
Abstract
The endocannabinoid system (ECS) is a complex cell-signaling system. To address the growing need of analytics capturing endocannabinoid levels to investigate the ECS, we developed and validated an assay for the quantitative analysis of 14 endocannabinoids and congeners. A simple extraction using protein precipitation with acetonitrile followed by online-trapping high-performance liquid chromatography–tandem mass spectrometry (LC/LC-MS/MS) was used to monitor the levels of 14 endocannabinoids in plasma. The assay was validated and intra-run and inter-run accuracies and imprecisions as well as matrix effects, recoveries, and sample stabilities were determined. As a proof of concept, a subset of study samples after naturalistic administration of Cannabis flower and concentrate was analyzed. With the exception of N-oleoyl dopamine and oleamide, all endocannabinoids fulfilled the predefined acceptance criteria. Reproducible recoveries and no significant matrix effects were observed. Sample stability was an issue. Analysis of the proof-of-concept study samples revealed a significantly (p = 0.006) higher concentration of docosatetraenoyl ethanolamide in concentrate users (300 ± 13 pg/mL) compared to flower users (252 ± 11 pg/mL). A robust, sensitive high-throughput assay for the quantitation of 14 endocannabinoids and congeners was successfully validated. Our study showed that it is mandatory to (A) appropriately stabilize samples and (B) separate and separately quantify 1-AG and 2-AG; otherwise, study results are unreliable. The analysis of study samples from Cannabis flower users versus Cannabis concentrate users revealed higher levels of docosatetraenoyl ethanolamide and anandamide (n.s.) in high THC concentrate users in accordance with the existing literature, supporting the validity of the assay measurements.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At the beginning of this century, the endocannabinoid system was primarily defined by three components: the endocannabinoids, the enzymes that synthesize and degrade the endocannabinoids, and the cannabinoid receptors CB1 and CB2 [1, 2]. The first two endocannabinoids discovered, anandamide (N-arachidonoylethanolamine; AEA) and 2-arachidonoylglycerol (2-AG), are still the most studied so far [3]. Unlike classical neurotransmitters and neuropeptides, endocannabinoids are not stored in vesicles in the cell, rather they are produced on demand from membrane phospholipids by a series of intracellular enzymes and released from cells, followed by immediate action as signaling molecules [4]. After reuptake by cells, they are rapidly inactivated by enzymatic hydrolysis. An influx of calcium (Ca2+) in the postsynaptic cell activates phospholipase D (PLD), which acts on N-arachidonoyl phosphatidylethanolamine (NAPE) to produce anandamide. 2-AG is synthesized by diacylglycerol lipase (DAGL)-α and DAGL-β [2]. Endocannabinoids exert their effects mainly through the G protein–coupled cannabinoid receptors type 1 (CB1) and type 2 (CB2). These receptors are guanine nucleotide–binding (G) protein–coupled receptors coupled to Gi/o transduction pathways, and their activation leads to inhibition of adenylate cyclase (AC), opening of presynaptic potassium (K+) channels, and inhibition of presynaptic Ca2+ channels ([5, 6]. These effects result in inhibition of releases of glutamate or γ-aminobutyric acid (GABA) from presynaptic terminals. Anandamide is transported into the cell via the endocannabinoid transporter (ECT) and is degraded by the fatty acid amide hydrolase (FAAH) to arachidonic acid and ethanolamine. Similarly, 2-AG is degraded by monoacylglycerol lipase (MAGL) [5, 7, 8]. Additionally, they are also able to interact with non-CB1/non-CB2 targets, including the transient receptor potential vanilloid type 1 (TRPV1) channel [1], peroxisome proliferator-activated receptor (PPAR)-α, and PPAR-γ [9] as well as the G protein–coupled receptor GPR55 [2, 10]. However, since the introduction of anandamide and 2-AG, numerous additional endocannabinoids and endocannabinoid congeners have been discovered. These compounds that directly or indirectly bind to the cannabinoid receptors include docosatetraenoyl ethanolamide (DEA), dihomo-γ-linoleoylethanolamide (DH-g-LEA), N-arachidonoyl dopamine (NADA), 2-arachidonoylglycerol ether (2-AGE), O-arachidonoylethanolamide (O-AEA), N-oleoyl dopamine (ODA), oleamide (OLA), linoleoyl ethanolamide (LEA), oleoyl ethanolamine (OEA), palmitoyl ethanolamide (PEA), and stearoyl ethanolamide (SEA) [8].
