
Abstract
A rapid and sensitive high-performance liquid chromatography–high-resolution orbitrap mass spectrometry method was developed for the simultaneous screening of 354 organic poisons and metabolites in blood and urine, including drugs, medications, pesticides, rodenticides, veterinary drugs, alkaloids, and mycotoxins with a multi-toxicant chromatography-mass spectrometry information library. The method and library showed good prospects in clinical poisoning screening and forensic toxicological identification. Blood and urine samples were extracted successively with ethyl acetate in acidic and alkaline conditions; then, the extract was blown to nearly dry by nitrogen gas and redissolved with methanol-aqueous solution (v:v, 50:50), and the dissolved solution was analyzed by LC-MS/MS after filtering. Precursor ions’ m/z was set for identification, retention time, fragment ions, and isotopic pattern which were used for confirmation. No interference peaks were found in the blank samples, showing good specificity. The LODs of toxicants in urine and blood were 1.00×10−3–50.0 ng/mL and 2.07×10−3–50.0 ng/mL, respectively, while the LOQs were 3.30×10−3–1.67×102 ng/mL and 6.91×10−3–1.67×102 ng/mL. The intra-day precision and inter-day precision of urine samples were 2.31–9.13% and 4.75–12.3%, respectively, which were 1.92–10.8% and 2.01–12.1% in blood samples. The established method was applied to analyze 9 cases of clinical poisoning patients, and bromadiolone, carbofuran, and amanitins were detected, respectively. A total of 382 biospecimens from drug abusers were analyzed with the proposed method, which indicated that some drugs were detected in 62 cases, mainly including methamphetamine, heroin, and MDMA. The results were consistent with the information from traditional liquid chromatography-triple quadrupole mass spectrometry.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With the development of industry, agriculture, and medicine, the types and quantities of chemicals have been increasing rapidly in recent years. It is reported that more than 350,000 compounds have been registered for industrial production and used in our daily life, which raises the risk of toxic exposure to people and causes health damage. Chemicals may cause health damage and thus lead to poisoning when they enter the human body through different pathways such as inhalation, ingestion, and skin and mucous membrane contact. In 2019, more than 2.0 million people died of chemical poisoning, and the loss of disability-adjusted life expectancy due to toxic chemicals exposure was 53 million years, which increased by 25% and 17.8% respectively compared to 2016 in the world [1].
There has been an increasing number of poisonings caused by organic chemicals in recent years, mainly including drugs, medications, pesticides, rodenticides, veterinary drugs, alkaloids, and mycotoxins. In China, accidents caused by organic poisons account for more than 70% of all chemical poisonings, and most of them were induced by drug overdose, alcoholism, and pesticide and rodenticide intake [2]. Similarly, in the USA, this problem was becoming more serious than before. According to the information from the American Association of Poison Control Centers [3], more than 2 million people experienced various degrees of poisoning in 2020 due to drug abuse and overdose. In Germany, organic toxicants are also becoming a major cause of poisoning, especially among children. Poison Information Centre- North indicated that there were 40,078 cases of poisoning or suspected poisoning in northern Germany in 2019, including 15,242 cases in children under 4 years old, in which medicine, toxic plants and animals, poisonous mushrooms, drugs, pesticides, and veterinary drugs were mainly responsible for the poisoning [4].
When poisoning occurs, timely identification of toxic substances is of great help for targeted treatment. Liquid chromatography (LC) [5,6,7], liquid chromatography-mass spectrometry (LC-MS) [8,9,10], gas chromatography (GC) [11, 12], gas chromatography-mass spectrometry (GC-MS) [13,14,15], immunoassay [16, 17], and spectrophotometry [18, 19] are common methods for chemicals analysis. Chromatography-mass spectrometry is the most commonly used laboratory confirmation method in toxicology analysis, but most of the existing methods are used for targeted analysis. When an unknown substance poisoning occurs, the targeted analysis method needs to identify the suspected poisons one by one, which could be inefficient and time-consuming, and it is difficult to meet the requirements of clinical poisoning diagnosis and forensic toxicological identification. How can we identify and confirm the suspected poisons from varieties of toxins and provide accurate poisoning information in time? Non-targeted screening, a method that enables broad-spectrum toxins identification efficiently in complex sample matrix, gives solutions. High-resolution mass spectrometry is one of the most popular techniques in non-targeted analysis, which can attain better resolution and accuracy with GC or HPLC, as well as provide precise molecular mass of substances and analyze the structure of unknown or suspected compounds by cooperating with professional software [20, 21]. Pan et al. [13] developed a new high-throughput screening method based on GC-Orbitrap-HRMS to identify 288 toxicants in human blood, and realized rapid screening of poisons by comparing with the previously developed poisons database. Roman et al. [20] established an LC-TOF-MS method for semi-targeted screening of toxicants in cadaver blood, and built a database involving 240 drugs and metabolites by reference standards and literature at the same time. The method was applied to analyze real cadaver blood samples, and the results presented that this method increased the types of screening compounds by 50% and had higher accuracy and sensitivity compared with the previous GC-nitrogen phosphorus detector screening method. Although the detection efficiency is higher than traditional methods, sometimes it is still not enough for practice, and the accurate mass spectrometry database for organic poisons is also restricted. Some libraries and databases supplied in manufacturer software packages can be used, but the compounds included are limited or lack some key information, such as fragment ions and retention time, which can greatly improve screening accuracy and reduce false positives. Moreover, retention time in packages usually obtained in different chromatographic conditions, which cannot support screening of multiple substances at one time. In literature, most of the methods focused on a specific type of poisons, and there are few high-resolution mass spectrometry screening methods for multiple types of organic poisons in biospecimen, limiting their application and popularization in clinical and forensic medicine to some extent.
