
Abstract
An automated microextraction by packed sorbent followed by liquid chromatography–tandem mass spectrometry (MEPS-LC–MS/MS) method was developed for the determination of four endocrine disruptors—parabens, benzophenones, and synthetic phenolic antioxidants—in wastewater samples. The method utilizes a lab-made repackable MEPS device and a multi-syringe robotic platform that provides flexibility to test small quantities (2 mg) of multiple extraction phases and enables high-throughput capabilities for efficient method development. The overall performance of the MEPS procedure, including the investigation of influencing variables and the optimization of operational parameters for the robotic platform, was comprehensively studied through univariate and multivariate experiments. Under optimized conditions, the target analytes were effectively extracted from a small sample volume of 1.5 mL, with competitive detectability and analytical confidence. The limits of detection ranged from 0.15 to 0.30 ng L−1, and the intra-day and inter-day relative standard deviations were between 3 and 21%. The method’s applicability was successfully demonstrated by determining methylparaben, propylparaben, butylated hydroxyanisole, and oxybenzone in wastewater samples collected from the São Carlos (SP, Brazil) river. Overall, the developed method proved to be a fast, sensitive, reliable, and environmentally friendly analytical tool for water quality monitoring.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Endocrine disruptors are a group of chemical compounds that can disrupt the normal functioning of the endocrine system by mimicking or antagonizing hormone properties [1, 2]. These compounds include various organic substances, such as parabens, benzophenones, and which are commonly found in consumer products like food, personal care items, cosmetics, pharmaceuticals, and sunscreens [3]. Consequently, endocrine disruptors can contaminate water sources through processes such as wastewater discharge, runoff, and leaching, and their prevalence in the environment has given rise to significant public concerns regarding the potential risks they pose to human and animal health, as well as the ecological balance.
The determination of endocrine disruptors (EDs) is of utmost importance in monitoring the quality of water and facilitating the development of remediation processes [4]. However, EDs, like other pollutants, are typically found in trace concentrations within water samples. Consequently, their sensitive analysis necessitates the use of efficient analytical tools capable of high sample throughput while ensuring reliable analytical confidence [5]. In this regard, the continued development of rapid, sensitive, and efficient sample preparation strategies for the accurate determination of EDs in water samples remains both relevant and necessary [6].
Nowadays, accurate detection of trace pollutants in water samples is efficiently achieved thanks to modern advancements in liquid chromatography–tandem mass spectrometry (LC–MS/MS). LC–MS/MS is a highly influential technique capable of providing enhanced sensitivity and specificity. However, conducting water quality surveillance via LC–MS/MS still presents significant challenges due to the presence of numerous interfering compounds that can compromise sensitivity and selectivity, leading to inadequate limits of detection (LODs) and quantification (LOQs) [7]. Therefore, despite the remarkable instrumental advancements, it remains crucial to develop efficient sample preparation methods capable of selectively extracting and pre-concentrating the target analytes, to achieve the requisite detectability and uphold the robustness of LC–MS/MS instruments.
EDs encompass a diverse range of compound families, exhibiting significant variations in molecular structures and physicochemical properties. So, selecting a technique or developing a new sample preparation method for the determination of EDs can depend on the specific class of compounds under analysis. Solid-phase extraction (SPE) has been widely utilized as an efficient technique for the extraction and preconcentration of EDs and various organic pollutants from water samples [8]. While SPE has shown remarkable recovery and enrichment factors, conventional methods employing this technique require the use of substantial amounts of toxic solvents and involve drying and resuspension stages. These characteristics render the methods unsustainable, labor-intensive, costly, and time-consuming [9]. To overcome these drawbacks, modern miniaturized versions of SPE have emerged as a beneficial alternative. These advanced techniques deliver comparable SPE performance while reducing reagent and sample consumption, minimizing waste generation, lowering costs, increasing sample throughput, and simplifying implementation and automation [10]. Consequently, contemporary methods employing miniaturized sorbent-based techniques for microextraction of EDs have gained prominence. Notable examples of such techniques include solid-phase microextraction (SPME), stir sorptive bar extraction (SBSE), and microextraction by packed sorbent (MEPS) [11,12,13,14,15,16].
Among the various miniaturized formats of SPE, MEPS emerges as a straightforward and highly efficient technique [8]. MEPS has demonstrated success across diverse matrices and applications, encompassing environmental surveillance as well as pharmaceutical and clinical analysis [17], forensic analysis, drugs of abuse [18,19,20,21], food composition [22,23,24], and metabolomics [25]. Introduced nearly two decades ago, MEPS has continuously evolved and is now available in a wide range of formats and extraction devices [26]. When coupled with gas chromatography, MEPS can be easily automated at-line using suitable autosampler instruments [27]. On the other hand, when used in conjunction with liquid chromatography, MEPS is typically performed in an offline and non-automated manner, which can be tedious and prone to errors due to the multiple draw/eject cycles required in each stage [28].
While several commercial solutions for semi- or fully automated MEPS are currently available, their high cost and limited accessibility pose challenges for many laboratories [29]. Consequently, researchers have actively pursued strategies to automate MEPS and increase its affordability. For instance, Serenjeh et al. successfully developed a semi-automated setup for MEPS, specifically focusing on the extraction of volatile polycyclic aromatic hydrocarbons in soil [30]. Our research group has also made significant contributions in this field by developing a Cartesian robot capable of complete automation and online coupling to high-performance liquid chromatography (HPLC) for various microextraction techniques, including MEPS [31,32,33,34]. Moreover, our team has designed a dedicated multi-syringe autosampler specifically tailored to enable the development of high-throughput MEPS methods. This innovative autosampler has demonstrated excellent performance and versatility, for example, in the MEPS of cannabinoids from human urine [35].
On the other hand, the commercial versions of MEPS face limitations due to the non-repackable nature of barrel insert and needle (BIN) devices, as well as the limited availability of extraction phases [36, 37]. So, the development of alternative MEPS devices that allows the exploitation of alternative or lab-made sorbents at reduced cost and with a high degree of reusability also becomes pertinent to make the technique more affordable and accessible. For example, our research group has introduced repackable MEPS devices, adaptable to gastight syringes [38], and cheaper versions in propylene Luer Slip syringes [39], allowing the exploration of diverse lab-made innovative sorbents.
