
Abstract
Lysophosphatidylcholine (LPC) can be used as a biomarker for diseases such as cancer, diabetes, atherosclerosis, and sepsis. In this study, we demonstrated the ability of nanozymes to displace the natural derived enzyme in enzyme-based assays for the measurement of LPC. Synthesized polyvinylpyrrolidone-stabilized platinum–ruthenium nanozymes (PVP/PtRu NZs) had a uniform size of 2.48 ± 0.24 nm and superb peroxidase-mimicking activity. We demonstrated that the nanozymes had high activity over a wide pH and temperature range and high stability after long-term storage. The LPC concentration could be accurately analyzed through the absorbance and fluorescence signals generated by the peroxidation reaction using the synthesized nanozyme with substrates such as 3,3′,5,5′-tetramethylbenzidine (TMB) and 10-acetyl-3,7-dihydroxyphenoxazine (Ampliflu™ Red). LPC at a concentration of 0–400 µM was used for the analysis, and the coefficient of determination (R2) was 0.977, and the limit of detection (LOD) was 23.1 µM by colorimetric assay. In the fluorometric assay, the R2 was 0.999, and the LOD was 8.97 µM. The spiked recovery values for the determination of LPC concentration in human serum samples were 102–115%. Based on these results, we declared that PVP/PtRu NZs had an ability comparable to that of the native enzyme horseradish peroxidase (HRP) in the enzyme-based LPC detection method.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lysophosphatidylcholine (LPC), also known as lysolecithin and lysoPC, is a type of lipid biomolecule produced from phosphatidylcholine via the enzymatic hydrolysis of one fatty acyl residue [1]. LPC constitutes the dominant part of oxidized low-density lipoprotein (oxLDL) and functions as an activator of several secondary messengers, including protein kinase C, protein tyrosine kinases, and Ca2+ [2, 3]. LPC is present in the plasma of healthy adults at a concentration of about 200–300 μM, and the concentration of LPC can be used as an indicator of various diseases [4]. This is because LPC concentrations in plasma correlate with various diseases such as several cancers [5,6,7,8], diabetes [9], pneumonia [10], atherogenic diseases [11], and sepsis [12]. Sepsis, the leading cause of death worldwide, has been reported to have a particularly strong correlation between patient plasma LPC concentration and mortality [13]. LPC has potential to serve as a biomarker for early diagnosis of various diseases, especially sepsis, and for this reason, investigation into accurate and rapid diagnosis of LPC concentration is being highlighted.
Established analytical technologies used to measure LPC are relatively complex, such as nuclear magnetic resonance (NMR) [14], matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS) [15], high-performance liquid chromatography–mass spectrometry (HPLC–MS) [16], and ultra-high-performance liquid chromatography–electrospray ionization–tandem mass spectrometry (UHPLC-ESI–MS) [17]. These methods require expensive equipment, skilled technicians who can handle the equipment and analyze the data, complex processing procedures, and long analysis times. For these reasons, hospitals and research institutes that can use these analytical methods are extremely limited. Simple analytical methods are emerging to overcome these technical and resource-driven limitations. An enzyme analysis method has been proposed that allows the determination of LPC concentration by simply obtaining an optical absorption signal using enzymes [18].
Most of the enzymes used in common enzymatic assays are derived from nature. Horseradish peroxidase (HRP), a representative natural enzyme, is a type of redox enzyme and has the ability to oxidize a substrate in the presence of hydrogen peroxide (H2O2) [19]. HRP is used in the fields of life science, to include medicine, biotechnology and biosensor systems, bioremediation, and biocatalysis [20]. Most enzyme assays that target biomolecules such as glucose and cholesterol and quantify the final product, H2O2, are inseparable from HRP. Nevertheless, as a protein derived from nature, HRP requires specialized labor to obtain. Sophisticated extraction steps, high costs, demanding working conditions, and exacting storage requirements are all part of acquiring HRP [21]. For this reason, over the past few decades, work has been conducted to develop an artificial enzyme that compensates for the shortcomings of natural enzyme. Compared to natural enzyme, artificial enzyme has several advantages such as capacity for large-scale synthesis at low cost through an easy method, operation in a wide temperature and pH range and high stability and activity [22].
Nanozymes that can replace natural enzymes reported so far include carbon-based nanoparticles [23], Co3O4 nanoparticles [24], CuO nanoparticles [25], Au nanoparticles [26], Fe nanoparticles [27, 28], Ir nanoparticles [29], Ru nanoparticles [30], Mn nanoparticles [31], and Pt nanoparticles [32,33,34]. Among these nanoparticles, Pt nanoparticles and their alloys exhibit outstanding catalytic properties because of their large surface areas. Additionally, Pt nanoparticles can be alloyed with transition metals such as Ru, which has low cost, good stability, and high activity, to maximize their catalytic activity [35]. Besides, the catalytic activities of Pt can be improved by the formation of nanoparticles with protective agents like polyvinylpyrrolidone (PVP) [36]. Our group recently fabricated PVP-stabilized Pt-Ru bimetallic nanozymes (PVP/PtRu NZs) and demonstrated that they have remarkable peroxidase-like activity and stability [37].
In this study, we applied PVP/PtRu NZs to LPC detection assay as a replacement for HRP. Using the ability of PVP/PtRu NZs to catalyze various substrates such as 3,3′,5,5′-tetramethylbenzidine (TMB) and 10-acetyl-3,7-dihydroxyphenoxazine (Ampliflu™ Red, AR), we determined LPC concentration in both absorbance and fluorescence systems, and they showed good linearity and high sensitivity.
