
Abstract
Epidemiological studies often call for analytical methods that use a small biospecimen volume to quantify trace level exposures to environmental chemical mixtures. Currently, as many as 150 polar metabolites of environmental chemicals have been found in urine. Therefore, we developed a multi-class method for quantitation of biomarkers in urine. A single sample preparation followed by three LC injections was optimized in a proof-of-approach for a multi-class method. The assay was validated to quantify 50 biomarkers of exposure in urine, belonging to 7 chemical classes and 16 sub-classes. The classes represent metabolites of 12 personal care and consumer product chemicals (PCPs), 5 polycyclic aromatic hydrocarbons (PAHs), 5 organophosphate flame retardants (OPFRs), 18 pesticides, 5 volatile organic compounds (VOCs), 4 tobacco alkaloids, and 1 drug of abuse. Human urine (0.2 mL) was spiked with isotope-labeled internal standards, enzymatically deconjugated, extracted by solid-phase extraction, and analyzed using high-performance liquid chromatography-tandem mass spectrometry. The methanol eluate from the cleanup was split in half and the first half analyzed for PCPs, PAH, and OPFR on a Betasil C18 column; and pesticides and VOC on a Hypersil Gold AQ column. The second half was analyzed for tobacco smoke metabolites and a drug of abuse on a Synergi Polar RP column. Limits of detection ranged from 0.01 to 1.0 ng/mL of urine, with the majority ≤0.5 ng/mL (42/50). Analytical precision, estimated as relative standard deviation of intra- and inter-batch uncertainty, variabilities, was <20%. Extraction recoveries ranged from 83 to 109%. Results from the optimized multi-class method were qualified in formal international proficiency testing programs. Further method customization options were explored and method expansion was demonstrated by inclusion of up to 101 analytes of endo- and exogenous chemicals. This exposome-scale assay is being used for population studies with savings of assay costs and biospecimens, providing both quantitative results and the discovery of unexpected exposures.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the United States (US), individuals have > 400 detectable exposure biomarkers with about 150 observed in urine per the latest National Health and Nutrition Examination Survey (NHANES) report from March 2021 [1]. Human exposures are common to highly prevalent chemicals [2], such as environmental phenols (EP) in personal care products (PCPs), phthalates in plastic additives (PHTH), polycyclic aromatic hydrocarbons (PAHs), flame retardants (OPFRs), pesticides, volatile organic compounds (VOCs), and tobacco smoke. A wide range of public health concerns and health effects have been associated with their exposures [3]. The vast range of exposures to environmental chemical mixtures is complex, but they represent the real-life scenario that humans face [4]. Exposomics include a broad range of xenobiotic and endogenous biomarkers of exposures and biological response [5]. Better definition of multiple exposures will improve research on health outcomes and on exposure source identification [6].
Most targeted analytical methods measure fewer than 15 biomarkers of exposure from a single chemical class in each biospecimen [7]. Class-specific extractions and instrumental analyses are used [8], for example, EP [9,10,11], PAH [12], OPFR and dialkyl phosphates (DAPs) [13], OP pesticides, pyrethroids, and herbicides [14], PHTH [15], VOC [16], and tobacco smoke [17]. Biospecimen availability is often limited in epidemiological studies restricting the number of possible targeted assays [18]. To overcome these barriers, multi-class techniques are gaining popularity using extractions that enrich different classes of chemicals in human specimens and simultaneous detection. Thus, multi-class chemicals can be measured without using separate conventional workflows, thereby reducing time, cost, and sample volume [19,20,21,22,23,24,25,26]. Reported multi-class methods cover a range of exposure biomarkers from two [21, 24,25,26] to three [19, 23], four [20], or six [22] broad chemical classes. An overview of multi-class methods relevant to this study is summarized in Table 1 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37], and salient features of recent ones are provided in Table S1 [19, 20, 22].
Our objective of developing a new analytical method was to achieve a broader range of biomarkers that represent prevalent exposures in the general population [1]. Because many common environmental biomarkers are measured in urine, we sought to combine available analytical methods and to make modifications that capture a possible range of common environmental exposures using one sample and one laboratory procedure. For the proof-of-approach of a multi-class method, 50 biomarkers of exposure in urine, also referred to as analytes, belonging to 7 environmental chemical classes and 16 sub-classes, were included in the method development [Table 2]. Analytes represent urinary metabolites of 12 EP, 5 PAH, 5 OPFR, 18 pesticides, 5 VOC, 4 tobacco alkaloids, and 1 drug of abuse. The resulting method can be applied to chemically similar endogenous and exogenous exposures, and other lifestyle choices, such as steroids, hormones, phytoestrogens, vitamins, pharmaceuticals, and drugs of abuse. Savings in assay costs and biospecimens result.
Materials and methods
Standards and reagents
Reference standards # 1–6, 8–12, 23–38, 40, and 46–47 were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA), whereas # 7, 39, and 48–50 from Sigma-Aldrich (St. Louis, MO, USA), and # 13–22 and 41–45 from Toronto Research Chemicals (North York, ON, Canada). Vendor and product information for native and labeled standards are given in Table S2. Purity for all standards ranged from > 90 to 99.9%. LC/MS grade acetic acid (≥ 99.7%), acetonitrile (≥ 99.9%), ammonium acetate (≥ 99 .0%), ethyl acetate (≥ 99.9%), formic acid (≥ 99.0%), methanol (≥ 99.9%), and water were purchased from Fisher Scientific (Hampton, NH, USA). Three commercial enzymes [38] were purchased from Sigma-Aldrich with their product number in parentheses: (i) H-1: β-glucuronidase from Helix pomatia, type H-1 (#G0751) with a β-glucuronidase activity ≥ 300,000 units/g solid and sulfatase activity 10,000 units/g solid. (ii) ALS: BGALA-RO β-glucuronidase/arylsulfatase from Helix pomatia (#10127698001) with a β-glucuronidase activity ≈100,000 units/mL and sulfatase activity ≈47,500 units/mL, and (iii) K-12: β-glucuronidase from E. coli-K12 (#03707601001) with a β-glucuronidase activity ≈140 units/mg protein. Standards for enzyme hydrolysis and deconjugation efficiency experiments were purchased from Toronto Research Chemicals with their abbreviation and product number in parentheses: 4-methylumbelliferone (MU, #M333000), 4-methylumbelliferone-13C4 (13C4-MU, #M333002), 4-methylumbelliferyl β-D-glucuronide (MUG, #M334550), 4-methylumbelliferyl sulfate (MUS, #M333100), bisphenol A β-D-glucuronide (BPAG, #B519510), bisphenol A-monosulfate (BPAS, #B519560), triclosan O-β-D-glucuronide (TCSG, #T774260), triclosan O-sulfate (TCSS, #T774265), and mono-2-ethylhexyl phthalate glucuronide (MEHPG, #M542500). All solvents, reagents, and synthetic urine (UTAK, Valencia, CA, USA) were tested for presence of analytes of interest, and none was above the limits of detection (LODs). Working and intermediate stock solutions of all native standards and labeled internal standards were prepared as separate mixtures at 1 mg/mL and 1 μg/mL in acetonitrile and stored at −20 °C.
Method development
Urine preparation, deconjugation, and one-step extraction
Urine processing for total forms (sum of free, aglycone, and conjugates of individual analyte) was based on analytical method of the Centers for Disease Control and Prevention (CDC) [39], with some modifications [40]. We customized the pretreatment step to provide clean baseline and adequate recoveries. All specimens, quality controls, and working standard solutions were thawed and equilibrated to room temperature. Sample tubes were vortexed at 1300 rpm for 5 min on Multi Reax vibrating shaker (product # 545-10000-00, Heidolph North America, Wood Dale, IL) and centrifuged at 4000 rpm for 15 min (Eppendorf centrifuge 5810, Eppendorf, Hauppauge, NY). Urine sample preparation and pretreatment was automated using a liquid handler (epMotion 5075vtc; Eppendorf, Hauppauge, NY). Urine (0.2 mL) was transferred to a 96-deep well plate (DWP) with 2 mL well volume (product #951033600, Eppendorf, Hauppauge, NY), spiked with 20 μL of labeled internal standards mixture at a concentration of 200 ng/mL. After vortexing for 5 min at 700 rpm, the urine sample was buffered with 100 μL of 1.0 M ammonium acetate solution at pH 5.0 adjusted with acetic acid, and 25 μL of β-glucuronidase/arylsulfatase enzyme from Helix pomatia that had an approximate specific activity of 100,000 units/mL β-glucuronidase and 47,500 units/mL sulfatase [38]. The DWP was incubated overnight at 37 °C at 300 rpm on a Mixmate vortexer (product # 022674200, Eppendorf, Hauppauge, NY) to hydrolyze the conjugates.
