sorting the peaks by intensity. Many of the peaks were part of the same compound, so up to 60 peaks were annotated until ten different metabolites were obtained. MetaboAnalyst [13] was used for statistical analysis and all data were transformed with normalization by sum, mean centering, and pareto scaling. Principle component analysis (PCA) was used to assess technical variability among QC replicates (Figures S5–S7) and to examine the RM suitability for differential analysis. Where separation between materials using PCA was observed, a further unsupervised analysis, partial least squares discriminant analysis (PLS-DA), was used for each comparison: WC FF vs. AQ FF, WC FD vs. AQ FD, WC FF vs. WC FD, AQ FF vs. AQ FD. The PLS-DA was used to obtain a variable importance projection (VIP) score for each of the intelligent bins to rank resonances based on their contribution to the observed variance between materials and processing methods. The top 20 bins with a VIP score > 2.0 were considered for annotation.
Metabolites were assigned using both the 1D 1H and 2D 1H-13C HSQC spectra and were putatively identified as Metabolomics Standard Initiative Level 2 [14]. Metabolite assignments were based on chemical shift comparisons using reference spectra from the human metabolome database (HMDB) [15], the Biofluid Reference Compound Database (bbiorefcode_0_1_2; Bruker Biospin, Inc., Billerica, MA), an in-house compiled database, and the database in Chenomx NMR Suite Profiler v8.5 (Chenomx Inc., Edmonton, Alberta).
LC-HRMS/MS
Full method details and instrument settings for the metabolomic and lipidomic analyses can be found in the Supplementary Information. Briefly, samples were analyzed using a Vanquish UPLC coupled to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Metabolite separation was achieved using an Acquity HSS T3 (1.8 µm, 2.1 mm id × 150 mm length; Waters, Milford, MA, USA) C18 column following the gradient program outlined in Table S2 in both positive and negative data acquisition modes. Individual sample extracts were then pooled and analyzed in Acquire X data acquisition mode to provide MS2 spectra. Lipid separation was achieved using an Acquity UPLC BEH (1.7 μm, 2.1 mm id × 100 mm length; Waters, Milford MA, USA) C18 column following the gradient program outlined in Table S3 in both positive and negative data acquisition modes. Individual sample extracts were then pooled and analyzed in Acquire X data acquisition mode to provide MS2 and MS3 spectra. All raw data files are available on MassIVE [12], dataset MSV000092716.
Following data acquisition, resulting raw files were processed and searched with Compound Discoverer (Thermo Fisher, Ver 3.3 SP1) using both local (NIST20, LipidBlast_V68, curated mass lists) and online databases, including mzCloud™ and Chemspider (Royal Society of Chemistry). To conduct the Compound Discoverer searches, retention time alignment was used with the ChromAlign model. The detect compounds node was set to a mass tolerance of 5 ppm, intensity threshold of 100,000 S/N threshold of 1.5, base ions of [M + H]1, [M—H]1-. The assign compound annotation node was used with 5 ppm mass tolerance with mzCloud, mzValult, ChemSpider, mass list search, and predicted compounds. All library searches of mzVault and mzCloud included a mass tolerance of 10 ppm of precursor and product ions with the HighChem-HighRes (mzCloud) and NIST (NIST20 and LipidBlast_V68) algorithms. Chemspider and local mass lists were searched with a mass tolerance of 5 ppm. Total mass features were reduced, background features filtered from the final analysis, and results exported in.csv format for further data comparison. Signal intensities were used to infer feature abundances for sample comparisons.
To evaluate measurement reproducibility among triplicate sample injections, the exported feature list generated in Compound Discoverer was used to normalize the peak area of each feature to the total peak area of features in that injection (sum normalization) for both the positive and negative data acquisition modes. The average normalized peak area from individual injections (n = 3) was then calculated for each prepared sample (n = 4) and the % CV calculated for individual features to evaluate measurement reproducibility.
A reduced feature list was exported from Compound Discoverer using a Sample Group % CV ≤ 20% and a full match annotation filter for both the positive and negative data acquisition modes. Each annotated feature list was indexed and imported into MetaboAnalyst (ver. 5.0) for further processing. Within MetaboAnalyst, interquartile range (IQR) filtering, normalization by sum, log transformation, and pareto scaling were used to produce all PCA plots.