In this study, we demonstrate the association of an easily repackable lab-made MEPS device with the Arduino-controlled multi-syringe lab-made robot as an alternative method for the development of sensitive, reliable, and high-throughput MEPS methods for environmental surveillance. To illustrate its effectiveness, we selected methylparaben (MeP), propylparaben (PrP), butylated hydroxyanisole (BHA), and oxybenzone (Oxi) as model compounds representing endocrine disruptors from the families of parabens, benzophenones, and synthetic phenolic antioxidants. A MEPS-UHPLC-MS/MS method was developed to determine these compounds in wastewater samples, demonstrating to be a competitive, fast, and sustainable analytical tool for environmental analysis.
Material and methods
Standards and reagents
MeP, PrP, BHA, and Oxi were procured from Sigma-Aldrich (St. Louis, MO, USA). The standard stock solutions were prepared by dissolving the analytes (1 mg mL−1) in methanol and stored in amber bottles at − 20 °C. Working solutions were freshly prepared on a daily basis. For detailed information on the physicochemical characteristics and compound structures, please refer to Table S1, which is available in the supplementary material.
Methanol and acetonitrile of chromatographic analysis grade were obtained from Tedia (Fairfield, OH, USA). Ammonium hydroxide (MS grade, 98%) was obtained from Fluka (Buchs, Switzerland), and ammonium acetate was purchased from J.T. Baker (Phillipsburg, NJ, USA), both used as a buffer in the aqueous mobile phase. Ultrapure water was generated using a Milli-Q purification system (Millipore, USA). Formic acid (98%) was obtained from Fluka (Darmstadt, Germany).
For the development of the MEPS method, several commercial extraction phases were evaluated, including Strata-X (33 µm particle size, from Phenomenex), Strata C18-E (55 µm particle size, from Phenomenex), Oasis MAX (30 µm particle size, from Waters), and BAKERBOND spe Amino (40 µm particle size, from Phenomenex).
Samples
Lab-made wastewater samples were prepared following the procedure described by Gomes et al. [40]. These samples were spiked with the compounds and used to create controlled samples for method development and subsequent analytical validation. As for the real sample, water was collected from Rio Monjolinho in São Carlos (SP, Brazil), a creek that receives both treated and untreated sewage discharges. The collected water was filtered through a 0.22-µm cellulose membrane and stored in amber flasks at − 20 °C.
Automated MEPS
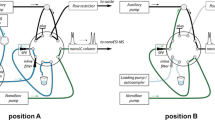
The extraction procedure using MEPS was carried out with a Hamilton 500 µL gastight syringe (Nevada, USA) with a removable needle. A modified ferrule nut was inserted between the needle and the syringe, which housed a lab-made repackable MEPS device. This extraction microdevice (Fig. 1a) consists of three PTFE detachable parts (base, body, and lid) and two stainless steel screens of 10-µm porosity (patent BR1020130254517) [41]. This repackable microcartridge can accommodate approximately 2.0 mg of any desired sorbent phase, commercial or lab-synthesized. Once the microdevice is sealed with the sorbent phase inside, the syringe can be integrated in a programmable robotic apparatus.

Instrumental setup for the performance of robot-assisted MEPS. a Schematic representation of the lab-made replaceable MEPS device. b Schematic representation of the lab-made multi-syringe robot
The automated MEPS procedures were performed using a lab-made multi-syringe autosampler (patent pending BR1020180046080), which consists of a Cartesian robot built of aluminum parts (Fig. 1b). The prototype had a horizontal rack platform with six syringes trails, and each trail could hold eight different containers to use for solvents or samples. Linear actuators and stepper motors were employed for spatial positioning. The commanding electronic circuit contains an Arduino® Mega 2560 microcontroller board, which controls the eight stepper motors: one for each syringe, plus one motor for the horizontal platform, and another to move the array of syringe drivers. The prototype was controlled via USB from a personal computer, utilizing pre-programmed instructions written in the Arduino Integrated Development Environment (IDE) [31, 35].
Optimization of the automated MEPS procedure
Univariate and multivariate analyses were performed to thoroughly assess the influence of key parameters on the performance of MEPS.
The selection of the MEPS sorbent was carried out through a univariate comparison of extraction performance using Strata-X, Strata C18-E, Oasis MAX, and Amino. Repackable MEPS devices were filled with approximately 2.0 mg of each sorbent, and extractions of lab-made wastewater samples fortified with 200 µg L−1 of each analyte were performed in triplicate to assess the relative extraction efficiency of the tested sorbents. The MEPS protocol used for evaluating the sorbent phases is provided in Table S2.
Subsequently, a 26–2 fractional factorial experimental design was employed to identify the steps of the automated MEPS procedure that significantly influenced the extraction of EDs (Tables S3-S6). The volumes of each step were maintained at a constant 500 µL, while the number of draw/eject cycles was varied between 3 ( −) and 20 ( +). To determine the optimal conditions (volumes and number of cycles) for the most influential step (sampling), a central composite design was conducted over an experimental range of 100 (− √2) to 500 (√2) microliters for volume and 2 (− √2) to 20 (√2) for the number of cycles. The complete experimental design is presented in Table S7, and the data obtained from these analyses were processed using Statistica 13 software (StatSoft Inc., Tulsa, USA, 2013).
HPLC–UV-Vis method
The development of the MEPS method was conducted using HPLC–UV-Vis. This encompassed the selection of the sorbent phase, determination of the most influential step in MEPS, and subsequent optimization. The HPLC–UV-Vis consisted of a Shimadzu (Kyoto, Japan) LC system consisting of a CBM-20A communication bus module, SIL-20AC autosampler, DGU-20AS degasser, two LC 20-AD pumps, CTO-20A column oven, and SPD-20A UV–Vis detector set to monitor at wavelengths of 250 nm and 290 nm. The mobile phases selected were water and acetonitrile, both containing 0.1% formic acid. The flow rate was set to 0.250 mL min−1. Initially, an isocratic elution with 12% acetonitrile was programmed for 5 min. From 5 to 15 min, gradient elution was performed by linearly increasing the acetonitrile content from 12 to 90%. Finally, in the subsequent 5 min, a linear change was made to return the organic modifier to 12%.