Solid-phase extraction
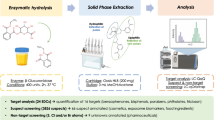
The enzymatic deconjugation provides a solution of free polar analytes, the total of free plus bound metabolites from the original urine sample. The hydrolysates containing total forms of analytes were extracted by solid-phase extraction (SPE) using an Oasis HLB hydrophilic-lipophilic balanced reversed-phase 96-well plate (30 mg sorbent per well, 30 μm particle size; Waters Corporation, Milford, MA). The procedure was automated using a liquid handler (epMotion 5075vtc; Eppendorf, Hauppauge, NY). The first step was to equilibrate the wells by adding 1 mL of methanol and conditioning with 1 mL of water. The second step was to acidify the enzymatic digestates of urine with 750 μL of 0.67% formic acid, vortex, and load onto the preconditioned 96-well SPE plate. Third, the native and corresponding labeled analytes were eluted twice with 0.75 mL methanol. The two eluates for each sample were pooled in a fresh DWP, vortexed, split into two equal volumes, and transferred to two separate fresh DWPs. Each half was evaporated to dryness under a gentle nitrogen stream with a SPE Dry 96 evaporator (Biotage, LLC; Charlotte, NC). Extract 1 was reconstituted with 0.1 mL of acetonitrile:water (50:50, v:v). Extract 2 was reconstituted with 0.1 mL of 0.1% acetic acid in water. The two extracts were analyzed by LC-MS/MS instrumentation as described in the following sections.
Multi-class separation and liquid chromatography (LC)
The two extracts were analyzed with three LC injections optimized for analytical separations of the mixture of 50 standards from seven environmental chemical classes (Graphical abstract, Fig. 1). Chromatographic separation was achieved using an Exion ultrahigh-performance liquid chromatographic (UHPLC) system from Sciex (Framingham, MA, USA). Injection 1 with 20 μL of extract 1 was made on a Betasil C18, 5 μm, 2.1 × 100 mm analytical column with 2.1 × 10 mm guard column (Thermo Scientific, Waltham, MA, USA) to resolve compounds and metabolites of EP, PAH, and OPFR (chemical class # 1–3). Injection 2 with 20 μL of extract 1 was on a Hypersil Gold AQ, 3 μm, 3.0 × 150 mm analytical column with 4.0 × 10 mm guard column (Thermo Scientific, Waltham, MA, USA) to resolve compounds and metabolites of pesticides and VOCs (chemical class # 4–5). Injection 3 with 10 μL extract 2 was on a Synergi Polar RP, 2.5 μm, 2.0 × 100 mm analytical column with 2.0 × 4.0 mm guard column (Phenomenex Inc., Torrance, CA, USA) to resolve compounds and metabolites of tobacco smoke and drugs of abuse (chemical class # 6–7). Mobile phases, flow rate, and gradient details are provided in Table S3. Calibration levels and SPE extracts were maintained at 4 °C in the autosampler, while the LC columns were maintained at 40 °C. Autosampler rinsing solution was 0.5 mL of acetonitrile:water (70:30, v:v).

Multi-class urine assay for multi-analyte exo- and endogenous polar metabolites. *Cortisol and cortisone are the endogenous biomarkers for psychosocial stress in our multi-class method
Tandem mass spectrometry (MS/MS)
The mass spectrometry in multiple-reaction monitoring mode (MRM) was used for data acquisition of each target analyte. MRM method with quantifier and qualifier ion transitions provides detection/quantification and identification/confirmation, which is essential to targeted analyses. A Sciex 6500+ triple quadrupole mass spectrometer (MS) equipped with electrospray ionization (ESI) source (SCIEX, Framingham, MA, USA) was operated in positive or negative ionization mode, sequentially or concurrently (detailed later), for the detection and quantitation of analytes of interest. Nitrogen served as curtain and collision gas. Ion source and gas parameters were set to the following values: curtain gas flow = 25 psi; nebulizer gas (ion source gas 1) = 50 psi; heater gas (ion source gas 2) = 55 psi; source temperature = 500 °C; and collision gas value = 8. Ion source voltage was set at 5500 V or −4500 V in the positive and negative ESI mode, respectively. The most prominent ion transition was used for quantitation and the most intense second ion transition was used for confirmation. Analyte specific parameters such as declustering potential, entrance potential, collision exit potential, and collision energy were individually optimized by direct syringe infusion of each compound into the mass spectrometer. Optimized instrumentation parameters for each individual analyte are provided in Table 3.
Method validation and application
Method validation steps included determining LOD, limit of quantification (LOQ), LOD and LOQ by reproducibility, calibration range, recovery of blank and matrix spikes, matrix effect on LOD, matrix effect on recovery and reproducibility, sample storage stability, accuracy, and intra- and inter-batch precision (repeatability). Instrument calibration range was determined by injecting 20 μL of 0.1 to 1000 ng/mL of native standards mixture solutions, which yielded a linear regression curve with r2 > 0.99. Calibration curves were based on plotting the ratio of the target ion area of the quantitation MRM transition to that of the ion area of corresponding isotope-labeled internal standard against the spiked concentration of the target analyte (ng/mL) with 1/x weighting. The LOD (signal/noise ≥ 3) and LOQ were calculated as 3S (three times standard deviation) [41] and 10S [42], respectively, of ten replicate analyses of a synthetic urine (matrix-blank) spiked with 1.0 ng/mL of native standards mixture. Although the matrix-matched calibration curve was linear, the instrument response was not proportional and noise level was not consistent in the lower end particularly between LOD and LOQ in the samples. Hence, a signal-to-noise ratio of 5 ± 10% CV (coefficient of variation) was considered acceptable in this concentration range.
Quality controls (QC) included in each batch were procedural (reagent-based), instrumental (no reagent, no matrix), and matrix pool blanks, in-house urine QC pool spikes of native standards mixture at low (0.5–5.0 ng/mL), medium (10–50 ng/mL), and upper range (100–1,000 ng/mL) of assay validation. Urine QC pool was prepared by mixing urine samples collected from anonymous volunteers, which were individually screened for analytes of this study interest prior to mixing to achieve a low-level QC with at least 50% of analytes < 1.0 ng/mL. Higher level QC pools were obtained by spiking with native standards. Efficiency of the deconjugation step was assessed and optimized using MU and its conjugates as test substrates [43]. MUG and MUS were spiked at five levels (0.1, 1, 10, 100, and 1,000 ng/mL) in water (reagent-blank) and synthetic urine, and followed the multi-class urine SPE analytical procedure to determine recovery of MU and efficiency of deconjugation step. Our lab, the Senator Frank R. Lautenberg Laboratory, is part of the Children’s Health Exposure Analysis Resource (CHEAR), Human Health Exposure Analysis Resource (HHEAR), and P30 Transdisciplinary Center on Early Environmental Exposures (TCEEE). We participated and qualified in proficiency testing (PT) programs for biomarkers of exposures to environmental chemicals conducted by G-EQUAS (The German External Quality Assessment Scheme for analyses in biological materials) (http://www.g-equas.de/) and OSEQAS (Organic Substances in urine Quality Assessment Scheme) by the Centre de Toxicologie du Quebec (CTQ) (https://www.inspq.qc.ca/en/ctq/eqas/oqesas/description) [44]. To present the proof-of-approach of this method, 15 urine samples, from children and adults, both genders, donated by volunteers and purchased from Lee Biosolutions, Inc. (Maryland Heights, MO, USA), were analyzed for the 50 analytes.
Results and discussion
Global analysis of external exposomic breadth is a challenge due to complexity of exposures to environmental chemical mixtures, diversity of their chemical properties, differences in metabolism, and trace level concentrations found in humans. Hence, typical class-specific and separate analytical methods are in use. Recent multi-class methods report a single sample preparation for simultaneous extraction of urinary metabolites of EP, PHTH and pesticides [19], or EP and PAH [20], or OPFR and pesticides [21], respectively. However, to the best of our knowledge, there is no reported method for the simultaneous extraction of seven classes of this study’s interest, which include VOC, tobacco smoke, and drugs. Steps and challenges involved in developing and validating a method for all these analytes are discussed below.
Automated low-volume and high-throughput assay
High throughput for bioanalytical methods is rate-limited by the number of biological and liquid handling steps. A user-friendly automated sample preparation was achieved with the epMotion 5075vtc liquid handler. The unit supports low-volume aliquoting of reagents and matrix, pipetting range of 0.2 μL–1 mL, diluting, adding internal standards mixture, various extractions (liquid-liquid, solid-phase, supported liquid extraction etc.), thermal incubation range of 0–110 °C, and mixing range of 300–2000 rpm. The platform also supports online sample extraction (liquid-liquid extraction, solid-phase extraction, protein precipitation, etc.) using single and 8 channel dispensing tools. The developed high-throughput automated method is rugged and enabled us to process 96 samples per batch per day, improved overall efficiency, and reduced tedious sample handling steps and human errors if any. Moreover, it enabled us to work with a low-volume urine sample (0.2 mL) and labeled standards mixture (20 μL), with a systematic measurement error < 1.0% at low volumes in the range of 1–50 μL, compared with the reported urine volume of 0.5–3 mL used for relevant multi-class analysis elsewhere [19, 20].
Enzyme selection for hydrolysis and use of a deconjugation probe
Upon exposure, environmental chemicals undergo phase I and II metabolism [45]. Biomarkers in the chemical classes of our study undergo mainly phase II metabolism. Previous studies have reported urinary excretion of phase II conjugates (aglycone, conjugated glucuronide, and sulfate forms) for EP [46, 47], PAH [48], OPFR [49], and OP and pyrethroid pesticides [50]; aglycone and glucuronide conjugates for phthalates [51], tobacco smoke [52], opioids [53], and cannabinoid [54]; mercapturic acid conjugates for VOC metabolites [55]; and aglycone forms for phenoxy acid herbicides [56], neonicotinoids [57], and DAPs [58]. Accordingly, the choices of enzyme for deconjugation of urine metabolites in the CDC methods were (i) H-1 enzyme for EP [9], OPFR and DAPs [13], tobacco smoke [17], and OP pesticides, pyrethroids, and herbicides [14]; (ii) ALS enzyme for PAH [12]; (iii) K-12 enzyme for phthalates and phthalate alternatives [15]; and (iv) no enzyme for VOC [16]. Complete hydrolysis of conjugates is not only dependent on the enzyme type but also amount, buffer pH, incubation temperature, and duration [19, 38, 59]. Thus, the choice and amount of β-glucuronidase and (aryl) sulfatase varied for biomarker panels. Enzymes with sulfatase activity resulted in unreproducible results of the non-oxidative, primary phthalate monoester metabolites (discussed later). This limited the choice of biomarkers that could be included in our multi-class method.