The term “endocannabinoid system” (ECS) in a strict sense refers to the two cannabinoid receptors CB1 and CB2, the endocannabinoids per se, and a group of enzymes involved in their synthesis and breakdown [3]. However, the ECS should not be considered a specific and distinct biological system. It has become increasingly clear how much the ECS is intertwined with other lipid- or non-lipid signaling pathways and connected to other regulatory networks. From a biochemical point of view, classical endocannabinoids can be seen as part of a large family of structurally related amides, esters, and ethers of fatty acids, which are continuously formed and degraded in a dynamic equilibrium [3]. Endocannabinoids show an overlapping activity for molecular targets that go far beyond the classical cannabinoid receptors and interact with other receptors besides CB1 or CB2, with potencies that differ little from those with which they interact with the cannabinoid receptors [3, 11, 12]. In addition, the biochemical pathways for synthesis and degradation of endocannabinoids and their congeners show several crossroads and connections with those of other bioactive lipids resulting in a number of regulatory nodes [9, 13]. There is a growing consensus that an “expanded’” view of the ECS, also referred to as “endocannabinoidome,” would be more appropriate to study and understand its full dimensions [1, 3, 14]. There have been several assays described in the literature for the quantitation of endocannabinoids using HPLC-MS; most of these were focused on the analysis of anandamide, 2-AG, and up to 3 endocannabinoid congeners [15,16,17,18,19,20,21,22,23,24] and often used more extensive sample workup procedures; some of these have used simple sample workup with column switching technology [15, 16, 24]. To address the growing need of analytics capturing endocannabinoid levels to probe the ECS/endocannabinoidome, we have developed and validated an assay for the quantitative analysis of 14 endocannabinoids and congeners. This includes several new endogenous cannabinoid receptor ligands as well as several endocannabinoid congeners that have been recently discovered. To probe the main endocannabinoids of the ECS, the assay focuses on the analysis of AEA, 2-AG, DEA, DH-g-LEA, NADA, 2–2-AGE, O-AEA, ODA, OLA, LEA, OEA, PEA, and SEA. It was the goal of this study to develop a comprehensive endocannabinoid panel assay and perform an internal validation. Furthermore, the assay was validated externally using samples from Cannabis users in a proof-of-concept sub-study, during which samples were collected from 2 different groups of Cannabis users (flower users and concentrate users).
Methods
Development, analytical parameters, and assay procedures
Chemicals and reagents
Solvents and reagents (HPLC grade acetonitrile, methanol, water, formic acid) used for mobile phases and sample extraction in this study were purchased from Fisher Scientific (Fair Lawn, NJ, USA) and used without further purification. The solvent (ethanol) used for the preparation of stock solutions and phosphate-buffered saline (PBS) was purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA). All study compound reference materials including anandamide (arachidonoyl ethanolamide, AEA), 2-arachionoylglycerol (2-AG), 1-arachionoylglycerol (1-AG), docosatetraenoyl ethanolamide (DEA), dihomo-γ-linoleoylethanolamide (DH-g-LEA), N-arachidonoyl dopamine (NADA), 2-arachidonoylglycerol ether (2-AGE), O-arachidonoylethanolamide (O-AEA), N-oleoyl dopamine (ODA), oleamide (OLA), linoleoyl ethanolamide (LEA), oleoyl ethanolamine (OEA), palmitoyl ethanolamide (PEA), and stearoyl ethanolamide (SEA) were received from Cayman Chemical Company (Ann Arbor, MI, USA). All isotope-labeled internal standards including 2-arachidonoylglycerol-d5 (2-AG-d5), anandamide-d4 (AEA-d4), N-arachidonoyl dopamine-d8 (NADA-d8), linoleoyl ethanolamide-d4 (LEA-d4), oleoyl ethanolamide-d4 (OEA-d4), palmitoyl ethanolamide-d4 (PEA-d4), and stearoyl ethanolamide-d3 (SEA-d3) were received from Cayman Chemical Company (Ann Arbor, MI, USA).