UHPLC-MS/MS method
The analytical validation of the method and the analysis of the real sample were carried out using a UPLC Acquity Waters coupled to a triple quadrupole XEVO TQ-MS system. Chromatographic separation was achieved using a Kinetex EVO C18 analytical column (150 mm × 2.1 mm, 5 µm) from Phenomenex (Torrance, CA, USA), which was maintained at a temperature of 40 °C. Water with acetate buffer (25 mM ammonium acetate + 25 mM ammonium hydroxide) and acetonitrile were used as mobile phases A and B, respectively, at a flow rate of 0.25 mL min−1. Initially, an isocratic elution was programmed with 10% of acetonitrile for 2 min. Gradient elution was then performed from 2 to 3 min, with a linear increase in acetonitrile content from 10 to 90%. From 3 to 6 min, an isocratic elution was kept with 90% of acetonitrile, then changing linearly to 10% of organic modifier in 1 min, and finally kept in 10% of acetonitrile for 5 min (7 to 12 min) for conditioning of the analytical column.
For MS/MS detection, the operation and data acquisition were performed using Waters MassLynx 4.1 software (Milford. MA, USA). The optimal parameters for ED detection were selected by direct infusion of aqueous solutions. The analysis was carried out using the multiple reaction monitoring (MRM) acquisition mode, with negative electrospray ionization (ESI −) employed. Monitored transitions and selected operational parameters are shown in Table 1. Nitrogen was used as a desolvation gas at a temperature of 400 °C and a flow rate of 800 L h−1. The source temperature was set to 150 °C.
Method validation
The developed method was validated using the matrix-matching calibration approach and lab-made wastewater samples. Method selectivity was ensured through MS/MS detection, where precursor ions and two transitions provided at least four identification points (IPs) for each of the investigated analytes [41]. The calibration curves were generated in triplicate by utilizing six levels of concentration. To determine the LODs and LOQs, a series of successively diluted samples were prepared and injected until reaching the lowest concentrations that yielded a signal three times and ten times higher than the noise, respectively. Precision was assessed under both inter- and intra-day conditions at two concentration levels, with a sample size of n = 6, employing the LC–MS/MS method.
The matrix effect (ME), total process efficiency (PE), and recovery (RE) were studied following the approach outlined by Matuszewski et al. [42]. In this case, the ME was calculated as (B/A) × 100, PE as (C/A) × 100, and RE as (C/B) × 100. Here, A represents the analytical response for the direct injection of a standard solution of the analytes at a concentration equivalent to 100% MEPS efficiency. B represents the analytical response for the injection of extracts obtained after MEPS of spiked matrix samples (1.0 ng L−1). C represents the analytical response for the injection of a solution prepared by spiking an extract obtained by MEPS of blank matrix samples at a concentration equivalent to 100% MEPS efficiency.
Results and discussion
Development of the MEPS method
MEPS has been widely utilized as a sample preparation technique for the determination of EDs. For instance, Silvera et al. recently presented a method for analyzing 16 EDs (such as parabens, benzophenones, bisphenols, and triclocarban) in human urine using commercial MEPS syringes with C18 as the extraction phase [42]. Similarly, Matin et al. investigated the effectiveness of a montmorillonite-reinforced polystyrene nanocomposite coated onto cellulose filter paper as an extraction sorbent in MEPS for the layered extraction of fluoxetine from environmental water and wastewater samples [43]. In both studies, the development of the MEPS method involved assessing the extraction phase, determining the necessary MEPS stages (conditioning, sampling, washing, drying, elution, and clean-up), and defining the volumes and number of draw/eject cycles to be employed in each stage. This rational workflow closely aligns with the approach we have adopted for the development of our MEPS method.
Selection of the extraction phase
As previously mentioned, EDs comprise a wide range of compound families with diverse molecular structures and properties. Therefore, the selection of an appropriate sorbent for extracting these compounds depends on the specific compound family under investigation. Currently, active research is focused on synthesizing and evaluating new extraction sorbents to develop advanced strategies for detecting EDs in water samples.
One notable trend in this field is the emergence of magnetic solid-phase extractions (MSPE), which employ magnetic sorbents like porous carbons [44, 45], graphene [46], covalent organic frameworks (MOFs) [47], chitosan [48], and histamine [49], among others. Additionally, other sorbent-based microextraction methods are being explored, including the use of coated devices with molecularly imprinted polymers [50] and packed bed-based techniques like sorptive stir bar microextraction with packed membranes. Some examples of these techniques include the use of cyclodextrin-based sorbents in stir bar sorptive extraction with packed fibers [51] and the use of some commercial phases such as divinylbenzene grafted polyvinylpyrrolidone (DVB@PVP) in SPE [52] and C18 in SPE [53] and MEPS [54].
In this study, to assess the performance of a proposed automated setup, utilizing a repackable MEPS device for the determination of EDs in wastewater samples was assessed. The commercial sorbents Strata-X, C18, Oasis Max, and aminopropyl silica were evaluated. 2.0 mg of each extraction phase was packed in the MEPS device, and extractions were performed as described in the “Optimization of the automated MEPS procedure” section. Subsequently, the obtained extracts underwent analysis using HPLC–UV-Vis, and the resulting chromatographic areas for each compound were plotted in Fig. 2a.

Selection of the best MEPS conditions. a Selection of the extraction phase; normal probability plot of the effects for the 22 factorial experimental design for b methylparaben; c propylparaben; d butylated hydroxyanisole; and e response surface for oxybenzone
To investigate the statistical significance of the chromatographic areas obtained with each tested sorbent, a two-way analysis of variance (ANOVA) test was conducted (Table S8). In all cases, the calculated F values exceeded the critical F value, and the corresponding p-values were below the pre-determined significance level (α = 0.05). These findings indicate the presence of statistically significant differences in the recovery of each analyte with each extraction phase and the extraction efficiency of the different analytes by each sorbent.
Further analysis of the ANOVA results was performed using a Tukey test (Table S9). The test revealed that, with an honestly significant difference observed in the chromatographic area of 1268.3, all the chromatographic areas exhibited statistically significant differences, except for the extractions of MeP with the amino and C18 phases, BHA with Strata-X and C18, and Oxi with amino and Oasis Max.