Proteomic sample preparation and analysis
Approximately 1 mg (exact mass known) of each FF RM was subsampled from eight jars (randomly sampled across the production batch) into separate 1.5 mL LoBind microcentrifuge tubes. Additionally, 1 mg (exact mass known) of each FD RM was subsampled from three jars (randomly sampled across the production batch) into separate 1.5 mL LoBind microcentrifuge tubes. The proteins were then isolated using the RapiGest TCEP/CAA method outlined in Davis et al. [16]. Each sample was processed through the C18 spin columns twice (maximum binding capacity of 30 µg of peptide) and the eluted fractions were combined yielding a maximum of 60 µg of peptide. These solutions were evaporated to dryness in a vacuum centrifuge and the two samples were combined in 75 µL 5% acetonitrile in water for analysis.
Samples were analyzed using an UltiMate 3000 Nano LC coupled to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) and separation was achieved using an Acclaim PepMap RSLC 2 µm C18 column (75 µm id × 25 cm length; Thermo Fisher Scientific) according to the gradient program outlined in Table S5. Full LC–MS/MS method details and instrument settings can be found in the Supplementary Information and all raw data files are available on MassIVE [12], dataset MSV000092716.
Following data acquisition, resulting raw files were processed and searched with Proteome Discoverer (Thermo Fisher, Ver 2.5) using Sequest HT algorithm. Two sets of searches included a database of coho salmon (Oncorhynchus kisutch, Taxonomy ID = 8019) retrieved from the 2023_2 release of the UniProtKB SwissProt database, along with Penaeoidea taxonomy (shrimp species SuperFamily Taxonomy ID = 111,520) retrieved from the 2023_2 release of the UniProtKB SwissProt database. All searches also included the common Repository of Adventitious Proteins database (cRAP; 2012.01.01; the Global Proteome Machine; 107 sequences).
The following search parameters were used: trypsin was specified as the enzyme allowing for two mis-cleavages; carbamidomethyl (C) was fixed and acetylation (protein n-term), deamidated (NQ), pyro-Glu (n-term Q), and oxidation (M) were variable modifications; 10 ppm precursor mass tolerance and 0.02 Da fragment ion tolerance. Within Sequest HT, the peptide length was specified as a minimum of six and maximum of 144 amino acids. Resulting peptide spectral matches were validated using the percolator algorithm, based on q-values at a 1% false discovery rate (FDR) and a minimum of one unique peptide ID was required to validate protein identification. Exported protein lists were indexed and imported into MetaboAnalyst (ver. 5.0) for further processing. Within MetaboAnalyst, normalization by median, log transformation, and pareto scaling were used to produce all PCA plots.
Results and discussion
Genetic analysis
Though the seafood materials were acquired from trusted sources, genetic analysis was completed on the RMs to corroborate morphological species identification. The sequences for salmon RM 8256 and RM 8257 clustered with the Oncorhynchus kisutch (coho salmon) clade in 100% of the bootstrap replicates (Figures S1 and S2, respectively). The sequences for WC shrimp (RM 8258) clustered with the Penaeus aztecus (syn. Farfantepenaeus aztecus, brown shrimp) clade in 100% of the bootstrap replicates (Figure S3) and the sequences for AQ shrimp (RM 8259) clustered with the P. vannamei (syn. Litopenaeus vannamei, white-legged shrimp) clade in 100% of the bootstrap replicates (Figure S4). The results were consistent among the FD materials (data not shown), suggesting the regions amplified by PCR were not substantially altered during the freeze-drying process.
Of note, RM 8258 was later determined by Next Generation Sequencing (NGS) to be primarily generated from brown shrimp (P. aztecus), but a minor component of P. setiferus (Atlantic white shrimp) was detected (in preparation). Species composition of the domestic WC shrimp fishery varies seasonally, and mixed species catches are not uncommon. PCR-based Sanger sequencing, employed in this study, is not the most appropriate method for detecting species mixtures in products. Various shrimp species can occupy the same environmental niche, allowing for a mixed catch during harvesting. However, depending on the ratio of species present in a mixed catch, Sanger sequencing may not detect trace species in small quantities in a mixture and, in cases where contamination is apparent, Sanger sequences cannot be reliably deconvoluted to identify the contributing species. More sensitive methods (such as Next Generation Sequencing) are required for identification of species within mixtures. In addition to genetic analysis, other analytical techniques must also be employed for product verification to assist in identifying source.