Experimental
Materials
Chloroplatinic acid hydrate (H2PtCl6 · xH2O), ruthenium(III) chloride hydrate (RuCl3 · xH2O), calcium chloride dihydrate (CaCl2 · 2H2O), 10 kDa polyvinylpyrrolidone (PVP), Triton™ X-100, sodium acetate trihydrate, sodium hydroxide (NaOH), H2O2, TMB, terephthalic acid (TA), Ampliflu™ Red (AR), dimethyl sulfoxide (DMSO), L-α-lysophosphatidylcholine (LPC), L-α-phosphatidic acid sodium salt (PA), L-α-phosphatidylcholine (PC), sphingomyelin (SM), and human serum were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium borohydride (NaBH4) and Spectra/Por® 3 dialysis tubing were acquired from Alfa Aesar (Ward Hill, MA, USA) and Repligen (Rancho Dominguez, CA, USA), respectively. Buffer solution (pH 1.0–12.0) was supplied by Samchun Chemical Co., Ltd (Pyeongtaek, South Korea). 1 M Tris–HCl buffer (pH 7.4) and acetic acid were obtained from Biosesang, Inc. (Sung Nam, South Korea) and Junsei Chemical Co., Ltd (Tokyo, Japan), respectively. Glycerophosphorylcholine phosphodiesterase (GPCP) and lysophospholipase (LYPL) were purchased from Asahi Kasei Corp. (Tokyo, Japan). Choline oxidase (COD) was acquired from Toyobo Co., Ltd (Osaka, Japan).
Preparation of PVP/PtRu NZs
PVP/PtRu NZs were produced according to a previous report [37]. Briefly, 49 mL of an aqueous solution containing 5 μmol of PVP and 20 μmol each of H2PtCl6 and RuCl3 was prepared and stirred for 30 min. Then, the temperature of the solution was raised to 50 °C, and 1 mL of 80 mM NaBH4 in DI water was added. After stirring for 3 h, the reaction of the mixture was complete. Finally, the PVP/PtRu NZs obtained by dialyzing the solution for 48 h were stored at 4 °C until use.
Characterizations
The morphology of PVP/PtRu NZs was observed by transmission electron microscopy (TEM, JEOL JEM-2010, Japan). The crystal structure of nanoparticles was characterized by an X-ray diffractometer (XRD, D/MAX-2500, Rigaku, USA). The valence state and surface composition of PVP/PtRu NZs were investigated by X-ray photoelectron spectroscopy (XPS, VG SCIENTA R3000, UK). Inductively coupled plasma atomic emission spectroscopy (ICP-AES, SPECTRO ARCOS) was used to confirm the concentration of synthesized nanoparticles. All optical experiments for obtaining absorbance and fluorescence spectra were carried out by a UV–visible spectrometer (OPTIZEN Alpha, K LAB Co. Ltd., South Korea) and a multimode microplate reader (BioTek Instruments, Inc., USA).
Kinetics of PVP/PtRu NZs
A kinetic assay was performed to investigate the peroxidase-like activity of PVP/PtRu NZs. The pH and temperature conditions were optimized for the assay, and the efficiency on how quickly the nanoparticles catalyze TMB and H2O2 was evaluated by varying the concentration of each substrate. Twenty μL of PVP/PtRu NZs (20 μg/mL) was added to 780 μL of 0.1 M sodium acetate–acetic acid (NaAc-HAc) buffer (pH 5.0), and 100 μL of each substrate was added to initiate the oxidation reaction. The change in absorbance of the solution with time was measured, and the initial reaction rate (V0) (1) was calculated using the Beer–Lambert law (2).
where A means absorbance, ε means the molar absorption coefficient, c means the molar concentration, and l means the optical path length. Based on the calculated V0 value, kinetic parameters were obtained using the Michaelis–Menten Eq. (3) and the Lineweaver–Burk reciprocal Eq. (4):
where Vmax means the maximum velocity, [S] means the substrate concentration, and Km means the Michaelis–Menten constant. Vmax and Km represent the reaction rate of the nanozyme and its affinity for the substrates, respectively.
Determination of LPC
The method of enzyme analysis for LPC quantification was described in a previous report [18]. The mechanism of the reaction involved in the measurement is described in Fig. 1. First, by the action of the lysophospholipase (LYPL), fatty acid was released from LPC in human plasma or serum to become glycerophosphorylcholine (GPC). After that, GPC was decomposed into glycerophosphate and choline by glycerophosphorylcholine phosphodiesterase (GPCP) and then oxidized by choline oxidase (COD) to generate H2O2. The generated H2O2 was used during the peroxidation process of substrate by peroxidase-mimic PVP/PtRu and was quantified by the change in absorbance or fluorescence.