We tested the deconjugation efficiency of the multi-class analytes by including conjugate forms in water and synthetic urine samples [43], i.e., MUG, MUS, BPAG, BPAS, TCSG, TCSS, and MEHPG (glucuronide of mono-2-ethylhexyl phthalate, MEHP), in the range of 0.1–1,000 ng/mL. Aglycone and conjugate forms were quantified before and after enzymatic treatments to calculate deconjugation efficiency [43]. ALS enzyme was used in this study for its ability to completely deconjugate O-glucuronides and sulfates of EP, and N-glucuronide of TCC [38]. ALS enzyme was the choice in recent multi-class methods [19, 20]. ALS enzyme activity was evaluated at five stages, as follows. Aglycone forms of the test analytes were extracted from 0.2 mL urine with Oasis HLB SPE and quantified by LC–MS/MS. Stage (i) test samples were spiked with glucuronide or sulfate conjugates of test analytes at 0.1, 1, 10, 100, or 1,000 ng/mL and incubated to assess spiked levels of conjugate on recoveries (Fig. 2). Complete hydrolysis was observed for conjugate levels up to 1,000 ng/mL. Sample dilution or a smaller aliquot (≤ 0.1 mL) is suggested for analytes > 1,000 ng/mL, which are atypical in a general population and non-smokers [1], for a satisfactory deconjugation recovery under these enzymatic conditions. (ii) the conjugate spiked samples were incubated for 1, 8, 16, or 24 h to assess incubation time effect on recoveries (Fig. S1B). Measured aglycone levels did not change significantly with incubation time between 8 and 16 h. Hence, hydrolysis time of 16 h was selected to perform an overnight incubation that fit post-deconjugation sample cleanup and extraction steps during typical work hours the following day. (iii) The conjugate spiked samples were incubated with ALS enzyme using 5, 25, or 50 μL enzyme/0.2 mL urine (i.e., ALS of 2.5, 12.5, 25 units/μL urine) to assess enzyme amount for satisfactory hydrolysis (Fig. S1C). Aglycone levels increased with higher enzyme units. (iv) the conjugate spiked samples were incubated in 1.0 M ammonium acetate buffer at pH 4.5, 5.0, or 5.5 (Fig. S1D). No difference was observed between pH 5.0 and 5.5; therefore, 5.0 was used for the rest of this work per manufacturer recommended pH range, while 5.5 was used by [19, 20], and (v) the conjugate spiked samples were incubated at room temperature (25 °C, 37 °C, or 50 °C) to assess enzyme activity at different temperatures (Fig. S1E). Hydrolysis was optimal at 37 °C. We found that incubation with 25 μL ALS/0.2 mL urine (i.e., ALS of 12.5 units/μL urine) for 16 h in 1.0 M pH 5.0 ammonium acetate buffer at 37 °C ensured the complete deconjugation of biomarkers in the chemical classes we report.

Deconjugation efficiency based on known glucuronide and sulfate conjugate levels (ng/mL), showing range of concentrations. The range of recoveries indicates the necessity to monitor deconjugation. See Fig. S1 for more information on deconjugation efficiency based on the duration of incubation (h), amount of enzyme (ALS units/μL urine), pH, and incubation temperature (°C). Conjugates were spiked at 0.1, 1, 10, 100, or 1,000 ng/mL into synthetic urine, and incubated with 25 μL ALS enzyme/0.2 mL urine (i.e., ALS enzyme of 12.5 units/μL urine) for 16 h in 1.0 M pH 5.0 ammonium acetate buffer at 37 °C. Aglycone forms of the test analytes were extracted from 0.2 mL urine with Oasis HLB SPE and quantified by LC–MS/MS to estimate percent deconjugation and spike recovery. “MEHPS: N/A” Sulfate conjugate of MEHP was unavailable.
NIST Standard Reference Materials (SRMs) 3672 and 3673 for organic contaminants in smokers’ and non-smokers’ urine, respectively, have certified or reference mass fraction values for conjugated forms of EP, PHTH, PAH, and VOC metabolites [60]. Hydrolysis of EP and PAH metabolites in NIST SRMs using the optimized enzyme conditions were in the satisfactory range of 80–120% and > 130% for MEHP. We observed a significant variation in recoveries of MNBP, MIBP, and MEHP from NIST SRM 3672, attributable to arylsulfatase activity in the ALS enzyme (Fig. 3). Enzymes with (aryl)sulfatase activity can hydrolyze phthalate diesters in the lab environment to monoesters through lipase/esterase activity, depending on the deconjugation conditions, resulting in artificial elevation of phthalate biomarkers [61, 62]. Because of the relatively high MEHP in blanks and higher recoveries > 130% from spiked MEHPG in the lower spike range 0.1–10 ng/mL in samples incubated with ALS enzyme for ≥ 8 h, and inconsistent QC performance for MNBP, MIBP, and MEHP, we have excluded PHTH from the multi-class assay. This was not the case with a shorter incubation of 1 h, similar to the other multi-class method with a 2 h step [19]. However, 1 h incubation was not sufficient for complete deconjugation of sulfates of EP. Lipase-mediated alkyl/aryl sulfatase hydrolysis of methyl paraben (MEPB) to p-hydroxybenzoic acid resulted in recovery < 80–50% at ALS ≥ 30 units/μL urine [38], which was not observed in our study range of ALS enzyme 2.5–25 units/μL urine. Methanol addition prior to enzymatic deconjugation quenched lipase activity and resulted in full recovery of EP including MEPB [38]. We plan in future to test methanol addition to prevent lipase hydrolysis of phthalate diesters in order to add this class to the assay. Further, an enzymatic deconjugation is required for a method to be applied in human biomonitoring of most common environmental classes, a step not used in [22].

Effect of deconjugation enzyme on phthalate metabolites recovery from NIST SRM 3672. Enzymatic hydrolysis using arylsulfatase-free β-glucuronidase K-12 enzyme from E. coli resulted in excellent recoveries (averages in the range of 94–100%). ALS enzyme with β-glucuronidase and sulfatase activity is essential to completely deconjugate glucuronides and sulfates of multi-class urinary metabolites included in this study, but not for phthalate metabolites. Lipase/esterase activity of ALS enzyme may lead to hydrolysis of extraneous, not relevant to exposure and non-metabolized, phthalate diesters, and result in significant variation in recoveries, especially MNBP, MBP, and MEHP. (a) NIST SRM 3672 was incubated with 25 μL ALS/0.2 mL urine (i.e., ALS enzyme of 12.5 units/μL urine) for 16 h in 1.0 M pH 5.0 ammonium acetate buffer at 37 °C. Aglycone forms of the test analytes were extracted from 0.2 mL urine with Oasis HLB SPE and quantified by LC–MS/MS to estimate percent deconjugation and spike recovery. Reference mass fraction values (μg/kg) for selected phthalate metabolites in NIST SRM 3672 are 2.99 (MCPP), 94.5 (MEP), 8.37 (MBZP), 10.6 (MNBP), 6.40 (MIBP), 4.13 (MEHP), 35.2 (MECPP), 14.9 (MEOHP), and 24.8 (MEHHP). (b) MCPP, mono-(3-carboxypropyl) phthalate; MEP, monoethyl phthalate; MBZP, monobenzyl phthalate; MNBP, mono-n-butyl phthalate; MIBP, mono-isobutyl phthalate; MEHP, mono-(2-ethylhexyl) phthalate; MECPP, mono-(2-ethyl-5-carboxypentyl) phthalate; MEOHP, mono-(2-ethyl-5-oxohexyl) phthalate; and MEHHP, mono-(2-ethyl-5-hydroxyhexyl) phthalate.
Analyte extraction, separation, detection, and quantification
SPE is generally considered to provide efficient cleanup, increased selectivity, and lowered solvent usage, suitable for large sample size and high throughput [7], compared to liquid-liquid extraction (LLE) in a multi-class method [22]. Oasis HLB SPE, a polymeric sorbent with hydrophilic–lipophilic balance [63], was determined to be suitable for this study, after testing various sorbent materials; it has also been a preferred universal sorbent in other multi-class methods [20, 35, 36]. Oasis HLB provided enhanced retention of low pKa analytes such as DAPs when enzymatic digestates were acidified prior to loading, following the “pKa-rule” [64], similar to observations in other multi-class methods [19, 20]. Exclusion of a water wash step improved DAPs recovery with no significant difference in extract matrix effects for the range of analytes. Wash with pure organic solvent (100% methanol) provided satisfactory recoveries for the study analytes, compared to the use of a combination of organic solvents in other multi-class methods such as acetonitrile:ethyl acetate (1:1, v:v) [19] or methanol:dichloromethane (50:50, v:v) [20]. In future expansion of the method, we plan to use a combination of organic solvents to elute analytes with higher logKow than those of this study’s analytes. Poor recovery of BP3 was reported when reconstituted in the pre-injection mobile phase, typically a high aqueous and low organic condition [24]. Therefore, we increased the organic content by using acetonitrile:water (50:50, v:v) for reconstitution and observed improved BP3 recovery. SPE recoveries of metabolites of EP, PAH, VOC, and tobacco smoke in NIST SRM 3672 from Oasis HLB were compared with sorbents used in representative single-class assays by following reported sample cleanup steps [40, 65,66,67] (Fig. S2). Recoveries of 81–110% were obtained for all analytes from Oasis HLB and considered satisfactory in comparison with 91–104% from SPE sorbents and protocols used in respective single-class assays. The optimized Oasis HLB SPE conditions achieved a satisfactory balance between recoveries and matrix effects, a desirable key feature for a multi-class with differing polarities, as demonstrated for all chemical classes of this study interest by using previous years’ proficiency test materials where reference values are available. NIST SRM 1507b was used to test THC-COOH recovery and found satisfactory.