Omics characterization of RMs
The data for each analysis were initially assessed for reproducibility using both replicate preparations and pooled control materials. Pooled control materials show the technical variance of sample extraction and NMR and MS measurement. The tight clustering of the replicates and pooled controls across each analytical platform indicates the technical variability was low (Figure S5–S7) and differences observed in the RMs were a result of metabolomic differences. In general, slightly greater variance was observed in the pooled control materials than in the RM replicates. This is likely because the pooled control materials were prepared by crude mixing of portions of each material immediately prior to sample preparation rather than rigorous homogenization of the materials, which is accomplished during RM production.
To utilize the seafood materials for matrix-matched QC applications, reference compounds must first be identified for use as benchmarks for comparability between studies. The top ten most abundant metabolites identified in the RMs using NMR are shown in Table S6. For the MS metabolite and lipid analyses, the top ten most abundant features identified in each RM according to analytical method and mode of detection are shown in Table S7. The ten most abundant proteins in each material are shown in Table S8 with the total number of protein groups, proteins, peptide groups, and peptide-spectrum matches (PSMs) identified in each RM shown in Table S9. Taurine, an amino acid commonly found in fish and shellfish, was identified using both NMR and MS methods. Additionally, analysis of the MS metabolite data indicates that many components of the ATP-degradation pathway previously described in fish meat were identified in both salmon and shrimp, such as adenosine diphosphate (ADP), adenosine monophosphate (AMP), inosine monophosphate (IMP), inosine, and hypoxanthine [17]. Lipids commonly associated with seafoods were also identified by MS (e.g., EPA, DHA, and arachidonic acid) as were seafood protein allergens such as tropomyosin and light- and heavy-chain myosin.
The consistent identification of features is highly dependent on several factors, including sample preparation method, instrument method and data processing method. Variation in any of these factors can affect the identification and relative ratios of the compounds present in a sample, emphasizing the importance of accurately reporting specific experimental details [18]. Additionally, the existence of a well-annotated reference dataset generated from the species in question is also required, as comparisons cannot necessarily be made across species. It is important to note that due to the sample preparation method utilized, the chromatographic method, and ion suppression that may occur during electrospray ionization during the MS analyses, the metabolites with the greatest relative abundance may not be representative of the most concentrated metabolites in the samples. However, this type of data in its entirety still provides a holistic view of the metabolite and lipid profiles of these materials and can serve as a mode of discovery to formulate research questions, demonstrating the use of untargeted analytical methods in identifying compounds of interest for future study.
Material differentiation by source/species
Metabolites
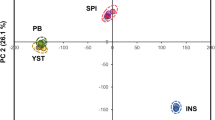
The metabolite profiles of seafood materials were compared for biochemical differences resulting from source (WC vs. AQ). The PCA comparison of WC and AQ materials for both salmon and shrimp (Fig. 1) revealed that each organism possessed distinct metabolic profiles based on source/species by NMR. A discriminant analysis revealed that 17 putative metabolite identifications were significantly different (VIP > 2.0 from PLS-DA; Q2 = 0.89 and R2 = 0.99) between WC and AQ FF salmon and 17 putative metabolite identifications were significantly different (VIP > 2.0 from PLS-DA; Q2 = 0.99 and R2 = 0.99) between WC and AQ FF shrimp (Table 1). Seven of these metabolites were common in both comparisons and exhibited the same directional change in relative concentration based on material source/species.

WC and AQ salmon (A) and shrimp (B) comparisons by PCA using NMR data. A clear source difference is shown in the metabolite profile of WC (RM 8256) and AQ (RM 8257) salmon RMs and WC (RM 8258) and AQ (RM 8259) shrimp RMs. The processing method, FD (dry) and FF (wet), did not appear to significantly influence the metabolite profile of the salmon materials, while processing method did influence the metabolite profile of the shrimp materials. Ellipses represent 95% confidence intervals
Metabolite analysis by LC-HRMS/MS also revealed that salmon and shrimp materials could be distinguished based on source in both positive (Fig. 2) and negative (Figure S9) ionization modes. Mass spectrometry feature differences are represented in Table 2. There were far more differences observed between the shrimp materials than the salmon materials, likely because the shrimp materials innately represent different species by source. Although an untargeted data acquisition method was used for the MS analysis, only betaine (salmon) and phenylalanine (shrimp) showed the opposite trend from the NMR observed differences. There were several metabolites that were not measured by MS due to their low m/z, but of the 11 that were measured by both analytical methods in the salmon, six demonstrated the same trend. The shrimp data was in slightly better agreement, with eight of the 12 commonly measured metabolites demonstrating the same trend by both NMR and MS (Table 1).