Scheme of enzymatic LPC assay mechanism
LPC assay with NZs (in absorbance system)
The method for determining LPC concentration using NZs in absorbance system was made through three steps. The concentrations of the elements constituting reagent 1 were GPCP 0.6 U/mL, COD 60 U/mL, and CaCl2 6 mM in a 0.1 M Tris–HCl solution at pH 8.0. Reagent 2 consisted of LYPL 60 U/mL in 0.1 M Tris–HCl solution at pH 8.0. Reagent 3 included 0.33 mM TMB and 26.67 μg/mL of PVP/PtRu NZs in a 0.1 M NaAc-HAc buffer solution at pH 5.0. In the first step, for LPC quantification, 20 μL of the LPC sample to be analyzed and 25 μL of reagent 1 were put into a 96-well plate and incubated at 37 °C for 2 min. As a second step, 25 μL of reagent 2 was added and incubated at 37 °C for 5 min. Finally for the third step, 150 μL of freshly prepared reagent 3 was added and incubated at 37 °C for 5 min, and then the absorbance at 652 nm was measured.
LPC assay with NZs (in fluorescence system)
Determination of LPC concentration using NZs in fluorescence system was also performed similarly to determining LPC concentration using NZs in absorbance system. Reagent 1 contained GPCP 0.2 U/mL, COD 20 U/mL, and CaCl2 2 mM in a 0.1 M Tris–HCl solution at pH 8.0. Reagent 2 consisted of LYPL 60 U/mL in 0.1 M Tris–HCl solution at pH 8.0. Reagent 3 included 10 μM AR and 20 μg/mL of PVP/PtRu NZs in a 0.1 M Tris–HCl solution at pH 8.0 containing 1% DMSO. First, 20 μL of the LPC sample and 75 μL of reagent 1 were put into a 96-well plate and incubated at 37 °C for 2 min. After that, 25 μL of reagent 2 was added and incubated at 37 °C for 5 min. Then, 100 μL of freshly prepared reagent 3 was added and incubated at 37 °C for 2 min, and after the fluorescence excitation at 563 nm, the emission at 587 nm was measured.
Results and Discussion
Characterization of PVP/PtRu NZs
To investigate the physicochemical properties of the synthesized PVP/PtRu NZs, various in-depth analyses were carried out. First, as a result of analyzing the fabricated PVP/PtRu NZs by ICP-AES, the concentration of nanozyme was determined at 85.6 mg/L, and it was confirmed that the prepared alloy had the same ratio of Pt and Ru elements. The calculated atomic proportions of Pt and Ru were 48.21% and 51.79%, respectively. Next, XRD analysis was performed to verify the crystal structure of the particles (Fig. 2A). The 2θ range of the scan for analysis was set from 30 to 90°. The resulting spectrum of PVP/PtRu NZs had the same pattern as the XRD analysis performed in previous studies [30, 38]. According to them, the spectrum of PVP/PtRu NZs was slightly shifted because the particles were dispersed by PVP to form a smaller crystal structure. The TEM images were obtained to investigate the shape and structure of the PVP/PtRu NZs. Figure 2B shows that well-dispersed PtRu alloys were uniformly synthesized by PVP acting as a stabilizer. The size of the synthesized nanoparticles was 2.48 ± 0.27 nm, and the inset indicates the size dispersion histogram of the PVP/PtRu NZs. Figure 2C is a high-resolution (HR) TEM image of the lattice pattern of each particle, and the spacing between the lattices was measured to be 0.23 nm. This value corresponds to the d-spacing of the (111) plane of the PtRu alloy. Figure 2D shows the selected-area electron diffraction (SAED) pattern of PVP/PtRu NZs. Each indexed ring in SAED pattern image reveals (111), (200), (220), and (311) planes, proving the polycrystalline structure of PVP/PtRu NZs.

A XRD profile of PVP/PtRu NZs. B TEM images of PVP/PtRu NZs. The inset shows a histogram of the average particle size. C HR-TEM images and D SAED pattern of PVP/PtRu NZs
Figure 3 shows the chemical composition of PVP/PtRu NZs through XPS analysis. Figure 3A is the survey spectrum of nanoparticles ranging from 0 to 700 eV. The peaks of Pt 4f and Ru 3p appeared at 66–82 eV and 450–495 eV, respectively. In addition, peaks of C 1 s, N 1 s, and O 1 s were observed, respectively, at 282–292 eV, 394–408 eV, and 527–540 eV, suggesting the presence of PVP. HR-XPS spectra were deconvolved to investigate the oxidation state of each element constituting PVP/PtRu NZs. The C 1 s spectrum in Fig. 3B included C–C (284.7 eV), C-N (285.7 eV), and O = C-N/O = C-O (287.1 eV) peaks [38]. These functional groups constitute PVP, and through these peaks, it was clearly confirmed that PVP was modified on PVP/PtRu NZs. Also, one peak (280.4 eV) was found in addition to the three peaks in the C 1 s spectrum. The additional peak corresponded to Ru3d whose binding energy range overlaps with that of C 1 s [39]. This peak indicated the presence of the oxidized Ru species, Ru4+. The Ru 3d peak detected was relatively weak because the surface of PVP/PtRu NZs was surrounded by a thick PVP layer. Figure 3C shows the HR spectrum of Pt 4f, and this doublet spectrum was formed by convolution of four peaks. They were two peaks (70.4 and 73.8 eV) consistent with metallic platinum (Pt0) and two peaks representing Pt2+ (71.5 and 75.2 eV). Figure 3D is the HR spectrum of Ru 3p, and peaks of Ru 3p3/2 (461.9 eV) and Ru 3p1/2 (484.1 eV) were observed. The oxidized species such as Ru4+ or Pt2+ were detected during the XPS analysis of PVP/PtRu NZs because of the oxidation of the sample by exposure to air, which inevitably occurs during the analysis process.