The cleanup of ours and other reported multi-class methods [19, 24,25,26] yields deconjugated monoesters and oxidative metabolites of phthalate diesters. However, our choice of enzyme ALS is ideal for EP [41, 46], PAH [48], OPFR [49], and OP and pyrethroid pesticides [50]. While it deconjugates other analytes, including monoesters of phthalates and phthalate alternatives, we observe elevated MEHP, MNBP, MIBP, and mono-2-ethylhexyl terephthalate (MEHTP), but not oxidative metabolites in the blanks. We believe this is due to the lipase/esterase activity of the sulfatase in ALS enzyme with an ability to hydrolyze extraneous phthalate diesters to monoesters and thus elevate their levels that are not due to human exposure [62]. Oxidative phthalate metabolites result from metabolism and are not compromised by extraneous diesters during sample handling or analysis [51]. Therefore, we perform a separate assay using K-12 enzyme for PHTH. In addition, glyphosate and its metabolite aminomethyl phosphonic acid (AMPA) are highly polar zwitterions and not retained or resolved by conventional reverse-phase SPE sorbents or LC columns [68]. Therefore, we currently perform a separate assay using a mixed-mode SPE sorbent and an ion chromatographic column for measuring underivatized polar ionic pesticides in urine [69]. We did not measure legacy long-chain per- and polyfluoroalkyl substances (PFAS) such as perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in this urine multi-class method. Although there have been a few limited reports including a multi-class [22], PFAS detection in urine is infrequent or below method detection limits. Therefore, PFAS are best studied in blood matrices and urinary assays are not considered validated quantitative biomarkers [70, 71]. By contrast, it is a possibility that the short-chain and more polar PFAS replacements such as perfluorobutanoate (PFBA), perfluorobutane sulfonate (PFBS), ADONA, and GenX with shorter biological half-lives have significant elimination in urine, which should be checked [70, 72].
Previous reports provide background information on suitable chromatography and mass spectrometry conditions for target chemical classes. We tested several LC columns and mobile phases to find maximize class coverage with a minimum number of injections of the same extract. Table S3 shows optimized LC gradients for the three injections. First, as expected EP were resolved satisfactorily on a Betasil C18 column (2.1 × 100 mm, 5 μm) [73]. However, in contrast to the acidified aqueous condition reported elsewhere [73], a combination of neutral water and acetonitrile mobile phases helped with inclusion of PAH and OPFR metabolites in the same LC run (Fig. 4A). Notably, the optimized Betasil LC gradient separated isomer peak pairs of PAH metabolites, e.g., NAP1 (metabolite of naphthalene and insecticide, carbaryl) and NAP2 (metabolite of naphthalene only) [74]; and OPFR, e.g., DOCP and DPCP (isomer metabolites of tri-cresyl phosphate) [75]. Similarly, the second injection, as expected, provided DAPs with long retention times, starting after 3 min. The 6 analytes in this sub-class separated well from each other and matrix ions, using Hypersil Gold AQ column (3.0 × 150 mm, 3 μm) [21]. In addition, the optimized Hypersil LC gradient also had good results for metabolites of other pesticides and VOC (Fig. 4B). Separation of isomer pairs of pesticides, e.g., CIS-DCCA and TRANS-DCCA (cis and trans isomer metabolites of permethrin) [76]; and VOC, e.g., 2-HPMA (propylene oxide biomarker) and 3-HPMA (acrolein biomarker) [77], was possible. Third, as expected, Synergi Polar RP (2.0 × 100 mm, 2.5 μm) resolved tobacco smoke [65] and drugs of abuse [78], and allowed simultaneous determination of NNAL with tobacco smoke metabolites (Fig. 4C) similar to [79]. Nicotelline [80], a minor tobacco alkaloid and a biomarker of combusted tobacco use [81], was included in the 3rd injection. We plan in future to add anatalline, another minor tobacco alkaloid and a biomarker of smokeless tobacco use [82]. Mass spectrometry parameters were further optimized in the ESI positive mode for insecticides (DEET) [36], tobacco smoke [65], and drugs of abuse [78], and in the ESI negative mode for the rest of the target analytes [21, 28, 36, 39]. Table 3 shows optimized MS/MS parameters for target analytes in three separate and sequential injections, and observed retention times. MRM provides a highly specific data acquisition, which is unique for each analyte, and multiple transitions can be run within a given time frame. Sensitivity in all three LC methods was further enhanced by using a Scheduled MRM program. Typical MRM chromatograms for all target analytes are shown from an unspiked (native) (Fig. S3) and a 1 ng/mL spiked QC urine pool (Fig. S4).

Extracted ion chromatograms of a standard mixture of 101 analytes under 3 different LC separation conditions described in “Materials and methods.” The 50 biomarkers represent exposures to multiple environmental chemical classes for which quantitative methods are shown [#1-50]. An additional 51 analytes are shown to illustrate expanded capability of the multi-class method to measure other biomarkers within a chemical class and other similar classes [#A-AY]. Panels A–C represent 3 separate and sequential injections of a 20 μL standards mixture, with 20 ng/mL of each analyte, on a Betasil [A], Hypersil [B], and Synergi [C] LC column [#1–50]. Fifty exposure biomarkers of interest. Betasil [3A]: (1) BPA, (2) BPF, (3) BPS, (4) BPZ, (5) TCS, (6) TCC, (7) BP1, (8) BP3, (9) BUPB, (10) ETPB, (11) MEPB, (12) PRPB, (13) NAP1, (14) NAP2, (15) PYR1, (16) FLUO2, (17) PHEN3, (18) BCETP, (19) BDCPP, (20) DPHP, (21) BCPP, and (22) DBUP. Hypersil [3B]: (23) CINA6, (24) AND (25) PNP, (26) TCP, (27) MDA, (28) DMP, (29) DMTP, (30) DMDP, (31) DEP, (32) DETP, (33) DEDP, (34) PBA, (35) FPBA, (36) CIS-DCCA, (37) TRANS-DCCA, (38) PCP, (39) 24D, (41) HEMA, (42) 2-HPMA, (43) 3-HPMA, and (44) SPMA. Synergi [3C]: (40) DEET, (45) NICT, (46) COTT, (47) HCOT, (48) NNAL, (49) NICTL, and (50) THC-COOH. Possible 51 exposure biomarkers measured in the same injections are additional 9 environmental phenols, 6 PAH metabolites, 3 OP flame retardants, 7 pesticides, 3 VOCs, 4 tobacco smoke, 17 substance abuse, and 2 psychosocial stress biomarkers [#A-AY]. Betasil [Fig. 4A]: (A) 4-hydroxybenzoic acid (HB4), (B) 2-hydroxyphenanthrene (PHEN2), (C) 1-hydroxyphenanthrene/9-hydroxyphenanthrene (PHEN1/PHEN9), (D) 9-hydroxyphenanthrene/1-hydroxyphenanthrene (PHEN9/PHEN1), (E) 4-hydroxyphenanthrene (PHEN4), (F) 3-hydroxyfluorene (FLUO3), (G) 9-hydroxyfluorene (FLUO9), (H) 4-hydroxybenzophenone (4OHBP), (I) 2,2′,4,4′-tetrahydroxybenzophenone (BP2), (J) 2,2′-dihydroxy-methoxybenzophenone (BP8), (K) di-benzyl phosphate (DBZP), (L) 3,4-dihydroxy benzoic acid (DHB34), (M) di-o-cresylphosphate (DOCP), and (N) di-p-cresylphosphate (DPCP). Hypersil [Fig. 4B]: (O) 2,4,5-trichlorophenoxyacetic acid (245T), (P) n-acetyl-s-(2-carbamoylethyl)-l-cysteine (AAMA), (Q) n-acetyl-s-(3,4-dihydroxybutyl)-l-cysteine (DHBMA), (R) 4-chlorophenol (MCP4), (S) 2,3,5,6-tetrachlorophenol (TECP2356), (T) 2,4,5-trichlorophenol (TCP245), (U) 2,4,6-trichlorophenol (TCP246), and (V) n-acetyl-s-(2-cyanoethyl)-l-cysteine (CEMA). Synergi (Fig. 4C): (W) n-nitroso anatabine (NAT), (X) 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), (Y) n-nitrosonornicotine (NNN), (Z) 4-hydroxy-4-(3-pyridyl)-butanoic acid (HyPyBut), (AA) cannabidiol (CBD), (AB) tetrahydrocannabinol (THC), (AC) n,n-diethyl-3-(hydroxymethyl) benzamide (DHMB), (AD) 2-isopropyl-4-methyl-pyrimidinol (IMPY), (AE) 3-(ethylcarbamoyl) benzoic acid (ECBA), (AF) clothianidin (CLOT), (AG) imidacloprid (IMID), (AH) 3-(diethylcarbamoyl) benzoic acid (DCBA), (AI) caffeine (CAFF), (AJ) acetaminophen (APAP), (AK) amphetamine, (AL) buprenorphine, (AM) cocaethylene, (AN) cocaine, (AO) codeine, (AP) ecgonine methyl ester (EME), (AQ) fentanyl, (AR) hydrocodone, (AS) 3,4-methylenedioxy-N-ethylamphetamine (MDEA), (AT) 3,4-methylenedioxymethamphetamine (MDMA), (AU) methamphetamine, (AV) morphine, (AW) oxycodone, (AX) cortisol, and (AY) cortisone.