For the enrichment of QC samples, blank human K2EDTA plasma from Bioreclamation IVT (Westbury, NY, USA) was used, and for calibrator standard preparation, charcoal-stripped plasma from BioCheMed (Winchester, VA, USA).
Calibrators and quality control samples
Stock solutions of the endocannabinoids AEA, O-AEA, DH-g-LEA, DEA, ODA, and NADA were prepared at 500 μg/mL in ethanol. All other endocannabinoids and congeners were prepared at 1 mg/mL in ethanol. A mixed stock solution of the endocannabinoids was prepared in ethanol at the concentration of 10 μg/mL for AEA, DH-g-LEA, DEA, ODA, NADA, SEA, O-AEA, 2-AGE, OEA, and LEA, at the concentration of 100 μg/mL for PEA, 2-AG, and 1-AG, and at the concentration of 50 μg/mL for OLA. This mix was used to generate calibration curves in the range between 10 pg/mL and 50 ng/mL (0, 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 25, 50 ng/mL) for most endocannabinoids in 1/10 diluted charcoal-stripped plasma (PEA, 2-AG and 1-AG were 10-fold higher and OLA was 5-fold higher). Quality control samples were prepared in a similar manner. However, all stocks and working solutions were prepared independently from the calibrators and human K2EDTA plasma was used instead of diluted charcoal-stripped plasma. Enriched plasma concentrations for QCs were 0.4, 4, 15, and 40 ng/mL (PEA, 2-AG and 1-AG were 10-fold higher and OLA was 5-fold higher). To cover the low range of the assay, mixed plasma lots containing the endogenous concentrations of endocannabinoids were diluted 1/3 and 1/9. The internal standards were prepared in ethanol at a concentration of 100 μg/mL AEA-d4, 2-AG-d5, PEA-d4, LEA-d4, and OEA-d4 in ethanol. The internal standards NADA-d8 and SEA-d3 were prepared at 1000 and 200 μg/mL, respectively. An internal standard mix was prepared and stored at −80 °C until preparation of the protein precipitation solution. One hundred milliliters of the protein precipitation solution (acetonitrile) was enriched with 200 μL of the internal standard mix resulting in a final concentration of 2 ng/mL for AEA-d5, 2-AG-d5, PEA-d4, LEA-d4, and OEA-d4, 8 ng/mL for SEA-d3, and 10 ng/mL for d8-NADA in the protein precipitation solution. Working solutions were always prepared freshly and had to be used within 1 day.
Sample extraction procedure
Aliquots of 200 μL of the calibrator, quality control, or study sample were transferred into a 1.5-mL polypropylene vial with conical bottom and snap-on lid. Eight hundred microliters of acetonitrile containing the internal standards (2 ng/mL for AEA-d5, 2-AG-d5, PEA-d4, LEA-d4, OEA-d4, 8 ng/mL for d3-SEA, and 10 ng/mL for d8-NADA) was added and samples were mixed for 10 min on a vortex mixer (Multitube vortexer, Fisher Scientific, Waltham, MA, USA). Samples were centrifuged at 25,000×g and 4 °C for 10 min (MR 23i centrifuge with a fiberlite rotor, Thermo Scientific, Waltham, MA, USA). Seven hundred fifty microliters of the supernatant was transferred into HPLC vials, 450 μL of 0.1% formic acid in water was added, and the extracts were briefly vortexed. The sample extracts were placed into the HPLC autosampler for subsequent analysis.