A XPS spectra of PVP/PtRu NZs. High-resolution XPS spectra of B C 1 s, C Pt 4f, and D Ru 3p
Enzyme-like activity of PVP/PtRu NZs
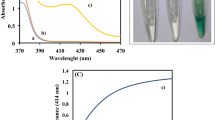
To analyze the enzyme-mimicking activity of the synthesized PVP/PtRu NZs, colorimetric analysis using TMB and H2O2 was also performed. The absorbance of 1 mL of the reaction solution was observed through UV–Vis analysis (Fig. 4A). The final concentration of the solution was 80 mM H2O2, 0.6 mM TMB, and 0.4 μg/mL PVP/PtRu NZs, in 0.1 M NaAc-HAc buffer (pH 5.0), and the reaction time was 90 s. In the absence of PVP/PtRu NZs (curve a) or TMB (curve b), no color change was observed because the reaction did not proceed. A very slight color change was observed when TMB and PVP/PtRu NZs were present simultaneously (curve c), indicating that PVP/PtRu NZs have the oxidase-mimic activity to oxidize TMB without H2O2. On the other hand, when H2O2, TMB, and PVP/PtRu NZs were all present (curve d), the reaction solution color changed significantly. This indicated that PVP/PtRu NZs have very potent peroxidase mimetic activity. Consequently, PVP/PtRu NZs are multi-functioned artificial enzymes with both weak oxidase-mimicking activity and strong peroxidase-mimicking activity.

A UV–Vis spectra of (a) H2O2 + TMB, (b) H2O2 + PVP/PtRu NZs (c) TMB + PVP/PtRu NZs, and (d) H2O2 + TMB + PVP/PtRu NZs. The inset shows a photographic image of four reaction solutions. B pH-dependent relative absorbance at 652 nm. C Temperature-dependent relative absorbance at 652 nm
Prior to obtaining the kinetic parameters of PVP/PtRu NZs using TMB and H2O2 substrates, the optimization conditions for PVP/PtRu NZs to peroxidize TMB were investigated. The substrates used for measurement were 0.2 mM TMB and 20 mM H2O2, and the reaction time was fixed at 90 s. First, to optimize the pH conditions, the activity was measured using a buffer solution having a pH range from 1.0 to 12.0, and as a result, high activity was shown to occur at pH 4.0 and 5.0 (Fig. 4B). The kinetic assay was performed at pH 5.0, which showed the highest activity. Changes in the activity of PVP/PtRu NZs with respect to temperature were also observed (Fig. 4C). As a result of measuring absorbance at intervals of 10 °C in the temperature range from 10 to 60 °C, there was no significant difference in activity over the whole temperature range measured. Experiments to determine kinetic parameters were carried out at room temperature using a buffer of pH 5.0 according to the condition optimization results.
Kinetics assay of PVP/PtRu NZs
Figs. 5A and B are Michaelis–Menten plots showing the reaction rate measured by changing the concentration of one substrate for another substrate with a fixed concentration. Figure 5A shows the results of measurements changing the concentration of TMB over 0.1–0.8 mM for 60 mM H2O2, and Fig. 5B shows the results of changing the concentration of H2O2 over 5–100 mM for 0.2 mM TMB. Figure 5C and D are Lineweaver–Burk plots obtained by taking the reciprocal of the corresponding values. Through this plotting, the Vmax and Km values of PVP/PtRu NZs were calculated. The synthesized NZs had Km and Vmax values of 0.25 mM and 5.95 \(\times\) 10−6 M s−1 for TMB and 33 mM and 5.80 \(\times\) 10−6 M s−1 for H2O2, respectively. The values were compared with the values of different catalysts with peroxidase-mimicking activity (Table 1). The Km value of PVP/PtRu NZs for TMB was 0.25 mM, which was about 1.7 times smaller than that of HRP, indicating that nanozymes had better affinity for TMB than HRP. However, it was confirmed that the Km value for H2O2 of PVP/PtRu NZs was higher than that of HRP and some catalysts. On the other hand, in the case of Vmax value, PVP/PtRu NZs had a huge value regardless of the type of substrate. This value was about 60 times greater than the Vmax value of HRP, which means that the nanozymes had the ability to react more substrates per unit time. Based on the above parameter values, the PVP/PtRu NZs could be considered to have sufficient potential to replace HRP.
Additionally, an investigation was carried out to discover whether the catalytic action of PVP/PtRu NZs through the ping-pong mechanism was the same as that of HRP [40]. The ping-pong mechanism means that enzyme reacts with substrates in sequence. That is, the enzyme reacts with the first substrate to release one product, and when it becomes an intermediate form, it reacts with the second substrate to produce another product. When comparing Lineweaver–Burk plots of substrates with several fixed concentrations, if each of them has a parallel shape, the response can be seen as following the ping-pong mechanism [41]. Since the Lineweaver–Burk plots for 40, 60, and 80 mM H2O2 (Fig. 5C) and the Lineweaver–Burk plots for TMB 0.15, 0.2, and 0.3 mM (Fig. 5D) were parallel, it was conceivable that the enzymatic reaction of PVP/PtRu NZs follows the ping-pong mechanism [42].