Customizing an assay, or panel tailoring, can be done to include additional analytes from a given class or other classes, and to regroup analytes that are compatible with more than one LC and/or MS condition. Possibilities to tailor our multi-class method are (i) expand one LC panel by adding other analytes within a chemical class, e.g., other EP analytes such as HB4, 4OHBP, BP2, and BP8 can be included in the Betasil assay (Fig. 4A); (ii) expand a LC panel by adding other chemical classes, e.g., biomarkers of lifestyle choices such as metabolites of alcohol, opiates/opioids, stimulants, prescription drugs, pain, and fever relievers. For example, biomarkers of psychosocial stress such as cortisol and cortisone can be included in the Synergi assay (Fig. 4C). Method expansion enabled inclusion of up to a total of 101 analytes in 3-tailored injections. The expanded method with an additional 51 analytes was optimized and validated by spiking QC urine at two levels (1 and 10 ng/mL) (data not shown); (iii) switch most of the pesticides and metabolites from Hypersil to Betasil panel to acquire EP, PAH, OPFR, and pesticides, except for DAPs, in a single injection (1st); and (iv) reduce the number of LC panels by simultaneous positive and negative ion scanning with 5 ms polarity switch time in the same injection, using concurrent acquisition. This detector technology helped to analyze biomarkers of psychosocial stress together with EP, PAH, and OPFR metabolites in Betasil panel (Fig. S5), and tobacco smoke and drugs of abuse together with pesticides and VOC metabolites in Hypersil panel (Fig. S6), thereby acquiring all 101 analytes in two tailored injections. Further, a 4th injection with extract 2 on a Zorbax SB column using the polarity switching feature allowed analysis of steroids and water-soluble vitamins (Fig. S7). New chemical classes in the 4th injection will require further validation and thus are not included in this report. In addition, there are biomarkers of interest in urine that did not perform in our multi-class method (Table S4).
Method performance
Matrix effects were resolved by using both stable isotope-labeled internal standards and matrix-matched calibration standard curves. An eleven-point calibration curve was used for quantification of target analytes and corresponding labeled ones prepared in acetonitrile:water (50:50, v:v), except for TCS, TCC, PCP, 24D, and 245T prepared in acetonitrile:water (70:30, v:v) based on their solubility. Acetonitrile in the calibration solutions prevented adhesion of analytes to the labware. Calibration curves were constructed with increasing levels up to 1000 ng/mL using water (reagent-based), synthetic urine (matrix-based), and a QC pool from real urine as matrices. A comparison was made between the slopes of the regression obtained from the three calibration curves, and no significant difference was observed (α = 0.1). Slope deviation of the calibration curves was calculated from the 10 batches and was < 20%. Signal suppression or enhancement (SSE) was evaluated as the ratio of slope of the calibration curve built in urine matrix and that from one built in a reagent-based water. SSE of the 50 analytes in this study was in the range of 0.8–1.2, which was considered acceptable. Satisfactory linearity (r2 > 0.95) was observed for calibration curves built in all three matrices. Correlations were r > 0.95 in urine matrices compared with those in water (r2 > 0.99). Matrix effects were evaluated by comparing recoveries of spiked target analytes in each matrix. Measured concentrations showed deviations of < 20% indicating no significant matrix effects.
A linear dynamic range of up to 6 orders of magnitude can be achieved on the Sciex 6500+ mass spectrometer, which helps quantification of a large number of diverse analytes whose concentration varies widely in urine in a single injection. In addition, this system with a multi-channel electron multiplier improves detection without raising signal/noise ratio at low levels (> LOQ–0.5 ng/mL), and a dead time correction algorithm with elevated pulse counting improves detection without saturation at high levels (>100–1000 ng/mL). Carryover was assessed at three stages: liquid handler, SPE, and LC. Carryover was nonexistent for most analytes and low for hydrophobic ones. Carryover was avoided by using an automated sample and reagent handling, a SPE setup with no crosstalk between reagent reservoirs, pipettor tools, tips, and DWP wells, and a LC setup with flow-through-needle design and an optimal pre-injection and post-run column equilibration program. A pure organic solvent injected after a run with the highest level of calibration curve or a measured concentration in urine extract did not show any carryover effects that are above LODs. A reagent-based blank was used to monitor and quantify background levels present in labware or inadvertent introduction during sample cleanup. In addition to the reagent blank, we prepared matrix-based blanks using synthetic urine and a QC urine pool. Given the high sensitivity of the instrumentation used and method developed, we were able to see trace levels of some analytes in procedural blanks. The following analytes were in the range of 0.5–1.0 ng/mL: TCS, BP3, NAP1, NAP2, BCETP, DMP, DMTP, DEP, DEDP, and CIS-DCCA. Therefore, no true blank for all analytes in one or more of the three matrices. However, all analytes in reagent blanks were below corresponding LODs. Moreover, because no urine analyzed was devoid of all analytes, and slope of calibration curves prepared in synthetic urine and QC urine pool was similar to those prepared in water, we chose a reagent-based calibration curve in water for quantification.
The QC urine pool analyte concentrations were characterized for each analyte (Table 4). Spiked QC urine pool at two levels (1 and 10 ng/mL) was analyzed in triplicate in 10 different analytical batches. Thus, we obtained a total of 30 measurements of each analyte from each spike to assess intra- and inter-batch variability. Accuracy was assessed from extraction efficiencies (EE) of spiked analytes in the QC pool and calculated as [Measured conc. (ng/mL) * 100] / [QC pool baseline conc. + Spiked conc. (ng/mL)]. Precision was assessed as the CV at each level. The inter-batch accuracy was reported as the relative error (RE) of an expected concentration [QC pool baseline conc. + Spiked conc. (ng/mL)], and the uncertainty in measurement (precision) as relative standard deviation (RSD) and CV. The 1 ng/mL spiked QC pool had analytes recoveries ranging from 83 to 109% EE, median 97% EE (Table 4). Corresponding inter-batch precision (range, median) had RSD of 0–18% (3.5%) and CV of 2–19% (9.5%). LOD for all analytes ranged from 0.01 to 1.0 ng/mL of urine, with the majority (42/50) ≤ 0.5 ng/mL (Table 4). Likewise, LOQ ranged from 0.02 to 3.2 ng/mL, with 33/50 analytes ≤ 1.0 ng/mL. As expected, %CV increased for analyte concentrations ≤ LOD. Samples were stable for 2–3 days at room temperature and 2–3 years stored at −80 °C when accuracies were calculated compared to the original concentrations. Similarly, extracts were found stable up to a week at 4 °C. We applied the multi-class method to participate in four successive PT rounds of G-EQUAS (rounds 67 and 68) and OSEQAS (rounds 1 and 2) in 2021 programs. The method fared well; the submitted results were typically within the tolerance range of the PT reference values (Table S5), and thus considered validated. As expected, the average percentage of satisfactory results increased with target concentration level in the reference materials [44, 83].
Our method is a high-throughput and robust method, and easily applicable to large sample sizes and studies. Using a single liquid handler and UHPLC-MS/MS instrument, we processed 96 samples cleanup in a day and completed LC injections of the 96 extracts each day, three LC replicate injections corresponding to three LC assays in 3 days, and obtained quantification results of biomarkers of exposure from 7 chemical classes and 16 sub-classes in a 5-day week. Several aspects of our method support its robustness including coverage of diverse chemical classes coverage, wide calibration range, reproducible sample cleanup, minimal cross contamination, cleaner LC guard column and MS frontend (curtain plate and orifice plate) for up to 1000 injections, no performance degradation of chromatography, inter- and intra-batch reproducibility, sensitivity, selectivity, consistent proficiency test qualification, and ability to analyze high volume of samples. For demonstration, we applied the multi-class method to 15 urine samples from a normal population, compared with median values from a latest NHANES survey where available (Table 5). Overall presence of all target analytes in normal urine indicates the utility of this method to survey and quantify biomarkers of common exposures in a single urine specimen. Although the number was small, the range of concentrations was consistent with that from NHANES and other studies.