In this paper, a multi-toxicant screening method for human blood and urine samples was established by high-performance liquid chromatography–high-resolution orbitrap mass spectrometry (HPLC-Q-orbitrap MS). A total of 354 kinds of organic toxicants and metabolites that often cause poisoning were selected, including drugs, medications, pesticides, rodenticides, veterinary drugs, alkaloids, and mycotoxins; moreover, a multi-toxin chromatography-mass spectrometry database was constructed using reference substances. Successfully applied in the clinical and forensic toxicology analysis, the method not only satisfies the demand of high-throughput screening for poisoning events but also provides an experimental basis and data for broad-spectrum screening of suspected toxicants.
Material and methods
Reagents and standards
Formic acid, ammonium formate, and ammonium acetate of LC-MS grade and ethyl ether, dichloromethane, and ethyl acetate of HPLC grade were supplied by Sigma-Aldrich (St. Louis, USA). Acetone, sodium hydroxide, and concentrated hydrochloric acid of analytical reagent grade were prepared from Chengdu Kelong Chemical Co., Ltd. (Chengdu, China). Methanol (LC-MS grade) was purchased from Thermo Fisher Scientific (Pittsburgh, USA). Ultrapure water (18.2 MΩ·cm) was prepared by Milli-Q purification system (Millipore, MA, USA). The solution of ammonium in water (1%, ACS grade) was obtained from Honeywell Forge (NJ, USA).
A total of 354 organic poisons include 22 drugs, 33 medications, 248 pesticides, 12 rodenticides, 4 veterinary drugs, 13 toxic alkaloids, and 22 mycotoxins, whose names and physicochemical properties are listed in Supplement Table, with information (e.g., polarity, boiling point, and acidity) obtained from http://www.chemspider.com, ACD/Labs, and https://pubchem.ncbi.nlm.nih.gov. Toxicity-related doses, for example, median lethal dose (LD50), lowest published toxic dose low (TDLo), and lowest published lethal dose (LDLo), were obtained from https://www.drugfuture.com/toxic/. The 354 reference standards were purchased from Dr. Ehrenstorfer (Augsburg, Germany), Bepure (Beijing, China), Beijing TanMo Quality Testing Technology Co., Ltd (Beijing, China), Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China), Toronto Research Chemicals (Toronto, Canada), AccuStandard (New Haven, USA), and Sigma-Aldrich (St. Louis, USA). Except for some standards of pesticides (stored in ethyl acetate/acetone) at a concentration of 50 μg/mL, the purity of other standards was ≥ 99.9%. The standard solution was prepared with methanol-aqueous solution (v:v, 50:50) to the desired concentration and stored at −80°C in a brown bottle.
The retention time of all toxicants was in the range of 0–15 min. The analytes for quality control solution were selected in different time ranges and the mixed standard solution prepared with methanol-aqueous solution (v:v, 50:50) was stored at −80°C in brown bottles, including phosphinothricin (RT=0.62 min), isoniazid (RT=1.63 min), omethoate (RT=2.60 min), metsulfuron-methyl (RT=3.57 min), caffeine (RT=4.63 min), salbutamol (RT=5.62 min), hexazinone (RT=6.63 min), fenamidone (RT=7.58 min), noscapine (RT=8.64 min), clozapine (RT=9.62 min), mabuterol (RT=10.69 min), phenylethanolamine A (RT=11.89 min), methadone (RT = 13.98 min), and bulleyaconitine (RT = 14.38 min).