Michaelis–Menten plot of A TMB and B H2O2. Lineweaver–Burk plot of C TMB and D H2O2

A Structures of TA and 2-hydroxy-TA. B Fluorescence spectra of (a) TA + H2O2, (b) TA + NZs, (c) TA + H2O2 + NZs, and (d) TA + H2O2 + NZs + DMSO
Catalytic mechanism of PVP/PtRu NZs
A TA assay was also performed to confirm that the synthesized PVP/PtRu NZs caused a peroxidation reaction in the same way as a natural peroxidase. Peroxidase first reacts with H2O2 to generate hydroxyl radicals (•OH), and the generated •OH reacts with TMB to produce oxidized TMB with a blue color as a result. The TA assay is an experiment to confirm whether •OH is generated during the enzyme mimic action of PVP/PtRu NZs. As shown in Fig. 6A, TA does not show fluorescence by itself, but when reacted with •OH, it becomes 2-hydroxy-TA with fluorescence. The composition of the solution used in the assay was TA 0.4 mM, H2O2 20 mM, and PVP/PtRu NZs 2 μg/mL in 0.1 M NaAc-HAc buffer (pH 5.0). The excitation and emission wavelengths were 315 nm and 420 nm, respectively. In Fig. 6B, when H2O2 and nanozyme were present at the same time (curve c), •OH was generated from H2O2 by the nanozyme, and thus 2-hydroxy-TA was generated. The reason that curve a shows higher fluorescence than curve b was because •OH generated when H2O2 was exposed to light during the experiment reacted with TA. Curve d shows the spectrum when DMSO, which is a scavenger of •OH, was added. It was confirmed that generated •OH was removed by DMSO, so that no fluorescence was detected.

A Time-dependent absorption change graph and B calibration curve in LPC colorimetric detection using PVP/PtRu NZs. C Fluorescence spectra of (a) H2O2 + AR, (b) H2O2 + PVP/PtRu NZs, (c) AR + PVP/PtRu NZs, and (d) H2O2 + AR + PVP/PtRu NZs. D Calibration curve in LPC fluorometric detection using PVP/PtRu NZs
Colorimetric and fluorometric detection of LPC
A colorimetric analysis method was performed to determine the concentration of LPC based on the absorption signal generated by oxidizing the TMB in the presence of H2O2 through PVP/PtRu NZs. Since the amount of H2O2, the final product of the enzymatic reaction, varies depending on the concentration of LPC present in the sample, the intensity of the signal that is finally generated varies. LPC samples were compared at six concentrations of 0, 10, 25, 50, 200, and 400 μM. TMB was used for detection with PVP/PtRu NZs, and the maximum absorption wavelengths were 652 nm. Figure 7A shows the change in absorption with time in LPC detection using PVP/PtRu NZs. From the time of addition of reagent 3, the absorption increased and saturated according to the concentration. The calibration curve was plotted based on 10 min at which the absorbance was sufficiently saturated, as shown in Fig. 7B. A result having a high coefficient of determination (R2) value of 0.977 and a limit of detection (LOD) value of 23.1 μM was obtained. The LOD value was calculated by dividing 3 times the standard deviation value (σ) of the blank by the slope (s) of the calibration plot (LOD = 3σ/s).

The selectivity test using A PVP/PtRu NZs in absorbance system and B fluorescence system. The concentration of each sample was fixed at 0.1 mg/mL. (PA, phosphatidic acid; PC, phosphatidylcholine; SM, sphingomyelin). C Change in the activity of PVP/PtRu NZs with long-term storage
An assay was also conducted to determine the LPC concentration in a fluorescence system utilizing PVP/PtRu NZs. In order to obtain a fluorescence signal through peroxidase-mimicking activity, a substrate such as AR that emits a fluorescence signal had to be used. AR itself does not fluoresce, but when it is oxidized to the form of resorufin, it is excited at 563 nm and emits fluorescence at 587 nm. Before LPC fluorescence analysis, the enzyme-mimicking activity of PVP/PtRu NZs using AR was investigated (Fig. 7C). The fluorescence of 200 μL of the reaction solution in a 96-well plate was measured. The final composition of the reaction solution was 80 mM H2O2, 10 µM AR, and 0.4 µg/mL PVP/PtRu NZs, in 0.1 M Tris–HCl buffer (pH 8.0) containing 1% DMSO. The pH 8.0 buffer was used to match the working range of AR, a substrate that emits fluorescence. It was confirmed that strong fluorescence was emitted only when the nanozymes, H2O2, and AR were all present (curve d) due to the excellent peroxidase-mimic activity of PVP/PtRu NZs. Weak fluorescence was detected in curve a because H2O2, which was inevitably exposed to light during the experiment, reacted with AR. As a result, it could be demonstrated that PVP/PtRu NZs are peroxidase-mimicking artificial enzymes that can be used not only for absorption reactions but also for fluorescence reactions and can act widely in both low and high pH ranges. Based on the revealed ability, an experiment to detect LPC concentration in a fluorescence system was also conducted. As in the absorption system, six concentrations of 0, 10, 25, 50, 200, and 400 μM were measured. As a result, the intensity of the emitted fluorescence is shown in Fig. 7D. The R2 and LOD values calculated through the derived calibration curve were 0.999 and 8.97 μM, respectively. LPC detection in the fluorescence system using PVP/PtRu NZs showed higher linearity and lower LOD values than LPC detection in the absorption system using PVP/PtRu NZs. This was because the fluorescence system was less affected by the oxidase-mimicking ability of the nanozymes.