Among the tested sorbent phases, Strata-X consistently demonstrated higher chromatographic areas for all the tested analytes and exhibited suitable precision with a relative standard deviation (RSD) below 10%. Consequently, Strata-X was selected for the subsequent stages of the extraction method development.
Determination of the most influential MEPS steps
The MEPS process involves several stages, including sorbent conditioning, sampling, washing, drying, desorption, and clean-up cartridge. To assess the impact of each stage on the process’s efficiency, a 26–2 fractional factorial experimental design was utilized (see Table S3-S6).
The influence of each MEPS stage was evaluated by constructing normal probability plots, which plotted the Z value against the standardized percentage effect. The effects were calculated by determining the difference between the responses obtained at the high and low levels for each variable, including their interactions. The percentage contribution of each variable’s effect was calculated by summing the squared effects and their contribution to the total sum. These calculated effects are associated with equal areas of a Gaussian curve. In a normal probability plot (Z value vs. % standardized effect), smaller effects tend to cluster around zero, while statistically significant effects deviate from zero.
To determine the magnitude of each MEPS stage’s impact on the chromatographic peak area, probability plots (Fig. 2b–e) were analyzed. Although other MEPS steps, such as water conditioning (v2), exhibited a relatively significant effect on the extraction performance of oxybenzone, the sampling (v3) stage emerged as the most influential. It accounted for approximately 60% of the increase in chromatographic area for MeP, PrP, and BHA and over 30% for Oxi. Consequently, we optimized the sampling stage using a multivariate experimental design.
Fine-tuning of the sampling step
To optimize the MEPS procedure and develop a fast and efficient method, the sampling step was meticulously refined using a surface response methodology. The conditioning, clean-up, elution, and washing steps were maintained at a lower level of 100 µL and three draw/eject cycles. A central composite design was employed, with the sample volume and the number of draw/eject cycles as the variables of interest. The experimental range for the sample volume was set between 100 and 500 µL, while the number of cycles ranged from 2 to 20. Figure 3 displays the surface responses obtained from the conducted experiments, and the ANOVA tables for the resulting models can be found in the supplementary information (Tables S10-S13).

Multivariate optimization of the sampling stage. a Response surface for methylparaben. b Response surface for propylparaben. c) Response surface for butylated hydroxyanisole. d Response surface for oxybenzone
The models derived from the experiments included six coefficients: an independent term, two linear coefficients for the number of cycles and volume, two square coefficients for these variables, and two coefficient products representing the interaction between the variables. However, not all coefficients were statistically significant (p < 0.05). Nevertheless, with a probability higher than 95%, none of the models exhibited a significant lack of fit. These results indicate that the obtained models effectively represent the experimental data and provide reliable predictability of the analytical responses, to obtain the experimental conditions capable of providing the maximum chromatographic area.
The highest analytical responses for all analytes were observed when passing 500-µL aliquots of the sample through the packed bed approximately 10 to 16 times. Based on these findings, the optimal conditions for the sampling stage were determined as a sample volume of 500 µL and 14 draw/eject cycles. Implementing these conditions in the MEPS procedure ensures a more effective and improved sampling step, resulting in better outcomes for all analytes.
Finally, the MEPS procedure was developed, which comprised seven steps: conditioning the extraction phase with organic solvent (methanol), conditioning the extraction phase with aqueous solvent (water), aspirating the sample (wastewater), washing the extraction phase (water), drying (air), eluting the analytes (acetonitrile), and cleaning the extraction phase (methanol). The specific volume and number of cycles for each step were determined and are presented in Table 2.
Analytical performance of the MEPS-LC–MS/MS method
Under the selected experimental conditions, the analytical performance of the proposed automated MEPS method was evaluated by considering some figures of merit. These include selectivity, LOD, LOQ, linearity, linearity range, precision, and enrichment factors (Table 3).
The selectivity of the developed method was successfully achieved through the utilization of MS/MS detection. Confident identification of the analytes was accomplished by obtaining a minimum of four identification points (IPs), encompassing known retention times from standard injections, precursor ions, and two transitions [41]. To ensure accurate quantification of the analytes with the necessary selectivity, black lab-made wastewater samples were subjected to extraction and analysis. No endogenous or exogenous interferences at the same retention time as the analytes were observed in any extracted ion chromatogram for every monitored MS/MS transition. This confirmation of selectivity for both quantification and confirmation ion transitions across all analytes guarantees the suitability of the chromatographic separation and the method's MS/MS detection selectivity for accurate quantification.
The LODs and LOQs were determined experimentally through a series of successive injections using consecutively diluted solutions. As a result, LODs and LOQs were established as the minimum concentrations capable of generating signal-to-noise ratios greater than 3 and 10, respectively, for the two monitored transitions of each analyte. The corresponding values can be found in Table 3.
This experimental determination ensures that each analyte can be detected with signal-to-noise ratios higher than 3 for both monitored transitions. Additionally, at the reported LOQ (which corresponds to the first point on the calibration curve), analytes not only can be identified with a signal-to-noise ratio higher than 10 for both transitions, but they could also be quantified with the appropriate level of precision, as indicated by the relative standard deviation (RSD). The obtained LOQs are in agreement with the criteria established for the regulatory agencies for determining EDs in wastewater samples, which depend on the ED compound range in the pg-ng L−1 order [55].
Matrix-matched calibration curves were established using MEPS of spiked lab-made synthetic wastewater samples, followed by UHPLC-MS/MS analysis (Table 3). The concentration range of the calibration curves spanned from the LOQ of each analyte to 5.0 ng/mL, and the experiments were performed in triplicate. The obtained linear correlations demonstrated acceptable relationships across the studied concentration range, with coefficients of determination (r2) ranging from 0.9807 to 0.9863.
Although the obtained r2 values explained only slightly over 98% of the data variability, residue analysis revealed that the proposed regression models could predict concentration values with an error below 20%. Supplementary information (Figure S1) presents the plots of relative residues (%). For MeP, PrP, and BHA, a 1/x2 weighted calibration was employed to minimize the absolute sum of residues (∑%RE) and ensure the heteroscedasticity of the data. Conversely, no improvement in residue behavior was observed for Oxi with the application of weighting, rendering a weighted calibration unnecessary for this analyte. Notably, relative residues remained below 20% for all cases, indicating a highly acceptable level of accuracy in predicting sample concentrations within the investigated concentration range.