Endocannabinoid LC/LC-APCI-MS/MS
An Agilent 1200 Series LC/LC system (Agilent Technologies, Palo Alto, CA, USA) with online-trapping column (Agilent Zorbax C18, 12 mm · 4.6 mm, 5-μm particle size) and an analytical column (Poroshell 120 EC-C18, 50 mm · 4.6 mm, 2.7-μm particle size, Agilent Technologies, Palo Alto, CA, USA) coupled to a Sciex API5500 triple quadrupole mass spectrometer (Sciex, Framingham, MA, USA) was used for separation and quantitative analysis of endocannabinoids. The use of the less sensitive Sciex API5000 mass spectrometer also resulted in sufficient sensitivity if only quantifier ions were monitored. However, the data presented in this manuscript derived from a system with a API5500 detector. The HPLC system consisted of the following components: CTC PAL autosampler (Leap Technologies, Carrboro, NC, USA) in combination with a G1316C thermostat and two G1312B binary pumps (Agilent Technologies, Palo Alto, CA, USA). The two columns and the two binary HPLC pumps were connected via a column switching valve that was mounted on a step motor (Rheodyne, Cotati, CA, USA) (Supplementary Information (ESM) Fig. S1). Four hundred microliters of the samples were injected onto a 4.6 · 12.5 mm extraction column (Zorbax XDB C18, Agilent Technologies, Palo Alto, CA, USA) with a particle size of 5 μm. Samples were loaded and washed with a mobile phase of 35% acetonitrile supplemented with 0.1% formic acid and 65% 0.1% formic acid in HPLC grade water. The flow was increased from 0.5 to 1.5 mL/min within 1 min. The extraction column was kept at room temperature. After 1 min, the switching valve was activated and the analytes were eluted in the backflush mode from the extraction column onto a 4.6 · 50 mm Poroshell Eclipse C18, 2.7-μm analytical column (Agilent Technologies, Palo Alto, CA, USA), which was kept at 60 °C. The analytical gradient started at a flow rate of 800 μL/min with 65% aqueous solvent (0.1% formic acid in water) and 35% organic solvent (0.1% formic acid in acetonitrile). After 1 min, the flow rate was increased to 1000 μL/min and organic solvent was increased to 65% within the next 0.3 min. At minute 2.5, the organic solvent was increased to 70%, and at minute 2.6 to 77%. For the following 1.3 min, the organic solvent was kept at 77%. This was followed by an increase to 90% organic solvent at minute 4. This was kept for 1.2 min and followed by an increase to 100% organic solvent within the next 0.3 min. The solvent was kept at 100% organic for the next 0.7 min (until minute 6.2). Hereafter, the column was re-equilibrated for 1.7 min to the initial conditions (800 μL/min and 65% aqueous solvent). The HPLC system was connected to the API5500 mass spectrometer via a turboV ion source (Sciex, Framingham, MA, USA) operated in positive atmospheric pressure chemical ionization mode (APCI). The nebulizer current was set to 3 kV and the temperature was set to 450 °C. The mass spectrometer was operated in positive multiple-reaction monitoring mode (MRM) and the compound-specific settings are listed in the ESM Table S1. The HPLC system, mass spectrometer, and column switching valve were controlled by Analyst software (Sciex, Framingham, MA, USA), and data was processed using Multiquant software version 3.0.2 (Sciex). Endocannabinoids were quantified by calculating the analyte area-under-the-peak/internal standard under-the-peak ratios and comparison to those of calibration curves constructed using 1/10 diluted charcoal-stripped plasma.
Internal validation
Validation strategy
The sample matrix for the validation was enriched human plasma and diluted human plasma. The dilution was chosen to extend the evaluated working range up to 9-fold below the average endogenous concentrations of endocannabinoids. The assay was completely validated using quality control samples, including inter-day performance and stability.
Specificity, selectivity, recovery, matrix effect (ion suppression/enhancement), and carry-over effect
A blank sample of each individual lot was extracted to show that there was no endogenous interference (signal needed to be less than 20% of the LLOQ). In this case, since endocannabinoids are endogenous compounds, the enrichment of blank samples and the increase in signal at the exact retention time of the endocannabinoid were evaluated. This experiment assessed specificity and selectivity. To exclude interferences in the matrices, plasma samples from 6 individuals (BioreclamationIVT, Westbury, NY, USA) were processed following the procedures described by Matuszewski et al. [25]. Briefly, samples were enriched at QC levels before (EB) and after extraction (EA) and were compared to enriched buffer samples (BF). The ratio of the EB sample compared to the EA sample reveals the recovery. The EA samples compared to the BF samples reflect the matrix effect in the samples from the different individuals. A potential carry-over was evaluated by analysis of buffer samples following the highest calibrator and quality control.