WC and AQ salmon (A) and shrimp (B) metabolite comparison by PCA using LC-HRMS/MS data in positive mode. A clear difference is shown in the metabolite profile of WC (RM 8256) and AQ (RM 8257) salmon RMs and WC (RM 8258) and AQ (RM 8259) shrimp RMs. The processing method, FD (dry) and FF (wet), also significantly influenced the metabolite profile of both the salmon and shrimp materials. Ellipses represent 95% confidence intervals
Since the seafood RMs were all stored and processed in a similar way, it is reasonable to suggest that the differential expression of metabolites may be due to dietary and/or species differences, as metabolism is greatly affected by nutritional input. Wild adult coho salmon typically feed on fish and squid, while AQ salmon are fed fish feed, which can include fishmeal as well as items not naturally found in the diet, such as poultry, nuts, and soy [19]. Wild shrimp, like P. aztecus (syn. Farfantepenaeus aztecus), are omnivorous bottom-feeders and eat mainly worms, algae, and other organic debris, while AQ shrimp, like P. vannamei (syn. Litopenaeus vannamei), are fed a feed consisting of plant products and fish meal [20]. Studies have shown that in addition to diet, age, sex, temperature, and chemical exposure [21,22,23] can also affect metabolite profiles in aquatic animal tissues. Here, AQ salmon were noticeably smaller than WC salmon and could have represented a younger cohort. Though specific aspects of the life history of the animals are unknown, variation in these factors could also have been attributed to the differences observed between the WC and AQ materials. Metabolite profiles have been used to differentiate between wild tuna by geographic origin [24]; however, metabolite profile variations have not yet been used to authenticate seafood products at import. Currently, this application is limited by a lack of verified reference databases for seafoods of interest. Because profiles change based on diet/origin, an extensive exploration of seafood metabolomes from a wide array of products from different regions would be required to ensure the variability across WC and AQ products available in a specified market is represented in the reference data, while differences between sources/species can still be detected. However, by starting with those products most regularly targeted for fraudulent activities, the field can begin to build reference databases to detect these activities.
Lipids
The lipid profiles of the NIST seafood materials were also compared for biochemical differences resulting from source (WC vs. AQ). Salmon and shrimp materials could be differentiated by source/species using lipid profile data in both positive (Fig. 3) and negative (Figure S10) ionization modes, with the number of feature differences represented in Table 2. Consistent with the metabolite data, the greater number of differences between the shrimp materials is likely explained by species composition.

WC and AQ salmon (A) and shrimp (B) lipid comparison by PCA using LC-HRMS/MS data in positive mode. A clear difference is shown in the lipid profile of WC (RM 8256) and AQ (RM 8257) salmon and WC (RM 8258) and AQ (RM 8259) shrimp RMs. The processing method, FD (dry) and FF (wet), also significantly influenced the lipid profile of both the salmon and shrimp materials. Ellipses represent 95% confidence intervals
Salmon have previously been differentiated by source and geographic origin using fatty acid profiles, including omega-3 and omega-6 fatty acids, with AQ salmon containing more omega-3 fatty acids than WC [25]. The untargeted lipid data were assessed in Skyline (University of Washington, version 22.2.0.351) to determine if some of the variability between the salmon could be attributed to the known differences in omega-3 and omega-6 fatty acid content. The ratio of the sum of the peak areas of the omega-6 fatty acids (linoleic acid and arachidonic acid) to the sum of the peak areas of the omega-3 fatty acids (α-linolenic acid, EPA, and DHA) was calculated to be 0.02 and 0.03 for the WC salmon FF and FD samples, respectively, and 0.10 and 0.20 for the AQ salmon FF and FD samples, respectively. Though the ratios are not in agreement with the quantitative analysis for the same fatty acids (0.05 and 0.41 respectively for the fresh-frozen salmon and shrimp RMs) [7], the ratios remain significantly different and can easily be distinguished by this untargeted lipid profiling method.