An unusual relationship between the intercept and slope values was observed in the calibration curve for oxybenzone. Various factors, including instrument-specific effects, matrix interferences, and experimental limitations, could contribute to this phenomenon. While there is limited literature explaining this fact, some of these factors can significantly impact the extraction and mass spectrometry detection of oxybenzone. For instance, during the development of an SPE method for determining household chemicals using GC and LC–MS/MS, Threnholm et al. reported that although oxybenzone was easily extracted from reagent water during method development, it exhibited poor recoveries in matrix spikes, which limited the analysis to qualitative observations [56]. The authors attributed the low recovery to the pH effect, noting that oxybenzone displayed improved recoveries at pH levels below 7 but poor recoveries at pH levels above 7. In their study, the pH of the reagent water ranged from 5.0 to 6.5, while the surface water and wastewater effluent had pH values ranging from 7.3 to 8.1, respectively. Similarly, although not providing a specific explanation, Chen et al., in their work on developing a method for screening chlorinated transformation products of aromatic pharmaceuticals and personal care products, reported the inability to detect oxybenzone using high-resolution mass spectrometry (HRMS) in spiked natural organic matter water samples [57]. These studies highlight the challenges associated with the detection and quantification of oxybenzone, particularly in complex matrices, and suggest that factors such as pH and the presence of organic matter can have a significant impact on its analysis.
Despite the atypical relationship between the intercept and slope values, the reliability and accuracy of the oxybenzone calibration model can be supported by considering other analytical parameters. Analysis of variance (ANOVA) confirmed the statistical significance of the regression for oxybenzone. The calculated F value of 815.13 for Oxi significantly exceeds the tabulated F value at a 0.05 probability level with 1 and 16 degrees of freedom (0.0041), indicating a highly significant regression. The ANOVA table for the oxybenzone linear regression can be found in the supplementary information (Table S14). Furthermore, as previously mentioned, the calibration curves were obtained in triplicate, resulting in relative standard deviation (RSD) values below 20% for all concentration levels and analytes. Additionally, the relative residues for the oxybenzone calibration curve were consistently below 20% across all concentration levels, further indicating the accuracy of the calibration model.
Precision was evaluated by calculating the intra- and inter-day relative standard deviations (% RSD) based on sextuplicate experiments at two concentrations for each analyte, as presented in Table 3. The intra-day RSD values ranged from 3.0 to 21.0%, while the inter-day RSD values ranged from 7.0 to 21.0%. These results demonstrate the method’s capability to accurately quantify the target analytes within the linear range studied.
In the field of analytical methods employing mass spectrometry, matrix effects play a crucial role in influencing the performance of sample preparation and the efficiency of analyte ionization in the ESI source. To assess these effects accurately, we employed the methodology proposed by Matuszewski et al. to determine the matrix effect (ME), extraction recovery (ER), and process efficiency (PE), as described in the “Optimization of the automated MEPS procedure” section [58].
The RE provides an estimate of the influence of matrix constituents on MEPS performance which ranges from 60 to 76%. These values indicate that certain matrix constituents can be adsorbed during the extraction phase, impeding the uptake of analytes. Similarly, the PE values ranged from 47 to 75%, suggesting a significant decrease in analyte ionization due to the presence of matrix constituents. Furthermore, the ME, which represents the combined effects of matrix constituents on both extraction performance and ionization efficiency, varied from 79 to 112% (Table 4). These RE, PE, and ME values offer an estimation of the impact of matrix constituents on method performance and emphasize the importance of employing matrix-matched calibration when developing methods for the treatment and analysis of complex samples.
Although notable effects of matrix constituents were observed during both the MEPS and ESI processes, these effects do not hinder the applicability of the robot-assisted MEPS setup. This is evidenced by the determination of LODs, LOQs, and other performance parameters using the matrix-matched approach, which ensures that matrix effects are considered during their estimation.
Comparison with recently previously reported methods
Table 5 presents a comparison between the method proposed in this study and the recently reported sorbent-based methods for the determination of EDs in water samples. The automated MEPS-LC–MS/MS method demonstrated competitive performance in terms of detectability, feasibility, and sample throughput. Its automated nature, along with the efficient utilization of sorbents, samples, and organic solvents, contributes to its economic and green attributes. Likewise, the extended reusability of the extraction devices further enhances its sustainability.
In comparison to conventional manual [53] and automated SPE [52] methods, the setup proposed in this study achieves competitive limits of quantification (LOQs) while consuming significantly lower amounts of sorbent, samples, and elution solvents. When compared to other miniaturized techniques such as MEPS [43], SPME [50], and some dispersive techniques such as the magnetic effervescence-assisted sorbent-based extraction (MNER-ME) [47], the MEPS-LC–MS method reported in this study demonstrates LOQs up to ten times lower. Additionally, it offers the advantages of automation and high-throughput capabilities provided by the robot, allowing for the simultaneous preparation of up to six samples. Some other magnetic dispersive extraction, such as the one using a Fe3O4/N-HCSCs sorbent [45], are also highly efficient and fast techniques capable of providing comparable LOQs with small amounts of sorbent and samples. However, the reusability of the sorbents can be limited, and for example, in the case of the Fe3O4/N-HCSCs sorbent, it was limited to only 12 times. In contrast, our method utilizing a single packed device with 2.0 mg of extraction phase achieved more than 100 reuses without appreciable loss in its extraction capabilities.
Application of the developed method to the analysis of real wastewater sample
To demonstrate the applicability of the proposed setup in analyzing real samples, we collected a flask of running water from Rio Monjolinho in São Carlos, SP. Four extractions were performed, yielding the following concentrations: MeP: 0.58 ± 0.07 ng L−1; PrP: 1.7 ± 0.06 ng L−1; Oxi: 5.1 ± 0.6 ng L−1; and BHA was not detected in the sample.