Instrumentation
HPLC conditions
HPLC analysis was conducted on a Dionex UltiMate 3000 RSLC system with a Hypersil Gold PFP column (2.1 mm × 100 mm, 3 μm, death volume of 0.10 mL, Thermo Fisher Scientific, USA). The injection volume was 5 μL, and the autosampler and column were maintained at 6 °C and 40 °C, respectively. The mobile phase consisted of methanol (A) and 5 mmol/L ammonium acetate solution (B, pH=6.5) was at a flow rate of 0.40 mL/min. The gradient elution procedure was as follows: 0–0.5 min, 5% A; 0.5–10 min, 5–95% A; hold with 95% A for 5 min, and equilibrate with 5% A for 5 min before the next injection.
HRMS conditions
The Q Exactive Plus Orbitrap mass spectrometer equipped with an ESI ion source (HESI-II, Thermo Fischer Scientific, Bremen, Germany) was used in the experiments with the alternating mode of positive and negative ions.
The parameters of the HESI-II source were set as follows: sheath gas flow rate of 50.00 arbitrary units (au), auxiliary gas flow rate of 12.50 au, purge gas flow rate of 0 au, S-Lens RF level of 55.00, electrospray voltage of 3.5 kV (+) and 3.0 kV (−). The capillary and auxiliary gas heater temperatures were both 350°C and the probe heater temp was 425°C. The exclusion list was set to “on” to avoid interference of plasticizer contamination from instrument pipe and sample preparation.
Data acquisition was carried out in full MS/dd-MS2 mode. For full MS, the mass-to-charge ratio (m/z) was in the range of 120–1300, with a resolution of 70,000, the automatic gain control (AGC) target was 3×106, and the maximum injection time (MIT) was set to 100 ms. For dd-MS2, the resolution was 17,500, the AGC target was 1×105, MIT was 50 ms, loop count was 10, TopN was 10, isolation window was 1.6 m/z, intensity threshold was 5×103, apex trigger was set to 0.1 to 10 s, and dynamic exclusion was set to 10s. Isotope exclusion was set to “on,” collision energy (CE) to 20, 30, and 40 eV, if idle was set to pick other and run time to 0–20 min. Mass calibration was performed on the instrument every 3 days by using pierce ESI positive and negative ion calibration solutions.
Instrument control and data acquisition were performed by Xcalibur 4.0 software, and target screening was done by TraceFinder 5.1 software (Thermo Fisher Scientific, MA, USA).
Sample preparation
All samples were obtained from the Sichuan Dingcheng Forensic Service, the West China Fourth Hospital of Sichuan University. Blood samples collected in vacuum tubes were centrifuged at 3500 rpm for 10 min to obtain the plasma, while urine samples were collected in polypropylene plastic tubes. All samples were stored at −80°C until analysis. Twenty microliters of 1.0 mol/L HCl solution was added to 1.0 mL samples to adjust the pH value; then, the mixture was extracted with 3.0 mL ethyl acetate by vortexing for about 2 min, and the ethyl acetate extract was collected after centrifuging at 4000 rpm for 2 min. The pH value of the remaining samples was adjusted to alkaline with 250 μL 0.10 mol/L NaOH solution, and then, the above extraction steps were repeated and the extract was collected. After being combined and blown to nearly dry with moderate nitrogen gas flow, the extract was redissolved with 100 μL (urine samples) or 200 μL (plasma samples) methanol-aqueous solution (v:v, 50:50). The reconstituted solution was injected into LC-MS/MS for analysis after being filtered through a 0.22-μm syringe filter. When the amount of sample is limited, a smaller amount can be used, and the concentration rate can be achieved by correspondingly reducing the volume of the redissolved solvent.
Toxin identification
The standard solution was diluted to 100 ng/mL and analyzed in full MS/dd-MS2 mode by the Q Exactive Plus Orbitrap HPLC-HRMS system. The accurate m/z, retention time (RT), and collision energy were obtained by Xcalibur 4.0 software, and a database for toxicant identification and confirmation was built by TraceFinder 5.1 software with the information of compound name, molecular formula, and chemical abstracts service (CAS) number.
Identification and confirmation settings are as follows: precursor ions’ m/z were used for identification, threshold override>5000, S/N ratio threshold>5, mass tolerance<5 ppm. RT was used for confirmation and the window override was ± 0.3min. Fragment ions were used to confirm, the minimum ion number of fragment ions was 1, the intensity threshold was set to 1000, mass tolerance <5 ppm, and MS order was MS2. Isotopic pattern was used to confirm, fit threshold >70%, allowed mass tolerance <5 ppm, and allowed intensity tolerance <20%.