Conclusion
Knowledge of actual broad exposures will improve research on health outcomes and on exposure source identification. Most targeted methods measure fewer than 15 biomarkers from a single chemical class in each biospecimen. Class-specific extractions and instrumental analyses are used. However, biospecimen availability is often limited in epidemiological studies restricting the number of single panel targeted assays. Development of new analytical methods to measure biomarkers across a range of environmental classes can significantly improve our capability to capture the totality of exposures using one sample and one laboratory procedure. Although previously reported multi-class methods have had similar sensitivity, selectivity, and other method performance benchmarks to ours, none of them provides the combination of broad coverage of environmental chemicals (7 classes and 16 sub-classes), low-volume specimen requirement (0.2 mL), satisfactory full recovery of polar conjugates (80–120%), and a high-throughput analysis with minimal manual steps (96 samples per batch per day). The slight disadvantage is a higher LOD (~0.5 ng/mL) and uncertainty at very low levels (CV > 20%) in comparison with the corresponding classical single-class assays. However, the limitations are outweighed by features like ability to tailor the assay, add other endo- and exogenous chemicals (as many as 101 analytes), and discovery of unexpected exposures. Thus, it is suitable as an exposome-scale assay with savings of assay costs and biospecimens. With continuous improvements in sample cleanup, chromatography, and mass spectrometry technologies, it will be possible to see further evolution in multi-class methods to overcome current challenges and improve studying human exposome.
References
Fourth National Report on Human Exposure to Environmental Chemicals. Volume two: NHANES 2011-2016. Updated Tables, March 2021. https://www.cdc.gov/exposurereport/pdf/FourthReport_UpdatedTables_Volume2_Mar2021-508.pdf. 2021. Accessed 10th Feb 2022.
Krowech G, Hoover S, Plummer L, Sandy M, Zeise L, Solomon G. Identifying chemical groups for biomonitoring. Environ Health Perspect. 2016;124(12):A219–a226. https://doi.org/10.1289/ehp537.
Calafat AM, Ye X, Silva MJ, Kuklenyik Z, Needham LL. Human exposure assessment to environmental chemicals using biomonitoring. Int J Androl. 2006;29(1):166–71; discussion 181-165. https://doi.org/10.1111/j.1365-2605.2005.00570.x.
Tsatsakis AM, Docea AO, Tsitsimpikou C. New challenges in risk assessment of chemicals when simulating real exposure scenarios; simultaneous multi-chemicals’ low dose exposure. Food and Chemical Toxicology. 2016;96:174–6.
Chung MK, Kannan K, Louis GM, Patel CJ. Toward capturing the exposome: exposure biomarker variability and coexposure patterns in the shared environment. Environmental Science & Technology. 2018;52(15):8801–10. https://doi.org/10.1021/acs.est.8b01467.
Dennis KK, Marder E, Balshaw DM, Cui Y, Lynes MA, Patti GJ, Rappaport SM, Shaughnessy DT, Vrijheid M, Barr DB. Biomonitoring in the era of the exposome. Environ Health Perspect. 2017;125(4):502–10. https://doi.org/10.1289/ehp474.
Yusa V, Ye X, Calafat AM. Methods for the determination of biomarkers of exposure to emerging pollutants in human specimens. Trends Analyt Chem. 2012;38:129–42. https://doi.org/10.1016/j.trac.2012.05.004.
Vorkamp K, Castaño A, Antignac J-P, Boada LD, Cequier E, Covaci A, Esteban López M, Haug LS, Kasper-Sonnenberg M, Koch HM, Pérez Luzardo O, Osīte A, Rambaud L, Pinorini M-T, Sabbioni G, Thomsen C. Biomarkers, matrices and analytical methods targeting human exposure to chemicals selected for a European human biomonitoring initiative. Environment International. 2021;146:106082. https://doi.org/10.1016/j.envint.2020.106082.
Bisphenol A and other environmental phenols and parabens in urine NHANES 2009-2010. https://www.cdc.gov/nchs/data/nhanes/nhanes_09_10/EPH_F_met_phenols_parabens.pdf. 2011. Accessed 10th Feb 2022.
Andra SS, Charisiadis P, Arora M, van Vliet-Ostaptchouk JV, Makris KC. Biomonitoring of human exposures to chlorinated derivatives and structural analogs of bisphenol A. Environ Int. 2015;85:352–79. https://doi.org/10.1016/j.envint.2015.09.011.
Andra SS, Austin C, Yang J, Patel D, Arora M. Recent advances in simultaneous analysis of bisphenol A and its conjugates in human matrices: exposure biomarker perspectives. Sci Total Environ. 2016;572:770–81. https://doi.org/10.1016/j.scitotenv.2016.07.062.
OH-PAH in urine NHANES 2011-2012. https://www.cdc.gov/nchs/data/nhanes/nhanes_11_12/pahs_g_met.pdf. 2013. Accessed 10th Feb 2022.
Organophosphate insecticides—dialkyl phosphate metabolites & flame retardants NHANES 2011-2012. https://wwwn.cdc.gov/nchs/data/nhanes/2011-2012/labmethods/OPD_G_MET.pdf. 2019. Accessed 10th Feb 2022.
Urinary pyrethroids, herbicides, and OP metabolites in urine NHANES 2009–2010. https://wwwn.cdc.gov/nchs/data/nhanes/2009-2010/labmethods/UPHOPM_F_met.pdf. 2013. Accessed 10th Feb 2022.
Metabolites of phthalates and phthalate alternatives NHANES 2015-2016. https://wwwn.cdc.gov/nchs/data/nhanes/2015-2016/labmethods/PHTHTE_I_MET.pdf. 2018. Accessed 10th Feb 2022.
Volatile organic compounds metabolites NHANES 2011-2012. https://www.cdc.gov/nchs/data/nhanes/nhanes_11_12/UVOC_G_MET_VOC_Metabolites.pdf. 2016. Accessed 10th Feb 2022.
Cotinine and hydroxycotinine (total) NHANES 2013-2014. https://wwwn.cdc.gov/nchs/data/nhanes/2013-2014/labmethods/UCOT_I_UCOTS_I_Urinary_Cotinine_and_Hydroxycotinine_MET.pdf. 2018. Accessed 10th Feb 2022.
Heffernan AL, Aylward LL, Toms L-ML, Sly PD, Macleod M, Mueller JF. Pooled biological specimens for human biomonitoring of environmental chemicals: opportunities and limitations. Journal of Exposure Science & Environmental Epidemiology. 2014;24(3):225–32. https://doi.org/10.1038/jes.2013.76.
Zhu H, Chinthakindi S, Kannan K. A method for the analysis of 121 multi-class environmental chemicals in urine by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2021;1646:462146. https://doi.org/10.1016/j.chroma.2021.462146.
Lin M, Ma S, Yu Y, Li G, Mai B, An T. Simultaneous determination of multiple classes of phenolic compounds in human urine: insight into metabolic biomarkers of occupational exposure to E-waste. Environmental Science & Technology Letters. 2020;7(5):323–9. https://doi.org/10.1021/acs.estlett.0c00187.
Jayatilaka NK, Restrepo P, Davis Z, Vidal M, Calafat AM, Ospina M. Quantification of 16 urinary biomarkers of exposure to flame retardants, plasticizers, and organophosphate insecticides for biomonitoring studies. Chemosphere. 2019;235:481–91. https://doi.org/10.1016/j.chemosphere.2019.06.181.
Preindl K, Braun D, Aichinger G, Sieri S, Fang M, Marko D, Warth B. A generic liquid chromatography-tandem mass spectrometry exposome method for the determination of xenoestrogens in biological matrices. Anal Chem. 2019;91(17):11334–42. https://doi.org/10.1021/acs.analchem.9b02446.
Asimakopoulos AG, Xue J, De Carvalho BP, Iyer A, Abualnaja KO, Yaghmoor SS, Kumosani TA, Kannan K. Urinary biomarkers of exposure to 57 xenobiotics and its association with oxidative stress in a population in Jeddah, Saudi Arabia. Environ Res. 2016;150:573–81. https://doi.org/10.1016/j.envres.2015.11.029.
Dewalque L, Pirard C, Dubois N, Charlier C. Simultaneous determination of some phthalate metabolites, parabens and benzophenone-3 in urine by ultra high pressure liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;949-950:37–47. https://doi.org/10.1016/j.jchromb.2014.01.002.
Chen M, Tao L, Collins EM, Austin C, Lu C. Simultaneous determination of multiple phthalate metabolites and bisphenol-A in human urine by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;904:73–80. https://doi.org/10.1016/j.jchromb.2012.07.022.
Heffernan AL, Thompson K, Eaglesham G, Vijayasarathy S, Mueller JF, Sly PD, Gomez MJ. Rapid, automated online SPE-LC-QTRAP-MS/MS method for the simultaneous analysis of 14 phthalate metabolites and 5 bisphenol analogues in human urine. Talanta. 2016;151:224–33. https://doi.org/10.1016/j.talanta.2016.01.037.
Mínguez-Alarcón L, Messerlian C, Bellavia A, Gaskins AJ, Chiu YH, Ford JB, Azevedo AR, Petrozza JC, Calafat AM, Hauser R, Williams PL. Urinary concentrations of bisphenol A, parabens and phthalate metabolite mixtures in relation to reproductive success among women undergoing in vitro fertilization. Environ Int. 2019;126:355–62. https://doi.org/10.1016/j.envint.2019.02.025.
Jayatilaka NK, Restrepo P, Williams L, Ospina M, Valentin-Blasini L, Calafat AM. Quantification of three chlorinated dialkyl phosphates, diphenyl phosphate, 2,3,4,5-tetrabromobenzoic acid, and four other organophosphates in human urine by solid phase extraction-high performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2017;409(5):1323–32. https://doi.org/10.1007/s00216-016-0061-4.