Conclusion
The utilization of an automated MEPS method for sample treatment has demonstrated its efficiency in determining some organic pollutants in environmental samples. Additionally, the application of factorial design proved to be practical in selecting the optimal extraction phase and parameters for sample treatment, resulting in enhanced extraction efficiency. Both the automated system and the MEPS device utilizing a commercial syringe offer excellent alternatives that can be implemented in the laboratory to meet the demands associated with the development of new analytical methods. The multi-syringe capability of the MEPS device offers the possibility of treating six samples simultaneously, enabling high-throughput characteristics to the developed method. Furthermore, the utilization of a microextraction technique makes it an excellent environmentally friendly option. Finally, the incorporation of MS/MS detection yielded exceptional LODs and LOQs even when using a reduced amount of sample. Consequently, the proposed approach represents an advantageous strategy for monitoring some organic pollutants as MeP, PrP, BHA, and Oxi in wastewater samples.
References
Witorsch RJ, Thomas JA, Witorsch RJ, Thomas JA, Witorsch RJ, Thomas JA Personal care products and endocrine disruption: a critical review of the literature. Crit Rev Toxicol. 2010;40(3):1–30: https://doi.org/10.3109/10408444.2010.515563.
Bergman Å, Heindel J, Jobling S, Kidd K, Zoeller RT, State-of-the-science of endocrine disrupting chemicals. World Health Organization. 2012;3–38. https://apps.who.int/iris/bitstream/handle/10665/78102/WHO_HSE_PHE_IHE_2013.1_eng.pdf?sequence=1.
Darbre PD, Harvey PW (2008) Paraben esters: review of recent studies of endocrine toxicity, absorption, esterase and human exposure, and discussion of potential human health risks. 561–578. https://doi.org/10.1002/jat.
Ebele AJ, Abou-Elwafa Abdallah M, Harrad S. Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment. Emerg Contam. 2017;3:1–16. https://doi.org/10.1016/j.emcon.2016.12.004.
Ternes TA, Joss A, Siegrist H. Peer reviewed: scrutinizing pharmaceuticals and personal care products in wastewater treatment. Environ Sci Technol. 2004;38:392A-399A. https://doi.org/10.1021/es040639t.
Daughton CG, Ternes TA. Pharmaceuticals and personal care products in the environment: agents of subtle change? Environ Health Perspect. 1999;107:907–38. https://doi.org/10.1289/ehp.99107s6907.
Pérez-Fernández V, Mainero Rocca L, Tomai P, Fanali S, Gentili A. Recent advancements and future trends in environmental analysis: sample preparation, liquid chromatography and mass spectrometry. Anal Chim Acta. 2017;983:9–41. https://doi.org/10.1016/j.aca.2017.06.029.
Faraji M, Yamini Y, Gholami M. Recent advances and trends in applications of solid-phase extraction techniques in food and environmental analysis. Berlin Heidelberg: Springer; 2019.
Andrade-Eiroa A, Canle M, Leroy-Cancellieri V, Cerdà V. Solid-phase extraction of organic compounds: a critical review. part ii. TrAC - Trends Anal Chem. 2016;80:655–67. https://doi.org/10.1016/j.trac.2015.08.014.
Filippou O, Bitas D, Samanidou V. Green approaches in sample preparation of bioanalytical samples prior to chromatographic analysis. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1043:44–62. https://doi.org/10.1016/j.jchromb.2016.08.040.
Soares da Silva Burato J, Vargas Medina DA, de Toffoli AL, Vasconcelos Soares Maciel E, Mauro Lanças F. Recent advances and trends in miniaturized sample preparation techniques. J Sep Sci. 2020:43(1):202–225. https://doi.org/10.1002/jssc.201900776
Soares Maciel EV, de Toffoli AL, Lanças FM. Recent trends in sorption-based sample preparation and liquid chromatography techniques for food analysis. Electrophoresis. 2018;39:1582–96. https://doi.org/10.1002/elps.201800009.
Celeiro M, Lamas JP, Vila M, Garcia-Jares C, Homem V, Ratola N, Dagnac T, Llompart M. Determination of multiclass personal care products in continental waters by solid-phase microextraction followed by gas chromatography-tandem mass spectrometry. J Chromatogr A. 2019;1607:460398. https://doi.org/10.1016/j.chroma.2019.460398.
Pérez-Lemus N, López-Serna R, Pérez-Elvira SI, Barrado E, Sample pre-treatment and analytical methodology for the simultaneous determination of pharmaceuticals and personal care products in sewage sludge. Chemosphere. 2020;258:127273. https://doi.org/10.1016/j.chemosphere.2020.127273.
Aparicio I, Martín J, Santos JL, Malvar JL, Alonso E. Stir bar sorptive extraction and liquid chromatography–tandem mass spectrometry determination of polar and non-polar emerging and priority pollutants in environmental waters. J Chromatogr A. 2017;1500:43–52. https://doi.org/10.1016/j.chroma.2017.04.007.
Pereira J, Gonçalves J, Alves V, Câmara JS, Microextraction using packed sorbent as an effective and high-throughput sample extraction technique: recent applications and future trends. Sample Prep 2013;1:38–53. https://doi.org/10.2478/sampre-2013-0005
Rani S, Malik AK. A novel microextraction by packed sorbent-gas chromatography procedure for the simultaneous analysis of antiepileptic drugs in human plasma and urine. J Sep Sci. 2012;35:2970–7. https://doi.org/10.1002/jssc.201200439.
Rosado T, Fernandes L, Barroso M, Gallardo E. Sensitive determination of THC and main metabolites in human plasma by means of microextraction in packed sorbent and gas chromatography–tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1043:63–73. https://doi.org/10.1016/j.jchromb.2016.09.007.
Rosado T, Gonçalves A, Margalho C, Barroso M, Gallardo E. Rapid analysis of cocaine and metabolites in urine using microextraction in packed sorbent and GC/MS. Anal Bioanal Chem. 2017;409:2051–63. https://doi.org/10.1007/s00216-016-0152-2.
Ares AM, Fernández P, Regenjo M, Fernández AM, Carro AM, Lorenzo RA. A fast bioanalytical method based on microextraction by packed sorbent and UPLC–MS/MS for determining new psychoactive substances in oral fluid. Talanta. 2017;174:454–61. https://doi.org/10.1016/j.talanta.2017.06.022.
Fernández P, González M, Regenjo M, Ares AM, Fernández AM, Lorenzo RA, Carro AM. Analysis of drugs of abuse in human plasma using microextraction by packed sorbents and ultra-high-performance liquid chromatography. J Chromatogr A. 2017;1485:8–19. https://doi.org/10.1016/j.chroma.2017.01.021.