Method validation
The method validation was performed with mixed blank plasma and urine samples from six different healthy volunteers without any objective drugs or medications taking history within 6 months. The precisions of target compounds (relative standard derivations, RSDs) were assessed by measuring the recoveries of spiked samples at the method quantification limit (LOQ) concentrations of urine and plasma repeatedly within the same day (n=5) and over five separate days. The method detection limits (LOD) and LOQ were calculated as 3 and 10 times of signal-to-noise (S/N) ratio by measuring S/N of the low-concentration spiked samples. The matrix effect was evaluated by comparing the slopes of the calibration lines prepared with the solvent and the blank matrix extracts. The quality control sample and blank sample were analyzed every 6 h or in every analysis batch. The analysis can be continued when the retention time difference of the quality control samples was within 2.5% compared with the mean value, the change of peak area was no more than 10%, and the target analytes were not detected in the blank sample. Otherwise, it is necessary to stop the analysis and find the cause, and then continue after eliminating the abnormality.
Results and discussion
Optimization of mass spectrometry conditions
One hundred nanograms per milliliter of standard solutions prepared with methanol-aqueous solution (v:v, 50:50) was injected into the HPLC-Q Exactive Plus Orbitrap MS system to optimize the mass spectrometry parameters and establish a mass spectrometry database. The optimized conditions are shown in “HRMS conditions.” Some of the database parameters are shown in Table 1. The inclusion list was “on” and all of the analytes were put into the list so that the mass system could preferentially pick up the precursor ions of the toxicants in the database. The interfering ions with m/z of 279.15909 and 149.02332 in the sample analysis coming from the plasticizer possibly were placed in the exclusion list to reduce the scanning of these ions by the mass spectrometer.
In the positive ion mode, adduct ions [M+H]+ and [M+NH4]+ can be observed, while only [M-H]- can be observed in the negative ion mode. Most of the poisons responded well in the [M+H]+ form, whereas some toxicants only have the [M+NH4]+ or [M-H]- form, such as [M-H]- for chlorophacinone and bromadiolone and [M+NH4]+ for fenvalerate. Some mycotoxins and pesticides have three ionized forms. To improve the sensitivity, the precursor ions of the highest response were selected in the establishment of the toxicant screening database and combined with 3–5 characteristic fragment ions in the top responding, while the collision voltage was further optimized. It is available for most poisons to get satisfactory mixed fragment ion spectrums under the stepped collision voltage of 20 eV, 30 eV, and 40 eV. For some compounds with fused ring structures like strychnine and sophoridine, however, higher collision voltages were required to obtain more sensitive results. Contrarily, lower collision voltages were required to achieve good spectrums for some compounds with many-branched chains like thebaine and barbital. The optimized collision voltages are shown in Table 1 and are added to the inclusion list.
Optimization of chromatographic conditions
There is a wide range of organic toxicants with huge differences between their properties; therefore, a PFP column (2.1 mm × 100 mm, 3 μm, Thermo Fisher Scientific, USA) was selected for separation, which takes pentafluorophenyl as a stationary phase to extend the retention time of polar components and has better separation efficiency for substances containing polar groups such as halogen, hydroxyl, carboxyl, nitro, and amino. The effects of methanol-aqueous solution and acetonitrile-aqueous solution as the mobile phases on response signal, peak shape, and separation of all toxicants were investigated. The results showed that methanol is a better choice because of its similar chromatographic behavior with acetonitrile, but lower toxicity. Better sensitivity and peak shape can be achieved by adding acidic and alkaline additives to the mobile phase. 0.1% ammonia, 0.1% formic acid, 5 mmol/L ammonium formate, and 5 mmol/L ammonium acetate were investigated as the modifier for improving chromatographic separation and mass spectrometry detection. The result indicated that most of the target compounds can obtain satisfactory sensitivity and peak shape in ammonium acetate solution, which is close to neutral and has good buffer capacity. The other acidic and basic additives conflict in the improvement of chromatographic behavior and the sensitivity of mass spectrometry. Moreover, to reduce the interference of the matrix effect while cleaning the column for continuous injection, gradient elution was selected and the elution procedure was optimized, which is shown in “HPLC conditions.”
Sample pretreatment optimization
Selection of extraction conditions
Liquid-liquid extraction was selected for sample pretreatment because of its simple operation and efficient extraction of a variety of substances [13, 22, 23]. Since target toxicants contain alkaline and acidic substances, 1mol/L HCl solution and 0.1mol/L NaOH solution were added to the samples to adjust the pH value to be acidic and alkaline respectively; then, the samples were extracted twice with the organic solvent, so that each target toxin can be extracted efficiently. Finally, the extract was dried using a nitrogen stream for concentration.
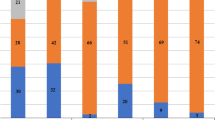
As seen from the logP value, the target toxicants are mostly compounds of medium polarity. As a result, three commonly used solvents of moderate polarity (ether, logP=0.98; ethyl acetate, logP=0.71; dichloromethane, logP=1.19) were examined as the extraction solvent. The spiked blank urine and plasma samples were used in the experiment, and the recoveries were calculated according to the following formula for evaluation. The spiked concentration was set in the range of 10–200 ng/mL according to the detection sensitivity and toxic dose, and the results are shown in Fig. 1.