Reemtsma T, Lingott J, Roegler S. Determination of 14 monoalkyl phosphates, dialkyl phosphates and dialkyl thiophosphates by LC-MS/MS in human urinary samples. Sci Total Environ. 2011;409(10):1990–3. https://doi.org/10.1016/j.scitotenv.2011.01.032.
Ao J, Zhang Q, Tang W, Yuan T, Zhang J. A simple, rapid and sensitive method for the simultaneous determination of eighteen environmental phenols in human urine. Chemosphere. 2021;278:130494. https://doi.org/10.1016/j.chemosphere.2021.130494.
Silveira RS, Rocha BA, Rodrigues JL, Barbosa F Jr. Rapid, sensitive and simultaneous determination of 16 endocrine-disrupting chemicals (parabens, benzophenones, bisphenols, and triclocarban) in human urine based on microextraction by packed sorbent combined with liquid chromatography tandem mass spectrometry (MEPS-LC-MS/MS). Chemosphere. 2020;240:124951. https://doi.org/10.1016/j.chemosphere.2019.124951.
Baker SE, Serafim AB, Morales-Agudelo P, Vidal M, Calafat AM, Ospina M. Quantification of DEET and neonicotinoid pesticide biomarkers in human urine by online solid-phase extraction high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2019;411(3):669–78. https://doi.org/10.1007/s00216-018-1481-0.
Behniwal PK, She J. Development of HPLC-MS/MS method for the simultaneous determination of metabolites of organophosphate pesticides, synthetic pyrethroids, herbicides and DEET in human urine. International Journal of Environmental Analytical Chemistry. 2017;97(6):548–62. https://doi.org/10.1080/03067319.2017.1325881.
Asimakopoulos AG, Wang L, Thomaidis NS, Kannan K. A multi-class bioanalytical methodology for the determination of bisphenol A diglycidyl ethers, p-hydroxybenzoic acid esters, benzophenone-type ultraviolet filters, triclosan, and triclocarban in human urine by liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2014;1324:141–8. https://doi.org/10.1016/j.chroma.2013.11.031.
Davis MD, Wade EL, Restrepo PR, Roman-Esteva W, Bravo R, Kuklenyik P, Calafat AM. Semi-automated solid phase extraction method for the mass spectrometric quantification of 12 specific metabolites of organophosphorus pesticides, synthetic pyrethroids, and select herbicides in human urine. Journal of Chromatography B. 2013;929:18–26.
Olsson AO, Baker SE, Nguyen JV, Romanoff LC, Udunka SO, Walker RD, Flemmen KL, Barr DB. A liquid chromatography–tandem mass spectrometry multiresidue method for quantification of specific metabolites of organophosphorus pesticides, synthetic pyrethroids, selected herbicides, and deet in human urine. Anal Chem. 2004;76(9):2453–61. https://doi.org/10.1021/ac0355404.
Vela-Soria F, Ballesteros O, Zafra-Gómez A, Ballesteros L, Navalón A (2014) A multiclass method for the analysis of endocrine disrupting chemicals in human urine samples. Sample treatment by dispersive liquid–liquid microextraction. Talanta 129:209-218. doi:https://doi.org/https://doi.org/10.1016/j.talanta.2014.05.016
Dwivedi P, Zhou X, Powell TG, Calafat AM, Ye X. Impact of enzymatic hydrolysis on the quantification of total urinary concentrations of chemical biomarkers. Chemosphere. 2018;199:256–62. https://doi.org/10.1016/j.chemosphere.2018.01.177.
Ye X, Kuklenyik Z, Needham LL, Calafat AM. Automated on-line column-switching HPLC-MS/MS method with peak focusing for the determination of nine environmental phenols in urine. Anal Chem. 2005;77(16):5407–13. https://doi.org/10.1021/ac050390d.
Gavin QW, Ramage RT, Waldman JM, She J. Development of HPLC-MS/MS method for the simultaneous determination of environmental phenols in human urine. International Journal of Environmental Analytical Chemistry. 2014;94(2):168–82. https://doi.org/10.1080/03067319.2013.814123.
Zhou X, Kramer JP, Calafat AM, Ye X. Automated on-line column-switching high performance liquid chromatography isotope dilution tandem mass spectrometry method for the quantification of bisphenol A, bisphenol F, bisphenol S, and 11 other phenols in urine. Journal of Chromatography B. 2014;944:152–6.
Yao Y, Shao Y, Zhan M, Zou X, Qu W, Zhou Y. Rapid and sensitive determination of nine bisphenol analogues, three amphenicol antibiotics, and six phthalate metabolites in human urine samples using UHPLC-MS/MS. Anal Bioanal Chem. 2018;410(16):3871–83. https://doi.org/10.1007/s00216-018-1062-2.
Grignon C, Dupuis A, Albouy-Llaty M, Condylis M, Barrier L, Carato P, Brunet B, Migeot V, Venisse N. Validation of a probe for assessing deconjugation of glucuronide and sulfate phase II metabolites assayed through LC-MS/MS in biological matrices. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1061-1062:72–8. https://doi.org/10.1016/j.jchromb.2017.07.013.
Kannan K, Stathis A, Mazzella MJ, Andra SS, Barr DB, Hecht SS, Merrill LS, Galusha AL, Parsons PJ. Quality assurance and harmonization for targeted biomonitoring measurements of environmental organic chemicals across the Children’s Health Exposure Analysis Resource laboratory network. International Journal of Hygiene and Environmental Health. 2021;234:113741. https://doi.org/10.1016/j.ijheh.2021.113741.
Kaivosaari S, Finel M, Koskinen M. N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica. 2011;41(8):652–69.
Völkel W, Colnot T, Csanády GA, Filser JG, Dekant W. Metabolism and kinetics of bisphenol A in humans at low doses following oral administration. Chemical research in toxicology. 2002;15(10):1281–7.
Sasso AF, Pirow R, Andra SS, Church R, Nachman RM, Linke S, Kapraun DF, Schurman SH, Arora M, Thayer KA, Bucher JR, Birnbaum LS. Pharmacokinetics of bisphenol A in humans following dermal administration. Environ Int. 2020;144:106031. https://doi.org/10.1016/j.envint.2020.106031.
Ramesh A, Walker SA, Hood DB, Guillén MD, Schneider K, Weyand EH. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. International Journal of Toxicology. 2004;23(5):301–33. https://doi.org/10.1080/10915810490517063.
Hou R, Xu Y, Wang Z. Review of OPFRs in animals and humans: absorption, bioaccumulation, metabolism, and internal exposure research. Chemosphere. 2016;153:78–90.
Barr DB, Olsson AO, Bravo R, Needham LL. Comprehensive approach for biological monitoring of pesticides in urine using HPLC-MS/MS and GC-MS/MS. In: Martínez Vidal JL, Frenich AG, editors. Pesticide protocols. Totowa, NJ: Humana Press; 2006. p. 61–78. https://doi.org/10.1385/1-59259-929-X:061.
Wittassek M, Angerer J. Phthalates: metabolism and exposure. International journal of andrology. 2008;31(2):131–8.
Benowitz NL, Jacob P, Fong I, Gupta S. Nicotine metabolic profile in man: comparison of cigarette smoking and transdermal nicotine. Journal of Pharmacology and Experimental Therapeutics. 1994;268(1):296–303.
Smith HS. Opioid metabolism. Mayo Clinic proceedings. 2009;84(7):613–24. https://doi.org/10.1016/S0025-6196(11)60750-7.
Huestis MA. Human cannabinoid pharmacokinetics. Chemistry & biodiversity. 2007;4(8):1770–804. https://doi.org/10.1002/cbdv.200790152.
De Rooij M, Jan NM, Commandeur Nico PE, Vermeulen B. Mercapturic acids as biomarkers of exposure to electrophilic chemicals:applications to environmental and industrial chemicals. Biomarkers: biochemical indicators of exposure, response, and susceptibility to chemicals. 1998;3(4-5):239–303. https://doi.org/10.1080/135475098231101.
Aylward LL, Morgan MK, Arbuckle TE, Barr DB, Burns CJ, Alexander BH, Hays SM. Biomonitoring data for 2,4-dichlorophenoxyacetic acid in the United States and Canada: interpretation in a public health risk assessment context using biomonitoring equivalents. Environmental Health Perspectives. 2010;118(2):177–81. https://doi.org/10.1289/ehp.0900970.
Harada KH, Tanaka K, Sakamoto H, Imanaka M, Niisoe T, Hitomi T, Kobayashi H, Okuda H, Inoue S, Kusakawa K. Biological monitoring of human exposure to neonicotinoids using urine samples, and neonicotinoid excretion kinetics. PloS one. 2016;11(1):e0146335.
Bravo R, Caltabiano LM, Weerasekera G, Whitehead RD, Fernandez C, Needham LL, Bradman A, Barr DB. Measurement of dialkyl phosphate metabolites of organophosphorus pesticides in human urine using lyophilization with gas chromatography-tandem mass spectrometry and isotope dilution quantification. Journal of Exposure Science & Environmental Epidemiology. 2004;14(3):249–59. https://doi.org/10.1038/sj.jea.7500322.
Calafat AM, Koch HM. BPA and risk assessment. Lancet Diabetes Endocrinol. 2020;8(4):269–70. https://doi.org/10.1016/s2213-8587(20)30070-x.