Adam M, Pavlíková P, Čížková A, Bajerová P, Ventura K. Microextraction by packed sorbent (MEPS) as a suitable selective method for l-ascorbic acid determination in beverages. Food Chem. 2012;135:1613–8. https://doi.org/10.1016/j.foodchem.2012.06.039.
Mercolini L, Protti M, Saracino MA, Mandrone M, Antognoni F, Poli F. Analytical profiling of bioactive phenolic compounds in argan (Argania spinosa) leaves by combined microextraction by packed sorbent (MEPS) and LC-DAD-MS/M S. Phytochem Anal. 2016;27:41–9. https://doi.org/10.1002/pca.2585.
Savastano ML, Losito I, Pati S. Rapid and automatable determination of ochratoxin A in wine based on microextraction by packed sorbent followed by HPLC-FLD. Food Control. 2016;68:391–8. https://doi.org/10.1016/j.foodcont.2016.04.016.
Silva C, Cavaco C, Perestrelo R, Pereira J, Câmara JS. Microextraction by packed sorbent (MEPS) and solid-phase microextraction (SPME) as sample preparation procedures for the metabolomic profiling of urine. Metabolites. 2014;4:71–97. https://doi.org/10.3390/metabo4010071.
Vargas Medina DA, Cardoso AT, Maciel EVS, Lanças FM, Current materials for miniaturized sample preparation: recent advances and future trends. TrAC - Trends Anal Chem. 2023;165:117120. https://doi.org/10.1016/j.trac.2023.117120.
Noche GG, Laespada MEF, Pavón JLP, Cordero BM, Lorenzo SM. Microextraction by packed sorbent for the analysis of pharmaceutical residues in environmental water samples by in situ derivatization-programmed temperature vaporizer-gas chromatography-mass spectrometry. J Chromatogr A. 2011;1218:9390–6. https://doi.org/10.1016/j.chroma.2011.10.094.
Kaur M, Rani S, Malik AK, Aulakh JS. Microextraction by packed sorbent-high-pressure liquid chromatographic-ultra violet analysis of endocrine disruptor pesticides in various matrices. J Chromatogr Sci. 2014;52:977–84. https://doi.org/10.1093/chromsci/bmt136.
Pereira J, Câmara JS, Colmsjö A, Abdel-Rehim M. Microextraction by packed sorbent: an emerging, selective and high-throughput extraction technique in bioanalysis. Biomed Chromatogr. 2014;28:839–47. https://doi.org/10.1002/bmc.3156.
Serenjeh FN, Hashemi P, Ghiasvand AR, Rasolzadeh F, Heydari N, Badiei A. Cooling assisted headspace microextraction by packed sorbent coupled to HPLC for the determination of volatile polycyclic aromatic hydrocarbons in soil. Anal Chim Acta. 2020;1125:128–34. https://doi.org/10.1016/j.aca.2020.05.067.
Medina DAV, Rodriguez Cabal LF, Lanças FM, Santos-Neto ÁJ. Sample treatment platform for automated integration of microextraction techniques and liquid chromatography analysis. HardwareX. 2019;5:e00056. https://doi.org/10.1016/j.ohx.2019.e00056.
Medina DAV, Rodríguez Cabal LF, Titato GM, Lanças FM, Santos-Neto ÁJ. Automated online coupling of robot-assisted single drop microextraction and liquid chromatography. J Chromatogr A. 2019;1595:66–72. https://doi.org/10.1016/j.chroma.2019.02.036.
da Silva LF, Vargas Medina DA, Lanças FM. Automated needle-sleeve based online hyphenation of solid-phase microextraction and liquid chromatography. Talanta. 2021;221:121608. https://doi.org/10.1016/j.talanta.2020.121608.
Cabal LFR, Medina DAV, Costa JL, Lanças FM, Santos-Neto ÁJ. Determination of ring-substituted amphetamines through automated online hollow fiber liquid-phase microextraction-liquid chromatography. Anal Bioanal Chem. 2019;411:7889–97. https://doi.org/10.1007/s00216-019-02196-0.
Sartore DM, Vargas Medina DA, Costa JL, Lanças FM, Santos-Neto ÁJ. Automated microextraction by packed sorbent of cannabinoids from human urine using a lab-made device packed with molecularly imprinted polymer. Talanta. 2020;219:121185. https://doi.org/10.1016/j.talanta.2020.121185.
Altun Z, Abdel-rehim M, Blomberg LG (2004) New trends in sample preparation : on-line microextraction in packed syringe ( MEPS ) for LC and GC applications Part III : determination and validation of local anaesthetics in human plasma samples using a cation-exchange sorbent, and MEPS – LC – MS – M. 813:129–135. https://doi.org/10.1016/j.jchromb.2004.09.020.
Mahdi M, Abdel-rehim A, Abdel-rehim M. Trends in analytical chemistry microextraction by packed sorbent ( MEPS). Trends Anal Chem. 2015;67:34–44. https://doi.org/10.1016/j.trac.2014.12.003.
Andrade FN, Santos-Neto AJ, Medina DA V, Lanças F.M. A molecularly imprinted polymer for microextraction by packed sorbent of sulfonylureas herbicides from corn samples. J Food Compos Anal. 2023;121:105388. https://doi.org/10.1016/j.jfca.2023.105388.
Jordan-Sinisterra M, Vargas Medina DA, Lanças FM. Microextraction by packed sorbent of polycyclic aromatic hydrocarbons in brewed coffee samples with a new zwitterionic ionic liquid-modified silica sorbent. J Food Compos Anal. 2022;114:104832. https://doi.org/10.1016/j.jfca.2022.104832.
Lima Gomes PCF, Tomita IN, Santos-Neto ÁJ, Zaiat M. Rapid determination of 12 antibiotics and caffeine in sewage and bioreactor effluent by online column-switching liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem. 2015;407:8787–801. https://doi.org/10.1007/s00216-015-9038-y.
Hernández F, Ibáñez M, Sancho JV, Pozo ÓJ. Comparison of different mass spectrometric techniques combined with liquid chromatography for confirmation of pesticides in environmental water based on the use of identification points. Anal Chem. 2004;76:4349–57. https://doi.org/10.1021/ac049768i.