Recovery results of 354 toxicants in urine and blood samples after extraction with ethyl ether, ethyl acetate, and dichloromethane
Most of the toxicants can get satisfactory recoveries with ethyl acetate, followed by ethyl ether, and the worst with dichloromethane, so ethyl acetate was selected as the extraction solvent and the influence of its volume on the analysis was studied in the range of 1–5 mL. The results suggested that the recoveries of most of the target toxicants were highest and stable in the range of 2–5 mL. Therefore, 3 mL of ethyl acetate in 1 mL of sample was chosen for toxicant extraction finally.
The pH value and ionic strength of samples were studied as important factors affecting the recoveries. In order to simplify the procedure, the addition amount of 1 mol/L HCl solution and 0.10 mol/L NaOH solution was examined to investigate the influence of sample pH value. At first, the spiked blank samples were extracted with 3 mL of ethyl acetate after adding 10~50 μL of 1mol/L HCl solution, and the recoveries were determined. The results showed that most of the toxicants obtained high and stable recoveries when 20μL of 1mol/L HCl solution was added and the pH value of the sample was about 2, except for alkaline substances such as theobromine, ephedrine, and colchicine. To achieve good recoveries of alkaline substances, it is necessary to adjust the pH value and extract again. Fifty to approximately three hundred microliters of 0.10mol/L NaOH solution was added to the sample solution after the above extraction, and the recoveries of toxicants were determined. The results showed that the alkaline toxicants obtained high and stable recoveries when 250μL of 0.10mol/L NaOH was added and the pH value of the sample was about 12.
NaCl solution was added to the sample to increase the ionic strength. The results indicated that the addition of NaCl had little effect on the recoveries, probably due to the fact that the salt concentration in urine and blood samples had reached a high level after pH was adjusted.
The recoveries of some toxicants were still lower than 40% even under the optimized extraction conditions (Fig. 1). As the sample matrix effect, the extraction, and the nitrogen blowing process would affect recoveries, the effects of these factors were investigated separately.
The sample matrix effects
After the samples were extracted, concentrated, and redissolved, the standard solutions were prepared with the redissolved solution, and another group at the same concentration was prepared with the solvent (methanol-aqueous solution, v:v, 50:50). The matrix effects were assessed by the ratio of the slopes of the two calibration lines, and the results are shown in Fig. 2.

Influence of matrix effect, extraction process, and concentration process on the recovery of toxicants
In general, the matrix effects of plasma were stronger than those of urine, and most toxicants tend to be ion suppression. The matrix spiked recoveries were in the range of 100%±40% for 93.2% and 79.1% poisons in urine and plasma samples, respectively. Six toxicants that isoniazid (14.1%), ribavirin (10.3%), tribenuron-methyl (18.9%), chlorophacinone (16.8%), diphacinone (12.1%), and delta-9-tetrahydrocannabinol (6.12%) were in strong matrix effects in urine, whose matrix spiked recoveries were less than 20%. Moreover, matrix effects of fifteen toxins were lower than 20% in plasma, including bifenthrin (18.9%), carbophenothion (3.40%), chlorthiophos (5.13%), cypermethrin (3.00%), dioxathion (0.51%), fenthion (16.64%), permethrin (1.33%), pyriproxyfen (5.76%), quinoxyfen (7.95%), 1,2,4-tributyl phosphorotrithioate (12.8%), taufluvalinate (15.9%), cyhalothrin (17.8%), ephedrine (18.8%), delta-9-tetrahydrocannabinol (2.32%), and esfenvalerate (19.9%). Although the recoveries of these poisons with high sample matrix effect were very low, their high mass spectrometry response signals enable sensitive and accurate screening at the toxic levels.
Extraction efficiency
For some toxicants, the sample matrix effects were not the main reason affecting recoveries. To find the reason for insufficient recoveries, the extraction efficiency was investigated separately using ultrapure water instead of samples, and calculated by comparing the peak areas of spiked ultrapure water after sample preparation with that of the standard solution of the same concentration prepared by methanol-aqueous solution (v:v, 50:50). The results are shown in Fig. 2. The recoveries of the most toxicants were in the range of 61–100% while four toxicants <40% in the extraction process, involving brodifacoum (38.9%), flocoumafen (37.5%), acrinathrin (33.6%), and fluvalinate (29.8%). It is possible that the low solubility of these four poisons in ethyl acetate resulted in inefficient extraction.