Schantz MM, Benner BA Jr, Heckert NA, Sander LC, Sharpless KE, Vander Pol SS, Vasquez Y, Villegas M, Wise SA, Alwis KU, Blount BC, Calafat AM, Li Z, Silva MJ, Ye X, Gaudreau É, Patterson DG Jr, Sjödin A. Development of urine standard reference materials for metabolites of organic chemicals including polycyclic aromatic hydrocarbons, phthalates, phenols, parabens, and volatile organic compounds. Anal Bioanal Chem. 2015;407(11):2945–54. https://doi.org/10.1007/s00216-014-8441-0.
Koch HM, Lessmann F, Swan SH, Hauser R, Kolossa-Gehring M, Frederiksen H, Andersson AM, Thomsen C, Sakhi AK, Bornehag CG, Mueller JF, Rudel RA, Braun JM, Harth V, Brüning T. Analyzing terephthalate metabolites in human urine as biomarkers of exposure: importance of selection of metabolites and deconjugation enzyme. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1100-1101:91–2. https://doi.org/10.1016/j.jchromb.2018.09.035.
Blount BC, Milgram KE, Silva MJ, Malek NA, Reidy JA, Needham LL, Brock JW. Quantitative detection of eight phthalate metabolites in human urine using HPLC−APCI-MS/MS. Analytical chemistry. 2000;72(17):4127–34.
Dias N, Poole C. Mechanistic study of the sorption properties of OASIS® HLB and its use in solid-phase extraction. Chromatographia. 2002;56(5):269–75.
Franke C, Westerholm H, Niessner R. Solid-phase extraction (SPE) of the fluorescence tracers uranine and sulphorhodamine B. Water Research. 1997;31(10):2633–7.
Scheidweiler KB, Shakleya DM, Huestis MA. Simultaneous quantification of nicotine, cotinine, trans-3′-hydroxycotinine, norcotinine and mecamylamine in human urine by liquid chromatography-tandem mass spectrometry. Clin Chim Acta. 2012;413(11-12):978–84. https://doi.org/10.1016/j.cca.2012.02.017.
Onyemauwa F, Rappaport SM, Sobus JR, Gajdošová D, Wu Ra, Waidyanatha S (2009) Using liquid chromatography–tandem mass spectrometry to quantify monohydroxylated metabolites of polycyclic aromatic hydrocarbons in urine. Journal of Chromatography B 877 (11):1117-1125. doi:https://doi.org/https://doi.org/10.1016/j.jchromb.2009.02.067
Zhang Y, Wang Q, Cheng J, Zhang J, Xu J, Ren Y (2015) Comprehensive profiling of mercapturic acid metabolites from dietary acrylamide as short-term exposure biomarkers for evaluation of toxicokinetics in rats and daily internal exposure in humans using isotope dilution ultra-high performance liquid chromatography tandem mass spectrometry. Analytica Chimica Acta 894:54-64. doi:https://doi.org/https://doi.org/10.1016/j.aca.2015.08.033
Adams S, Guest J, Dickinson M, Fussell RJ, Beck J, Schoutsen F. Development and validation of ion chromatography–tandem mass spectrometry-based method for the multiresidue determination of polar ionic pesticides in food. Journal of Agricultural and Food Chemistry. 2017;65(34):7294–304. https://doi.org/10.1021/acs.jafc.7b00476.
Karthikraj R, Kannan K. Widespread occurrence of glyphosate in urine from pet dogs and cats in New York State, USA. Science of The Total Environment. 2019;659:790–5. https://doi.org/10.1016/j.scitotenv.2018.12.454.
Calafat AM, Kato K, Hubbard K, Jia T, Botelho JC, Wong L-Y. Legacy and alternative per- and polyfluoroalkyl substances in the U.S. general population: paired serum-urine data from the 2013–2014 National Health and Nutrition Examination Survey. Environment International. 2019;131:105048. https://doi.org/10.1016/j.envint.2019.105048.
Stein CR, Wolff MS, Calafat AM, Kato K, Engel SM. Comparison of polyfluoroalkyl compound concentrations in maternal serum and amniotic fluid: a pilot study. Reprod Toxicol. 2012;34(3):312–6. https://doi.org/10.1016/j.reprotox.2012.05.039.
Kato K, Kalathil AA, Patel AM, Ye X, Calafat AM. Per- and polyfluoroalkyl substances and fluorinated alternatives in urine and serum by on-line solid phase extraction–liquid chromatography–tandem mass spectrometry. Chemosphere. 2018;209:338–45. https://doi.org/10.1016/j.chemosphere.2018.06.085.
Asimakopoulos AG, Wang L, Thomaidis NS, Kannan K. A multi-class bioanalytical methodology for the determination of bisphenol A diglycidyl ethers, p-hydroxybenzoic acid esters, benzophenone-type ultraviolet filters, triclosan, and triclocarban in human urine by liquid chromatography–tandem mass spectrometry. Journal of Chromatography A. 2014;1324:141–8. https://doi.org/10.1016/j.chroma.2013.11.031.
Meeker JD, Barr DB, Serdar B, Rappaport SM, Hauser R. Utility of urinary 1-naphthol and 2-naphthol levels to assess environmental carbaryl and naphthalene exposure in an epidemiology study. Journal of Exposure Science & Environmental Epidemiology. 2007;17(4):314–20. https://doi.org/10.1038/sj.jes.7500502.
Wang L-M, Luo D, Li X, Hu L-Q, Chen J-X, Tu Z-Z, Sun B, Chen H-G, Liu L, Yu M, Li Y-P, Pan A, Messerlian C, Mei S-R, Wang Y-X. Temporal variability of organophosphate flame retardant metabolites in spot, first morning, and 24-h urine samples among healthy adults. Environmental Research. 2021;196:110373. https://doi.org/10.1016/j.envres.2020.110373.
Personne S, Marcelo P, Pilard S, Baltora-Rosset S, Corona A, Robidel F, Lecomte A, Brochot C, Bach V, Zeman F. Determination of maternal and foetal distribution of cis- and trans-permethrin isomers and their metabolites in pregnant rats by liquid chromatography tandem mass spectrometry (LC-MS/MS). Anal Bioanal Chem. 2019;411(30):8043–52. https://doi.org/10.1007/s00216-019-02157-7.
Alwis KU, Blount BC, Britt AS, Patel D, Ashley DL. Simultaneous analysis of 28 urinary VOC metabolites using ultra high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI/MSMS). Anal Chim Acta. 2012;750:152–60. https://doi.org/10.1016/j.aca.2012.04.009.
de Castro A, Concheiro M, Shakleya DM, Huestis MA. Simultaneous quantification of methadone, cocaine, opiates, and metabolites in human placenta by liquid chromatography-mass spectrometry. Journal of analytical toxicology. 2009;33(5):243–52. https://doi.org/10.1093/jat/33.5.243.
Kotandeniya D, Carmella SG, Ming X, Murphy SE, Hecht SS. Combined analysis of the tobacco metabolites cotinine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in human urine. Analytical Chemistry. 2015;87(3):1514–7. https://doi.org/10.1021/ac504047j.
Jacob P, Goniewicz ML, Havel CM, Schick SF, Benowitz NL. Nicotelline: a proposed biomarker and environmental tracer for particulate matter derived from tobacco smoke. Chemical Research in Toxicology. 2013;26(11):1615–31. https://doi.org/10.1021/tx400094y.
Jacob P, St Helen G, Yu L, Nardone N, Havel C, Cheung P, Benowitz NL. Biomarkers of exposure for dual use of electronic cigarettes and combustible cigarettes: nicotelline, NNAL, and total nicotine equivalents. Nicotine & Tobacco Research: Official Journal of the Society for Research on Nicotine and Tobacco. 2020;22(7):1107–13. https://doi.org/10.1093/ntr/ntz235.
Benowitz NL, Bernert JT, Foulds J, Hecht SS, Jacob P, Jarvis MJ, Joseph A, Oncken C, Piper ME. Biochemical verification of tobacco use and abstinence: 2019 update. Nicotine & Tobacco Research: Official Journal of the Society for Research on Nicotine and Tobacco. 2020;22(7):1086–97. https://doi.org/10.1093/ntr/ntz132.
Vaccher V, Lopez ME, Castaño A, Mol H, Haji-Abbas-Zarrabi K, Bury D, Koch HM, Dvorakova D, Hajslova J, Nübler S, Kaur Sakhi A, Thomsen C, Vorkamp K, Göen T, Antignac J-P. European interlaboratory comparison investigations (ICI) and external quality assurance schemes (EQUAS) for the analysis of bisphenol A, S and F in human urine: results from the HBM4EU project. Environmental Research. 2022;210:112933. https://doi.org/10.1016/j.envres.2022.112933.
Acknowledgements
We gratefully acknowledge Dr. Dana B. Barr (Professor, Rollins School of Public Health, Emory University, Atlanta, GA, USA) for consultation for the umbelliferone validation steps and for DAPs analysis.
Funding
The Mount Sinai CHEAR/HHEAR laboratory hub acknowledges funding for this study from NIH/NIEHS: U2C ES026561 and P30 ES023515.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Source of biological material
The authors declare obtaining informed consent of the anonymous volunteers for providing urine samples to prepare the urine QC pool in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 2703 kb)
Rights and permissions
About this article

Cite this article
Jagani, R., Pulivarthi, D., Patel, D. et al. Validated single urinary assay designed for exposomic multi-class biomarkers of common environmental exposures. Anal Bioanal Chem 414, 5943–5966 (2022). https://doi.org/10.1007/s00216-022-04159-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-04159-4