Silveira RS, Rocha BA, Rodrigues JL, Barbosa F. Rapid, sensitive and simultaneous determination of 16 endocrine-disrupting chemicals (parabens, benzophenones, bisphenols, and triclocarban) in human urine based on microextraction by packed sorbent combined with liquid chromatography tandem mass spectrometry (MEPS-LC-MS/MS). Chemosphere. 2020;240:124951. https://doi.org/10.1016/j.chemosphere.2019.124951.
Parastou Matin, Zahra Ayazi, Kazem Jamshidi-Ghalehb Department of Physics F, Montmorillonite reinforced polystyrene nanocomposite supported on cellulose as a novel layered sorbent for microextraction by packed sorbent for determination of fluoxetine followed by spectrofluorimetry based on multivariate optimisation. Int J Environ Anal Chem. 2022;102:5150–5165. https://doi.org/10.1080/03067319.2020.1791333.
Wu H, Dong S, Huang G, Zheng Q, Huang T. The extraction of four endocrine disrupters using hollow N-doped mesoporous carbon spheres with encapsulated magnetite (Fe3O4) nanoparticles coupled to HPLC-DAD determination. Microchem J. 2021;164:105984. https://doi.org/10.1016/j.microc.2021.105984.
Lopes KL, de Oliveira HL, Serpa JAS, Torres JA, Nogueira FGE, de Freitas VAA, Borges KB, Silva MC. Nanomagnets based on activated carbon/magnetite nanocomposite for determination of endocrine disruptors in environmental water samples. Microchem J. 2021;168:106366. https://doi.org/10.1016/j.microc.2021.106366.
Tasmia Shah J, Jan MR. Eco-friendly alginate encapsulated magnetic graphene oxide beads for solid phase microextraction of endocrine disrupting compounds from water samples. Ecotoxicol Environ Saf. 2020;190:110099. https://doi.org/10.1016/j.ecoenv.2019.110099.
Tan C, Li J, Liu W, Zhao Q, Wang X, Li Y. Core-shell magnetic covalent organic framework nanocomposites as an adsorbent for effervescent reaction-enhanced microextraction of endocrine disruptors in liquid matrices. Chem Eng J. 2020;396:125191. https://doi.org/10.1016/j.cej.2020.125191.
Tasmia SJ, Jan MR. Microextraction of selected endocrine disrupting phenolic compounds using magnetic chitosan biopolymer graphene oxide nanocomposite. J Polym Environ. 2020;28:1673–83. https://doi.org/10.1007/s10924-020-01714-x.
Lopes D, Bernardi G, Pinheiro G, Ruschel Campedelli R, de Souza BS, Carasek E. Histamine functionalized magnetic nanoparticles (HIS-MNP) as a sorbent for thin film microextraction of endocrine disrupting compounds in aqueous samples and determination by high performance liquid chromatography-fluorescence detection. J Chromatogr A. 2019;1602:41–7. https://doi.org/10.1016/j.chroma.2019.05.032.
Wang X, Huang P, Ma X, Du X, Lu X. Enhanced in-out-tube solid-phase microextraction by molecularly imprinted polymers-coated capillary followed by HPLC for endocrine disrupting chemicals analysis. Talanta. 2019;194:7–13. https://doi.org/10.1016/j.talanta.2018.10.027.
Hafiz Rozaini MN, Saad B, Lim JW, Yahaya N, Ramachandran MR, Kiatkittipong W, Mohamad M, Chan YJ, Goh PS, Shaharun MS. Development of β-cyclodextrin crosslinked citric acid encapsulated in polypropylene membrane protected-μ-solid-phase extraction device for enhancing the separation and preconcentration of endocrine disruptor compounds. Chemosphere. 2022;303:135075. https://doi.org/10.1016/j.chemosphere.2022.135075.
Goeury K, Vo Duy S, Munoz G, Prévost M, Sauvé S, Assessment of automated off-line solid-phase extraction LC-MS/MS to monitor EPA priority endocrine disruptors in tap water, surface water, and wastewater. Talanta. 2022;241:123216. https://doi.org/10.1016/j.talanta.2022.123216.
Spataro F, Ademollo N, Pescatore T, Rauseo J, Patrolecco L. Antibiotic residues and endocrine disrupting compounds in municipal wastewater treatment plants in Rome, Italy. Microchem J. 2019;148:634–42. https://doi.org/10.1016/j.microc.2019.05.053.
Silveira RS, Rocha BA, Rodrigues JL, Barbosa F. Rapid, sensitive and simultaneous determination of 16 endocrine-disrupting chemicals (parabens, benzophenones, bisphenols, and triclocarban) in human urine based on microextraction by packed sorbent combined with liquid chromatography tandem mass spectrom. Chemosphere. 2020;240:124951. https://doi.org/10.1016/j.chemosphere.2019.124951.
Metcalfe CD, Bayen S, Desrosiers M, Muñoz G, Sauvé S, Yargeau V, Methods for the analysis of endocrine disrupting chemicals in selected environmental matrixes. Environ Res.2022;206:112616. https://doi.org/10.1016/j.envres.2021.112616.
Trenholm RA, Vanderford BJ, Drewes JE, Snyder SA. Determination of household chemicals using gas chromatography and liquid chromatography with tandem mass spectrometry. J Chromatogr A. 2008;1190:253–62. https://doi.org/10.1016/j.chroma.2008.02.032.
Chen WL, Cheng JY, Lin XQ. Systematic screening and identification of the chlorinated transformation products of aromatic pharmaceuticals and personal care products using high-resolution mass spectrometry. Sci Total Environ. 2018;637–638:253–63. https://doi.org/10.1016/j.scitotenv.2018.05.011.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–30. https://doi.org/10.1021/ac020361s.
Funding
This research project was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. The authors are grateful to FAPESP (Grants 2019/22724–7, 2017/02147–0, 2015/15462–5, and 2014/07347–9) and CNPq (307293/2014–9) for the financial support provided.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bocelli, M.D., Medina, D.A.V., Lanças, F.M. et al. Automated microextraction by packed sorbent of endocrine disruptors in wastewater using a high-throughput robotic platform followed by liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 415, 6165–6176 (2023). https://doi.org/10.1007/s00216-023-04888-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04888-0