The influence of concentration with nitrogen blowing
Ten nanograms per milliliter standard solution prepared with ethyl acetate was blown to dryness with nitrogen stream, and then redissolved with methanol-aqueous solution (v:v, 50:50) and concentrated 10 times for LC-MS/MS analysis. At the same time, 100 ng/mL standard solution prepared with methanol-aqueous solution (v:v, 50:50) was injected directly into LC-MS/MS for detection. Comparing their peak areas to evaluate the impact of nitrogen blowing and redissolution, the results are shown in Fig. 2. The results demonstrated that all recoveries of the toxicants were from 61 to 120% except dichlorvos (53.4%) and methomyl (58.9%) which are highly volatile (boiling points are 140°C and 144°C, respectively), so they may volatilize during the nitrogen blowing, resulting in the decline of recoveries.
In addition, the reason for the low recoveries of some poisons was the mixed effect of the matrix effect, the extraction, and the concentration method throughout the sample pretreatment, containing acephate, flucythrinate, and nivalenol in urine samples, and alpha-solanine, bifenox, reserpine, sinomenine, aclonifen, deltamethrin, dichlofenthion, diclofop-methyl, fenvalerate, pendimethalin, α-amanitin, and β-amanitin in plasma samples, as well as isofenphos-methyl, methoprene, methyl-parathion, naled, oxyfluorfen, parathion, terbufos, γ-amanitin, paraquat, phallacidin, phallisacin, and phalloidin in urine and plasma samples.
Method validations
There were not any interference peaks in the blank samples of plasma or urine. The LODs and LOQs of toxicants ranged from 1.00×10−3 to 50.0 ng/mL and from 3.30×10−3 to 1.67×102 ng/mL in the urine samples and 2.07×10−3–50.0 ng/mL and 6.91×10−3–1.67×102 ng/mL in the plasma samples, respectively. The LOD and LOQ results of some representative poisons are shown in Table 2. The results showed that the method was effective in identifying and confirming most target toxicants in the acute poisoning circumstances when using LD50, TDLo, and LDLo as the index.
For urine samples, the intra-day precisions were 2.31–9.13%, and the inter-day precisions were 4.75–12.3% at the spiked concentrations of LOQ, respectively. The intra-day precisions of plasma samples ranged from 1.92 to 10.8%, and inter-day precisions were 2.01–12.1% at the spiked concentrations of LOQ, respectively.
Sample analysis
The biospecimens of clinical poisoning and drug abusers in Sichuan Province of P.R. China in 2019–2020 were collected and analyzed with the proposed method.
Rapid screening for clinical poisoning
Blood samples from four acute poisoning patients were screened through the established HPLC-Orbitrap-MS method. By mapping with the database, two pesticides were detected in four samples, in which two cases were bromadiolone, and the other two cases were carbofuran. The result was in agreement with the detected results of liquid chromatography-triple quadrupole mass spectrometry (QqQ-MS). Meanwhile, the clinical symptoms of the four patients were consistent with the symptoms of the corresponding toxic poisoning. The extracted ion chromatograms, precursor mass spectra, and fragment ion mass spectra of the sample solutions are shown in Figs. 3 and 4.

Extracted ion chromatograms (a), precursor mass spectra (b), and fragment ion mass spectra (c) of bromadiolone (m/z= 526.07797) in blood sample

Extracted ion chromatograms (a), precursor mass spectra (b), and fragment ion mass spectra (c) of carbofuran (m/z= 221.10519) in blood sample
Blood and urine samples from 5 cases of mushroom poisoning were analyzed by the established method. The results showed that different types of amanitins were detected in 2 blood samples and 3 urine samples, respectively. The chromatograms of the three detected amanitins in positive samples are shown in Fig. 5. According to the literature [24,25,26], the half-life of mushroom toxins is short in both blood and urine, but in urine (usually lasts for several days) is relatively longer than in blood (usually lasts for 12–36 h). Therefore, in the case of toxic mushroom poisoning, it is important to collect and detect the leftover mushrooms. The patient’s biospecimen analysis can be used as an auxiliary support, especially when the leftover samples cannot be collected. In the experiment, the positive blood samples were collected 2–4 h after poisoning, which was 2–24 h for urine sample, and the negative samples for 4 days after the poisoning.

The chromatograms of three amanitins in positive samples
Drug screening for drug abuse
A total of 328 blood samples and 54 urine samples of drug abuse cases were detected with a positive rate of 16.5% by the proposed method. The highest rates of drugs were methamphetamine (53.3%), and then followed by heroin (21.4%) and MDMA (10.7%). The other identified drugs were ketamine (8%) and codeine (1.3%). The extracted ion chromatograms, precursor ion mass spectra, and fragment ion mass spectra of methamphetamine and 6-acetylmorphine (the metabolite of heroin) are shown in Figs. 6 and 7. Six cases were confirmed with mixed toxins (all of them were ketamine and MDMA).