Proteins
The protein profiles of the NIST seafood materials were compared and both salmon and shrimp materials could be differentiated by source (WC vs. AQ, Fig. 4). According to the associated RM information sheets, the concentration of total protein in the WC RMs (23.77% and 21.63% for the salmon and shrimp respectively) was slightly higher than that in the AQ RMs (20.60% and 19.30% for the salmon and shrimp respectively) for both species [7], which could be a contributing factor to the observed differences and may be due to their differing diets. Though ideally, fish feed should have a similar nutritional composition as the diet of wild-caught fish, the nutritional value is heavily impacted by the quality of the ingredients used to produce the feed and the production process itself. Fish feeds made from fish byproducts typically have a lower protein content than higher-quality feed made from whole fish, thereby affecting the protein composition of the animal ingesting it [26]. This could account for some of the observed differences. Additionally, commonly known protein allergens for salmon (e.g., parvalbumin, tropomyosin, triosephosphate isomerase, serum albumin) and shrimp (myosin, tropomyosin, paramyosin, troponin sarcoplasmic calcium-binding protein) were identified in each of the respective materials. As such, these materials may be useful in future allergen studies, though appropriate stability testing should be completed for each RM before utilizing the materials for this purpose to ensure allergen protein abundances and their ratio to the total protein mass fractions are maintained.

WC and AQ salmon (A) and shrimp (B) protein comparison by PCA using LC-HRMS/MS data in positive mode. A clear difference is shown in the protein profile of WC (RM 8256) and AQ (RM 8257) salmon and WC (RM 8258) and AQ (RM 8259) shrimp RMs. The processing method, FD (dry) and FF (wet), also significantly influenced the protein profile of the salmon and shrimp materials. Ellipses represent 95% confidence intervals
Material differentiation by preparation
Metabolites
To determine whether freeze-drying of the NIST RMs affected the metabolic composition of the seafood materials, the profiles of the dry materials were compared to their FF (wet) counterparts. NMR analysis of NIST seafood reference materials revealed no observable alteration in the metabolic profile of the WC and AQ salmon materials due to freeze-drying; however, the processing method altered the metabolic profile of both WC and AQ shrimp materials (Fig. 1). Seventeen putative metabolites were significantly different between processing methods in WC shrimp and 16 metabolites were significantly different between processing methods in AQ shrimp (Table 3). The freeze-drying process caused 14 of the same metabolites to be altered for both shrimp materials, and the directional change in relative concentration based on the processing method was the same for ten of those 14 metabolites (Table 3). Two unknown peaks noted in both shrimp materials also decreased with the freeze-drying method. Ten consistently altered metabolites, except leucine, had an increase in relative concentration due to the freeze-drying process. The four remaining metabolites in the shrimp materials that changed inconsistently between processing methods could be a result of the extraction method. The ratio of the solvents within the extraction system is important for complete phase separation of polar and nonpolar metabolites. Estimates of tissue water content were used for each organism based on previous studies, which may impact the extraction efficiency of polar metabolites, thereby influencing the metabolite changes due to processing method. Similar inconsistent changes in metabolites between species and between samples subjected to freeze-drying (as shown here with the shrimp) were also demonstrated in a plant tissue study [4]. However, previous NMR analysis of mussels has shown that wet tissue extracts can be less stable than dry tissue extracts [27]. Therefore, freeze-drying is still a beneficial technique for RMs intended for use as QC materials in differential metabolomics analysis.
The LC-HRMS/MS analysis revealed that the salmon and shrimp materials were differentiated by preparation method using PCA (Fig. 2), indicating that the freeze-drying process altered metabolite composition. Differences were seen in positive and negative ionization modes for both salmon and shrimp (negative mode, Figure S8), with the number of feature differences listed in Table 4. The patterns observed when comparing the FD and FF preparations were not consistent between the salmon and shrimp, with a greater number of features being more abundant in the FD salmon materials when compared to the FF salmon, while fewer features were more abundant in the FD shrimp materials when compared to FF shrimp. The variable effects of freeze-drying have been previously documented and were shown to be inconsistent among species [4]. Additionally, material processing effects also varied with analytical method (NMR vs. MS) which may be because HRMS/MS is a more sensitive analytical technique, so less abundant compounds could be contributing to the cumulative differences between processing methods. Quantitative analysis would better elucidate the effects of freeze-drying on specific metabolites of interest in these materials.