Selectivity, stability tests, and spike recovery
LPC colorimetric and fluorometric detection using PVP/PtRu NZs were all tested for the specificity of LPC measurements (Fig. 8A and B). The materials selected to investigate the selectivity of each method were phosphatidic acid (PA), phosphatidylcholine (PC), and sphingomyelin (SM), which have a structure similar to that of LPC. Each experiment was carried out in the same manner as the protocol used for LPC measurement, and the concentration of each sample used was 0.1 mg/mL. Both methods showed strong signals only in LPC. However, in the LPC colorimetric detection method using PVP/PtRu NZs, negative signal was generated for PA. This result was because reduced PA had an effect on the reduction of oxidized TMB. A stability test for long-term storage of PVP/PtRu NZs was conducted (Fig. 8C). For the stability test, the nanozymes were stored at 4 °C for 30 days, and activity was checked every 5 days. It was demonstrated that the activity of nanozyme did not change even after 30 days by PVP acting as a stabilizer. In addition, a spike recovery test was performed to evaluate the accuracy of the methods for detecting LPC present in human serum samples (Table 2). For evaluation, LPC samples (100 and 200 μM) were prepared by adding LPC to human serum. In absorbance system, the observed recovery values of 100 and 200 μM LPC were 104% and 103%, and relative standard deviations were 10.3% and 2.89%, respectively. In fluorescence system, the observed recovery values of 100 and 200 μM LPC were 115% and 102%, and relative standard deviations were 8.07% and 6.29%, respectively. These results in absorbance and fluorescence systems using PVP/PtRu NZs demonstrate that the proposed colorimetric and fluorometric methods are suitable for LPC determination in real samples.
Conclusions
We demonstrated that accurate LPC analysis in absorbance and fluorescence systems was also possible using PVP/PtRu NZs instead of HRP. LPC analysis using PVP/PtRu NZs could be performed at low cost and in a short time without using expensive equipment compared to LPC analysis methods that have been generally used in these days. The PVP/PtRu NZs used in this method, replacing HRP, a naturally occurring enzyme, had advantages such as low cost, easy synthesis, good stability, and ability to operate under various conditions. In addition, the corresponding nanozymes had a vigorous peroxidase-mimicking activity due to the synergistic effect of Pt and Ru, and it was well-dispersed and synthesized in an even form with the introduction of PVP so that it showed a very stable activity even during long-term storage. Considering the kinetic parameters obtained from the kinetic assay using TMB and H2O2, PVP/PtRu NZs had superb peroxidase-mimicking activity. Additionally, the reaction of the nanozymes followed a ping-pong mechanism and generated •OH as in the reaction of HRP. The results indicated that PVP/PtRu NZs are peroxidase-mimicking artificial enzymes that are not deficient compared to HRP, and based on this, absorbance and fluorescence detection of LPC using the nanozymes was conducted. The principle of the analysis was that the signal intensity of the oxidized substrates generated by the nanozymes and H2O2 was different because the amount of H2O2, the final product produced by the enzymatic reaction, varies according to the concentration of LPC. As a result, LPC in the range of 0–400 μM was measured, and in the case of absorbance detection, an R2 of 0.977 and an LOD of 23.1 μM were obtained, and in the case of a fluorescence detection method, an R2 of 0.999 and an LOD of 8.97 μM were obtained. Corresponding results suggested that PVP/PtRu NZs could be used as peroxidase for LPC enzyme analysis, as well as being multi-function nanozymes that catalyze TMB at pH 5.0 and AR at pH 8.0. In summary, PVP/PtRu NZs could be simply synthesized and had high stability and remarkable activity, which showed sufficient potential to replace naturally occurring enzymes. We expect that in the near future, this nanozyme will not only be used in the field of biomolecule detection, but will be suitably applied in the food industry, environmental field, and the pharmaceutical field where natural derived enzymes are currently used.
References
Baeza-Jimenez R, Lopez-Martinez LX, Otero C, Kim IH, Garcia HS. Enzyme-catalysed hydrolysis of phosphatidylcholine for the production of lysophosphatidylcholine. J Chem Technol Biot. 2013;88(10):1859–63.
Kabarowski JHS, Xu Y, Witte ON. Lysophosphatidylcholine as a ligand for immunoregulation. Biochem Pharmacol. 2002;64(2):161–7.
Matsumoto T, Kobayashi T, Kamata K. Role of lysophosphatidylcholine (LPC) in atherosclerosis. Curr Med Chem. 2007;14(30):3209–20.
Taylor LA, Arends J, Hodina AK, Unger C, Massing U. Plasma lyso-phosphatidylcholine concentration is decreased in cancer patients with weight loss and activated inflammatory status. Lipids Health Dis. 2007;6:17.
Zhang Q, Xu HR, Liu R, Gao P, Yang X, Jin W, et al. A novel strategy for targeted lipidomics based on LC-tandem-MS parameters prediction, quantification, and multiple statistical data mining: evaluation of lysophosphatidylcholines as potential cancer biomarkers. Anal Chem. 2019;91(5):3389–96.
Dong J, Cai XM, Zhao LL, Xue XY, Zou LJ, Zhang XL, et al. Lysophosphatidylcholine profiling of plasma: discrimination of isomers and discovery of lung cancer biomarkers. Metabolomics. 2010;6(4):478–88.
Zhao ZW, Xiao YJ, Elson P, Tan HY, Plummer SJ, Berk M, et al. Plasma lysophosphatidylcholine levels: potential biomarkers for colorectal cancer. J Clin Oncol. 2007;25(19):2696–701.