Extracted ion chromatograms (a), precursor mass spectra (b), and fragment ion mass spectra (c) of methamphetamine (m/z= 149.12045) in blood sample

Extracted ion chromatograms (a), precursor mass spectra (b), and fragment ion mass spectra (c) of 6-acetylmorphine (m/z=327.14706) in urine samples
In addition, compared with HPLC-QqQ-MS, HRMS has a high-resolution and unique data acquisition mode that enables it to quickly identify unknown poisons when the information is limited. In drug abuse cases, one of them was suspected of taking methamphetamine, whose blood samples were determined by HPLC-QqQ-MS, and the precursor and fragment ions of methamphetamine appeared; however, the retention time was inconsistent, and the relative abundance deviation of characteristic ions was not within the allowable range. In contrast, a chromatographic peak was found after screening in full scan mode using the proposed HRMS method, but no matching toxicant was found in the database. Then through the comparison of chemspider chemical database, the precursor ions of 4F-ABUTINACA were matched. Finally, the toxicant was further matched with the synthetic cannabinoid database established by the research group including RT and fragment ions and confirmed as 4F-ABUTINACA.
Conclusion
A method for the determination of 354 common organic toxicants in blood and urine by liquid-liquid extraction combined with high-performance liquid chromatography-quadrupole orbitrap mass spectrometry (LLE-HPLC-Orbitrap MS) has been established, which enables rapid analysis and is easily operated. At the same time, a database containing chromatographic-mass spectrometric information of 354 common organic toxicants has been built, improving the detection efficiency greatly. At the acute toxic doses, the method is effective in identifying and confirming 354 common organic toxicants in urine and blood samples within 60min including sample pretreatment and LC-MS analysis, which is characterized by high throughput, high sensitivity, and high accuracy, meeting the requirements for the screening of toxic substances in clinical and forensic poisoning events. Besides, it is superior in finding new types of toxins and unknown toxins that makes it applicable in a toxicological analysis.
Change history
06 December 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00216-022-04470-0
References
Organization WH. The public health impact of chemicals: knowns and unknowns - data addendum for 2019 2021 [cited 2021 12-26]. Available from: https://www.who.int/publications/i/item/WHO-HEP-ECH-EHD-21.01
Shengdi L, Bin H, Jinsong Z, Jinru L, Hua L, Yong M. An epidemiology analysis of acute poisoning in China in 2012-2016. J Clin Emerg. 2018;19(8):528–32.
Gummin DD, Mowry JB, Beuhler MC, Spyker DA, Bronstein AC, Rivers LJ, et al. 2020 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 38th Annual Report. Clin Toxicol. 2021;59(12):1282–501.
Centre-North PI. Annual report 2019(General report) 2020 [cited 2021 12-26]. Available from: https://www.giz-nord.de/php/images/downloads/jahresberichte/2019/jabe19d.pdf
Duverneuil C, de la Grandmaison GL, de Mazancourt P, Alvarez J-C. A high-performance liquid chromatography method with photodiode-array UV detection for therapeutic drug monitoring of the nontricyclic antidepressant drugs. Ther Drug Monit. 2003;25(5).
Herzler M, Herre S, Pragst F. Selectivity of substance identification by HPLC-DAD in toxicological analysis using a UV spectra library of 2682 compounds. J Anal Toxicol. 2003;27(4):233–42.
Valenta H. Chromatographic methods for the determination of ochratoxin A in animal and human tissues and fluids. J Chromatogr A. 1998;815(1):75–92.
Abdel-Hamid ME. Comparative LC–MS and HPLC analyses of selected antiepileptics and beta-blocking drugs. Il Farmaco 2000;55(2):136-145.
Walravens J, Mikula H, Rychlik M, Asam S, Devos T, Njumbe Ediage E, et al. Validated UPLC-MS/MS methods to quantitate free and conjugated Alternaria toxins in commercially available tomato products and fruit and vegetable juices in Belgium. J Agric Food Chem. 2016;64(24):5101–9.
Slobodchikova I, Vuckovic D. Liquid chromatography – high resolution mass spectrometry method for monitoring of 17 mycotoxins in human plasma for exposure studies. J Chromatogr A. 2018;1548:51–63.
Rezazadeh M, Yamini Y, Seidi S. Application of a new nanocarbonaceous sorbent in electromembrane surrounded solid phase microextraction for analysis of amphetamine and methamphetamine in human urine and whole blood. J Chromatogr A. 2015;1396:1–6.
Xiong J, Chen J, He M, Hu B. Simultaneous quantification of amphetamines, caffeine and ketamine in urine by hollow fiber liquid phase microextraction combined with gas chromatography-flame ionization detector. Talanta. 2010;82(3):969–75.
Pan M, Xiang P, Yu Z, Zhao Y, Yan H. Development of a high-throughput screening analysis for 288 drugs and poisons in human blood using Orbitrap technology with gas chromatography-high resolution accurate mass spectrometry. J Chromatogr A. 2019;1587:209–26.