Lipids
Salmon and shrimp materials were also differentiated by preparation method using lipid profile data in both positive (Fig. 3) and negative (Figure S10) modes, with the number of feature differences represented in Table 4. The majority of the lipid differences observed indicated greater signal in the FD materials compared to the FF materials. These differences could be due to moisture corrections, or changes in lipid composition due to the freeze-drying process. Other studies conducted on foodstuffs have not demonstrated a significant change in the concentration of some nutritional lipids [28]. However, the untargeted technique employed here may elucidate the cumulative effect of freeze-drying on the overall lipid profile, since such a large number and wide breadth of compounds were evaluated. Additionally, polar lipid and metabolite changes previously observed in FD plant material demonstrated the need to confirm recovery for each compound of interest [4]. This principle may also apply to nonpolar lipids, suggesting quantitative analysis of lipids of interest should be conducted.
Proteins
Salmon and shrimp materials could also be differentiated by preparation method using protein profile data (Fig. 4); however, the analysis of additional FD samples would provide more power to the analysis. To account for the digested sample mass difference due to water content, injection volumes for the FD samples were half of those for the FF (1 µL vs. 2 µL, with approximately 2 µg of peptides injected). It is possible that some of the separation could be influenced by water present in the material. However, it is well documented that freeze-drying and other preparation methods can cause conformational changes to proteins [29, 30], and some of those changes may have been detected using the current method. This should be taken into consideration when assessing FD materials for protein content.
Conclusions
The NIST WC and AQ salmon and shrimp RMs can be utilized to explore markers of source and/or species using the various omics platforms demonstrated here, and can also serve as individual, homogenous, matrix-matched control materials in untargeted omics applications to evaluate instrument precision, correct data due to instrument variation, and harmonize studies across laboratories. Additionally, WC and AQ salmon and shrimp were distinguishable with respect to metabolite, lipid, and protein signatures, supporting their use as differential RMs. In the omics field, differential RMs are intended to be used in tandem with experimental samples during extraction, instrument analysis, and data processing. Unlike targeted analysis, which is used to assign a quantitative value, this untargeted application can be used for qualitative assessment of analytes present in the material, where the relative difference between the analytes is used for reference, since it should not change if the methodology is consistent. By including these RMs in an analytical workflow, researchers can demonstrate their ability to differentiate between known materials, thereby providing more confidence in the analysis of test samples. In the future, this concept could potentially be applied to the differentiation of food products based on source/species.
Though some modifications were observed in metabolite, lipid, and protein profiles of materials based on production method, the freeze-drying process did not mask the differences observed between the materials based on source/species nor did it affect the expected genetic classification of the materials. This evidence provides a case study suggesting that applying a freeze-drying method to FF RMs for omics-based analyses may be acceptable for those not accustomed to receiving, storing or preparing such materials for analysis. However, because the full extent of the resultant changes due to freeze-drying is not understood, care should be taken when using FD materials for identifying markers of provenance. Taken together, the results promote the usefulness of these RMs as both individual QC materials and differential materials designed to advance the value of untargeted genomics, metabolomics, lipidomics, and/or proteomics studies.
References
Beauchamp CR et al. Metrological tools for the reference materials and reference instruments of the NIST Materials Measurement Laboratory. Special Publication (NIST SP), National Institute of Standards and Technology, Gaithersburg, MD. 2021. https://doi.org/10.6028/NIST.SP.260-136-2021.
Phillips MM, Sharpless KE, Wise SA. Standard reference materials for food analysis. Anal Bioanal Chem. 2013;405(13):4325–35.
Petrova I, Xu SY, Joesten WC, Ni SS, Kennedy MA. Influence of Drying Method on NMR-Based Metabolic Profiling of Human Cell Lines. Metabolites. 2019;9(11):256. https://doi.org/10.3390/metabo9110256.
Oikawa A, Otsuka T, Jikumaru Y, Yamaguchi S, Matsuda F, Nakabayashi R, Takashina T, Isuzugawa K, Saito K, Shiratake K. Effects of freeze-drying of samples on metabolite levels in metabolome analyses. J Sep Sci. 2011;34(24):3561–7. https://doi.org/10.1002/jssc.201100466.
Kirwan JA, Brennan L, Broadhurst D, Fiehn O, Cascante M, Dunn WB, Schmidt MA, Velagapudi V. Preanalytical Processing and Biobanking Procedures of Biological Samples for Metabolomics Research: A White Paper, Community Perspective (for “Precision Medicine and Pharmacometabolomics Task Group”-The Metabolomics Society Initiative). Clin Chem. 2018;64(8):1158–82. https://doi.org/10.1373/clinchem.2018.287045.