Zhou XC, Lawrence TJ, He Z, Pound CR, Mao JH, Bigler SA. The expression level of lysophosphatidylcholine acyltransferase 1 (LPCAT1) correlates to the progression of prostate cancer. Exp Mol Pathol. 2012;92(1):105–10.
Barber MN, Risis S, Yang C, Meikle PJ, Staples M, Febbraio MA, et al. Plasma lysophosphatidylcholine levels are reduced in obesity and type 2 diabetes. PLoS One. 2012;7(7):e41456.
Cho WH, Yeo HJ, Yoon SH, Lee SE, Jeon DS, Kim YS, et al. Lysophosphatidylcholine as a prognostic marker in community-acquired pneumonia requiring hospitalization: a pilot study. Eur J Clin Microbiol. 2015;34(2):309–15.
Schmitz G, Ruebsaamen K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis. 2010;208(1):10–8.
Lee EH, Shin MH, Park JM, Lee SG, Ku NS, Kim YS, et al. Diagnosis and mortality prediction of sepsis via lysophosphatidylcholine 16:0 measured by MALDI-TOF MS. Sci Rep. 2020;10:13833.
Drobnik W, Liebisch G, Audebert FX, Frohlich D, Gluck T, Vogel P, et al. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J Lipid Res. 2003;44(4):754–61.
Ahmmed MK, Carne A, Stewart I, Tian H, Bekhit AEA. Phosphorus-31 nuclear magnetic resonance (P-31 NMR) for quantitative measurements of phospholipids derived from natural products: effect of analysis conditions. LWT - Food Sci Technol. 2021;142:110991
Bresler K, Pyttel S, Paasch U, Schiller J. Parameters affecting the accuracy of the MALDI-TOF MS determination of the phosphatidylcholine/lysophosphatidylcholine (PC/LPC) ratio as potential marker of spermatozoa quality. Chem Phys Lipids. 2011;164(7):696–702.
Nishikimi M, Yagi T, Shoaib M, Takegawa R, Rasul R, Hayashida K, et al. Phospholipid screening postcardiac arrest detects decreased plasma lysophosphatidylcholine: supplementation as a new therapeutic approach. Crit Care Med. 2022;50(2):E199–208.
Wang JX, Lu XN, Zhang JE, Xiao ZH. Simultaneous quantification of the lipids phosphatidylcholine, 3-sn-phosphatidylethanolamine, sphingomyelin, and L-alpha-lysophosphatidylcholine extracted from the tissues of the invasive golden apple snail (Pomacea canaliculata) using UHPLC-ESI-MS/MS. Food Chemist. 2021;343:128427
Kishimoto T, Soda Y, Matsuyama Y, Mizuno K. An enzymatic assay for lysophosphatidylcholine concentration in human serum and plasma. Clin Biochem. 2002;35(5):411–6.
Lopes GR, Pinto DCGA, Silva AMS. Horseradish peroxidase (HRP) as a tool in green chemistry. Rsc Adv. 2014;4(70):37244–65.
Krainer FW, Glieder A. An updated view on horseradish peroxidases: recombinant production and biotechnological applications. Appl Microbiol Biot. 2015;99(4):1611–25.
Zhai GJ, Pelletier JP, Liu M, Aitken D, Randell E, Rahman P, et al. Activation of the phosphatidylcholine to lysophosphatidylcholine pathway is associated with osteoarthritis knee cartilage volume loss over time. Sci Rep 2019;9:9648
Fan SN, Jiang XX, Yang MH, Wang XG. Sensitive colorimetric assay for the determination of alkaline phosphatase activity utilizing nanozyme based on copper nanoparticle-modified Prussian blue. Anal Bioanal Chem. 2021;413(15):3955–63.
Song YJ, Qu KG, Zhao C, Ren JS, Qu XG. Graphene oxide: intrinsic peroxidase catalytic activity and its application to glucose detection. Adv Mater. 2010;22(19):2206–10.
Wang Q, Liu SW, Sun HY, Lu QF. Synthesis and intrinsic peroxidase-like activity of sisal-like cobalt oxide architectures. Ind Eng Chem Res. 2014;53(19):7917–22.
Hu LZ, Yuan YL, Zhang L, Zhao JM, Majeed S, Xu GB. Copper nanoclusters as peroxidase mimetics and their applications to H2O2 and glucose detection. Anal Chim Acta. 2013;762:83–6.
Wang S, Chen W, Liu AL, Hong L, Deng HH, Lin XH. Comparison of the peroxidase-like activity of unmodified, amino-modified, and citrate-capped gold nanoparticles. ChemPhysChem. 2012;13(5):1199–204.
Gao LZ, Zhuang J, Nie L, Zhang JB, Zhang Y, Gu N, et al. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat Nanotechnol. 2007;2(9):577–83.
Son SE, Gupta PK, Hur W, Choi H, Lee HB, Park Y, et al. Determination of glycated albumin using a Prussian blue nanozyme-based boronate affinity sandwich assay. Anal Chim Acta. 2020;1134:41–9.
Tran VK, Gupta PK, Park Y, Son SE, Hur W, Lee HB, et al. Functionalized bimetallic IrPt alloy nanoparticles: multi-enzyme mimics for colorimetric and fluorometric detection of hydrogen peroxide and glucose. J Taiwan Inst Chem E. 2021;120:336–43.