Woźniak MK, Wiergowski M, Aszyk J, Kubica P, Namieśnik J, Biziuk M. Application of gas chromatography–tandem mass spectrometry for the determination of amphetamine-type stimulants in blood and urine. J Pharm Biomed Anal. 2018;148:58–64.
Jinlei L, Wurita A, Xuejun W, Hongkun Y, Jie G, Liqin C. Supramolecular solvent (SUPRASs) extraction method for detecting benzodiazepines and zolpidem in human urine and blood using gas chromatography tandem mass spectrometry. Legal Med. 2021;48:101822.
Turner NW, Subrahmanyam S, Piletsky SA. Analytical methods for determination of mycotoxins: a review. Anal Chim Acta. 2009;632(2):168–80.
FAN TSL, ZHANG GS, CHU FS. An indirect enzyme-linked immunosorbent assay for T-2 toxin in biological fluids. J Food Prot. 1984;47(12):964–7.
Cheng J, Wang S, Zhang S, Wang P, Xie J, Han C, et al. Rapid and sensitive determination of clenbuterol residues in animal urine by surface-enhanced Raman spectroscopy. Sensors Actuators B Chem. 2019;279:7–14.
Armenta S, Garrigues S, de la Guardia M, Brassier J, Alcalà M, Blanco M. Analysis of ecstasy in oral fluid by ion mobility spectrometry and infrared spectroscopy after liquid–liquid extraction. J Chromatogr A. 2015;1384:1-8.
Roman M, Ström L, Tell H, Josefsson M. Liquid chromatography/time-of-flight mass spectrometry analysis of postmortem blood samples for targeted toxicological screening. Anal Bioanal Chem. 2013;405(12):4107–25.
Phillips MR, Yang G, Zhang Y, Wang L, Ji H, Zhou M. Risk factors for suicide in China: a national case-control psychological autopsy study. Lancet (London, England). 2002;360(9347):1728-36.
China MoJotPsRo. Gas chromatography-mass spectrometry method for the determination of 108 poisons (drugs) in blood and urine (SF/Z JD0107014-2015) 2015 [updated 2015-11-20; cited 2021 12-26]. Available from: http://www.moj.gov.cn/pub/sfbgw/zwfw/zwfwbgxz/202101/1565869778272044576.pdf
China MoJotPsRo. Determination of 238 poisons (drugs) in blood and urine by liquid chromatography-tandem mass spectrometry (SF/Z JD0107005-2016) 2016 [updated 2016-9-22; cited 2021 12-26]. Available from: http://www.moj.gov.cn/pub/sfbgw/zwfw/zwfwbgxz/202101/1565870205956007711.pdf
Jaeger A, Jehl F, Flesch F, Sauder P, Kopferschmitt J. Kinetics of amatoxins in human poisoning: therapeutic implications. J Toxicol Clin Toxicol. 1993;31(1):63–80.
Robinson-Fuentes VA, Jaime-Sánchez JL, García-Aguilar L, Gómez-Peralta M, Vázquez-Garcidueñas MS, Vázquez-Marrufo G. Determination of α- and β-amanitin in clinical urine samples by capillary zone electrophoresis. J Pharm Biomed Anal. 2008;47(4):913–7.
Shaozhen X, Feng L, Wusheng F, Daozhi W. Determination of amatoxins and phallotoxins in plasma and urine by ultra performance liquid chromatography-tandem mass spectrometry. Food Sci. 2018;39(22):7.
Funding
This study was supported by the Key Project of Sichuan Province Science and Technology (Grant No. 2020YFS0056).
Author information
Authors and Affiliations
Contributions
Zhuoya Li: methodology, writing — original draft. Xinyi Huang: methodology, writing — review. Xuan Wang: methodology, data curation. Jianwei Ren: methodology, data curation. Buyi Xu: resources, validation. Chunying Luo: validation. Yanjun Wang: resources. Lian Wang: instrument operation. Kouji Harada: Writing — review and editing. Xiaoli Zou: conceptualization, funding acquisition.
Corresponding authors
Ethics declarations
Ethical approval
As this study does not involve any work on humans or animals, no study-specific approval by the appropriate ethics committee was required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: First and last name of “Harada Kouji” were interchanged. Middle initial was also not included. It should be “Kouji H. Harada
Supplementary Information
ESM 1
(PDF 385 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Z., Huang, X., Wang, X. et al. Establishment and application of a screening method for 354 organic toxicants in blood and urine with high-performance liquid chromatography–high-resolution orbitrap mass spectrometry. Anal Bioanal Chem 415, 493–507 (2023). https://doi.org/10.1007/s00216-022-04432-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-04432-6