Lippa KA, Aristizabal-Henao JJ, Beger RD, Bowden JA, Broeckling C, Beecher C, Davis WC, Dunn WB, Flores R, Goodacre R, Gouveia GJ, Harms AC, Hartung T, Jones CM, Lewis MR, Ntai I, Percy AJ, Raftery D, Schock TB, Sun JC, Theodoridis G, Tayyari F, Torta F, Ulmer CZ, Wilson I, Ubhi BK. Reference materials for MS-based untargeted metabolomics and lipidomics: a review by the metabolomics quality assurance and quality control consortium (mQACC). Metabolomics 2022;18(4). https://doi.org/10.1007/s11306-021-01848-6.
Ellisor D, Place B, Phillips MM, Yen JH. Analysis of Seafood Reference Materials: RM 8256, RM 8257, RM 8258 and RM 8259. Special Publication (NIST SP), National Institute of Standards and Technology, Gaithersburg, MD. 2021. https://doi.org/10.6028/NIST.SP.260-214.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35(6):1547–9. https://doi.org/10.1093/molbev/msy096.
Saitou N, Nei M. The Neighbor Joining Method - A New Method for Reconstructing Phylogenetic Trees. Mol Biol Evol. 1987;4(4):406–25. https://doi.org/10.1093/oxfordjournals.molbev.a040454.
Schock TB, Duke J, Goodson A, Weldon D, Brunson J, Leffler JW, Bearden DW. Evaluation of Pacific White Shrimp (Litopenaeus vannamei) Health during a Superintensive Aquaculture Growout Using NMR-Based Metabolomics. Plos One. 2013;8(3):e59521. https://doi.org/10.1371/journal.pone.0059521.
Wu HF, Southam AD, Hines A, Viant MR. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem. 2008;372(2):204–12. https://doi.org/10.1016/j.ab.2007.10.002.
Wang MX, Wang J, Carver J, Pullman BS, Cha SW, Bandeira N. Assembling the Community-Scale Discoverable Human Proteome. Cell Syst. 2018;7(4):412–21.
Pang ZQ, Zhou GY, Ewald J, Chang L, Hacariz O, Basu N, Xia JG. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17(8):1735–61. https://doi.org/10.1038/s41596-022-00710-w.
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3(3):211–21. https://doi.org/10.1007/s11306-007-0082-2.
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li CR, Karu N, Sayeeda Z, Lo E, Assempour N, Berjanskii M, Singhal S, Arndt D, Liang YJ, Badran H, Grant J, Serra-Cayuela A, Liu YF, Mandal R, Neveu V, Pon A, Knox C, Wilson M, Manach C, Scalbert A. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–17. https://doi.org/10.1093/nar/gkx1089.
Davis WC, Kilpatrick LE, Ellisor DL, Neely BA. Characterization of a human liver reference material fit for proteomics applications. Scientific Data 2019;6(1). https://doi.org/10.1038/s41597-019-0336-7
Li KF, Luo YK, Shen HX. Postmortem Changes of Crucian Carp (Carassius auratus) During Storage in Ice. Int J Food Prop. 2015;18(1):205–12. https://doi.org/10.1080/10942912.2011.614986.
Kirwan JA, Gika H, Beger RD, Bearden D, Dunn WB, Goodacre R, Theodoridis G, Witting M, Yu LR, Wilson ID, Metabolomics Quality Assurance Q. Quality assurance and quality control reporting in untargeted metabolic phenotyping: mQACC recommendations for analytical quality management. Metabolomics 2022;18(9). https://doi.org/10.1007/s11306-022-01926-3
Schmidt V, Amaral-Zettler L, Davidson J, Summerfelt S, Good C. Influence of Fishmeal-Free Diets on Microbial Communities in Atlantic Salmon (Salmo salar) Recirculation Aquaculture Systems. Appl Environ Microb. 2016;82(15):4470–81. https://doi.org/10.1128/aem.00902-16.
Li EC, Xu C, Wang XD, Wang SF, Zhao Q, Zhang ML, Qin JG, Chen LQ. Gut Microbiota and its Modulation for Healthy Farming of Pacific White Shrimp Litopenaeus vannamei. Rev Fish Sci Aquac. 2018;26(3):381–99. https://doi.org/10.1080/23308249.2018.1440530.
Watson AM, Napolitano MP, Schock TB, Bowden JA, Frost J, Yost J, Denson MR. Evaluation of graded levels of soy oil as a fish oil replacement in high soy protein feeds for juvenile red drum, Sciaenops ocellatus. Aquaculture 2020;529. https://doi.org/10.1016/j.aquaculture.2020.735627.