Gupta PK, Son SE, Seong GH. Functionalized ultra-fine bimetallic PtRu alloy nanoparticle with high peroxidase-mimicking activity for rapid and sensitive colorimetric quantification of C-reactive protein. Microchimica Acta. 2021;188(4):119.
Qiao FM, Chen LJ, Li XN, Li LF, Ai SY. Peroxidase-like activity of manganese selenide nanoparticles and its analytical application for visual detection of hydrogen peroxide and glucose. Sensor Actuat B-Chem. 2014;193:255–62.
Fan J, Yin JJ, Ning B, Wu XC, Hu Y, Ferrari M, et al. Direct evidence for catalase and peroxidase activities of ferritin-platinum nanoparticles. Biomaterials. 2011;32(6):1611–8.
Arsalan M, Qiao XJ, Awais A, Wang YH, Yang SY, Sheng QL, et al. Enhanced sensitive electrochemical sensor for simultaneous catechol and hydroquinone detection by using ultrasmall ternary pt-based nanomaterial. Electroanal. 2021;33(6):1528–38.
Lee HB, Son SE, Gupta PK, Venkatesan J, Hur W, Park J, et al. Mesoporous platinum nanozyme-based competitive immunoassay for sensitive detection of 25-hydroxyvitamin D. Mater Lett. 2023;330:133286
Cao GJ, Jiang X, Zhang H, Croley TR, Yin JJ. Mimicking horseradish peroxidase and oxidase using ruthenium nanomaterials. Rsc Adv. 2017;7(82):52210–7.
Khan MA, Al Mamun MS, Ara MH. Review on platinum nanoparticles: synthesis, characterization, and applications. Microchem J. 2021;171:106840
Park Y, Gupta PK, Tran VK, Son SE, Hur W, Lee HB, et al. PVP-stabilized PtRu nanozymes with peroxidase-like activity and its application for colorimetric and fluorometric glucose detection. Colloid Surface B. 2021;204:111783
Liu C, Guo YQ, Zhang JM, Tian B, Lin OK, Liu YW, et al. Tailor-made high-performance reverse osmosis membranes by surface fixation of hydrophilic macromolecules for wastewater treatment. Rsc Adv. 2019;9(31):17766–77.
Hasa B, Kalamaras E, Papaioannou EI, Vakros J, Sygellou L, Katsaounis A. Effect of TiO2 loading on Pt-Ru catalysts during alcohol electrooxidation. Electrochim Acta. 2015;179:578–87.
Porter DJT, Bright HJ. The mechanism of oxidation of nitroalkanes by horseradish-peroxidase. J Biol Chem. 1983;258(16):9913–24.
Sychantha D, Clarke AJ. Peptidoglycan modification by the catalytic domain of Streptococcus pneumoniae OatA follows a ping-pong bi-bi mechanism of action. Biochemistry-Us. 2018;57(16):2394–401.
Nazifi M, Ahmadi R, Ramezani AM, Absalan G. Introducing hierarchical hollow MnO2 microspheres as nanozymes for colorimetric determination of captopril. Anal Bioanal Chem. 2021;413(28):7063–72.
Wang X, Tang CL, Liu JJ, Zhang HZ, Wang J. Ultra-small CuS nanoparticles as peroxidase mimetics for sensitive and colorimetric detection of uric acid in human serum. Chinese J Anal Chem. 2018;46(5):E1825–31.
Li J, Zhao J, Li SQ, Chen Y, Lv WQ, Zhang JH, et al. Synergistic effect enhances the peroxidase-like activity in platinum nanoparticle-supported metal-organic framework hybrid nanozymes for ultrasensitive detection of glucose. Nano Res. 2021;14(12):4689–95.
Zhou DD, Zeng K, Yang MH. Gold nanoparticle-loaded hollow Prussian Blue nanoparticles with peroxidase-like activity for colorimetric determination of L-lactic acid. Microchim Acta. 2019;186:121
Chen LJ, Wang N, Wang XD, Ai SY. Protein-directed in situ synthesis of platinum nanoparticles with superior peroxidase-like activity, and their use for photometric determination of hydrogen peroxide. Microchim Acta. 2013;180(15–16):1517–22.
Ming J, Zhu TB, Li JC, Ye ZC, Shi CR, Guo ZD, et al. A novel cascade nanoreactor integrating two-dimensional Pd-Ru nanozyme, uricase and red blood cell membrane for highly efficient hyperuricemia treatment. Small. 2021;17:2103645
Cai SF, Xiao W, Duan HH, Liang XX, Wang C, Yang R, et al. Single-layer Rh nanosheets with ultrahigh peroxidaselike activity for colorimetric biosensing. Nano Res. 2018;11(12):6304–15.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea (2018R1A6A1A03024231 and 2021R1A2C1003566).
Author information
Authors and Affiliations
Contributions
Ji Yeon Park and Han Been Lee, conceptualization, methodology, investigation, formal analysis, visualization, and writing, original draft. Seong Eun Son, investigation. Pramod K. Gupta, investigation. Yosep Park, investigation. Won Hur, investigation. Gi Hun Seong, supervision; visualization; writing, review and editing; and funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, J.Y., Lee, H.B., Son, S.E. et al. Determination of lysophosphatidylcholine using peroxidase-mimic PVP/PtRu nanozyme. Anal Bioanal Chem 415, 1865–1876 (2023). https://doi.org/10.1007/s00216-023-04590-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04590-1