Viant MR, Werner I, Rosenblum ES, Gantner AS, Tjeerdema RS, Johnson ML. Correlation between heat-shock protein induction and reduced metabolic condition in juvenile steelhead trout (Oncorhynchus mykiss) chronically exposed to elevated temperature. Fish Physiol Biochem. 2003;29(2):159–71.
Ekman DR, Teng Q, Jensen KM, Martinovic D, Villeneuve DL, Ankley GT, et al. NMR analysis of male fathead minnow urinary metabolites: A potential approach for studying impacts of chemical exposures. Aquat Toxicol. 2007;85(2):104–12.
Bodin N, Amiel A, Fouche E, Sardenne F, Chassot E, Debrauwer L, Guillou H, Tremblay-Franco M, Canlet C. NMR-based metabolic profiling and discrimination of wild tropical tunas by species, size category, geographic origin, and on-board storage condition. Food Chem. 2022;371. https://doi.org/10.1016/j.foodchem.2021.131094.
Masoum S, Malabat C, Jalali-Heravi M, Guillou C, Rezzi S, Rutledge DN. Application of support vector machines to 1H NMR data of fish oil: methodology for the confirmation of wild and farmed salmon and their origins. Anal Bioanal Chem. 2007;387:1499–510. https://doi.org/10.1007/s00216-006-1025-x.
Hua K, Cobcroft JM, Cole A, Condon K, Jerry DR, Mangott A, Praeger C, Vucko MJ, Zeng CS, Zenger K, Strugnell JM. The Future of Aquatic Protein: Implications for Protein Sources in Aquaculture Diets. One Earth. 2019;1(3):316–29. https://doi.org/10.1016/j.oneear.2019.10.018.
Watanabe M, Meyer KA, Jackson TM, Schock TB, Johnson WE, Bearden DW. Application of NMR-based metabolomics for environmental assessment in the Great Lakes using zebra mussel (Dreissena polymorpha). Metabolomics. 2015;11(5):1302–15. https://doi.org/10.1007/s11306-015-0789-4.
Cavazos-Garduno A, Serrano-Nino JC, Solis-Pacheco JR, Gutierrez-Padilaa JA, Gonzalez-Reynoso O, Garcia HS, Aguilar-Uscanga BR. Effect of Pasteurization, Freeze-drying and Spray Drying on the Fat Globule and Lipid Profile of Human Milk. J Food Nutr Res. 2016;4(5):296–302. https://doi.org/10.12691/jfnr-4-5-5.
Roy I, Gupta MN. Freeze-drying of proteins: some emerging concerns. Biotechnol Appl Bioc. 2004;39:165–77. https://doi.org/10.1042/ba20030133.
Wulff T, Nielsen ME, Deelder AM, Jessen F, Palmblad M. Authentication of Fish Products by Large-Scale Comparison of Tandem Mass Spectra. J Proteome Res. 2013;12(11):5253–9. https://doi.org/10.1021/pr4006525.
Jacob D, Deborde C, Lefebvre M, Maucourt M, Moing A. NMRProcFlow: a graphical and interactive tool dedicated to 1D spectra processing for NMR- based metabolomics. Metabolomics 2017;13(4). https://doi.org/10.1007/s11306-017-1178-y.
Acknowledgements
The authors would like to thank the manuscript reviewers for their thoughtful comments, the National Oceanic and Atmospheric Administration Northwest Fisheries Science Center Forensic Laboratory for assistance in identifying trusted suppliers of these seafood materials, and the NIST Biorepository team for their support in processing the bulk RMs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The NIST Research Protection Office reviewed the protocol for this project and determined it meets the criteria for research that does not require cognizant external Institutional Animal Care and Use Committee (IACUC) approval.
Conflict of interest
The authors declare no comepting interests.
Disclaimer
Official contribution of the National Institute of Standards and Technology. Not subject to copyright in the United States. Certain commercial equipment, instruments, software, or materials are identified in this document. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection Food Safety Analysis 2.0 with guest editor Steven J. Lehotay.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ellisor, D.L., Bayless, A.L., Schock, T.B. et al. Multi-omics characterization of NIST seafood reference materials and alternative matrix preparations. Anal Bioanal Chem 416, 773–785 (2024). https://doi.org/10.1007/s00216-023-04928-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04928-9