Range of reliable response
The lower limit of quantification (LLOQ) and range of reliable response was determined by analysis of 6 sets (n = 6) of quality control samples. Since endocannabinoids are endogenous samples, diluted blank samples were used to evaluate the LLOQ. The requirement for the LLOQ was a minimum mean accuracy between 75 and 125% and for at least 2/3 of all calibrators at this level. The RSD% had to be less than 20% and the correlation coefficient (r) >0.99.
Intra- and inter-day accuracy and imprecision
Intra-day and inter-day accuracy and imprecision were established for the duration of 4 days based on the analysis of QC samples. Each day, 6 QCs were analyzed per level (n = 6). Two calibration curves were generated, and one was analyzed at the beginning and one at the end of the analytical batch.
Stability assessment
Stock solution stability was assessed by analysis of a buffer stock solution at 200 ng/mL that was stored at −20 °C for the duration of 0, 1 week, 1 month, and 3 months. Autosampler stability was assessed by analyzing a set of QCs after 24 h and 72 h in the autosampler at +4 °C. These QCs were analyzed together with freshly prepared calibrator samples. Freeze-thaw cycle stability was assessed by exposing sets of QCs to 0, 1, and 3 freeze-thaw cycles (−80 °C) followed by analysis using a freshly prepared calibration curve. Benchtop stability was assessed by placing plasma QCs for the duration 3 h and 6 h and 24 h on the bench at ambient temperature prior to analysis. Short-term stability was assessed by storing QC samples at room temperature and 4 °C for 1 day and 7 days prior to analysis. Long-term stability was assessed by storing QC samples at −20 °C and − 80 °C for the duration of 3 months.
Acceptance criteria
For all experiments (specificity, selectivity, recovery, matrix effect, determination of the range of reliable response, intra- and inter-day accuracy and imprecision, and stability assessment), two calibration curves were generated. The requirements were a minimum mean accuracy between 80 and 120% within the range of the assay and for at least 3/4 of all calibrators with a correlation coefficient (r) >0.99. Two thirds of all quality control samples and stability samples had to be within 80–120% and the %CV had to be ≤20% [26].
External validation
Proof of concept study samples
Endocannabinoids were analyzed in a subset of samples collected during observational research investigating the effects of naturalistic Cannabis administration using different Cannabis products in Colorado. The research was approved by the University of Colorado Boulder Institutional Review Board and the primary outcomes have been published previously [27].
Sample stability sub-study
It was the aim of the first sub-study to investigate the stability of samples when stored with or without addition of 0.5% formic acid as stabilizing agent. This research involved a cohort design in which Cannabis flower users and concentrate users were randomly assigned to higher- vs lower-THC products within user groups. Of the 133 individuals enrolled and assessed, 55 regular flower Cannabis users and 66 regular concentrate Cannabis users complied with the study’s Cannabis use instructions and had complete data across primary outcomes. Flower users were randomly assigned to use either 16% or 24% THC flower and concentrate users were randomly assigned to use either 70% or 90% THC concentrate that they purchased from a state market dispensary. Potency effects were not seen in the primary study, so the different strain potencies were not evaluated here. Samples from both studies were used. In the first sub-study, a total of 130 samples from 34 subjects (enrolled in either study) were collected via venous blood draw into regular K2EDTA containers (Becton Dickinson, Franklin Lakes, NJ, USA). After plasma was generated, one aliquot of 0.9 mL of sample was stored in tubes containing 0.1 mL of 5% formic acid and mixed, and the second aliquot was stored without any additives. All samples were stored in glass containers at −70 °C or below in monitored freezers until analysis.
Cannabis potency effect sub-study
For the second endocannabinoid sub-study, a subset of 36 study subjects (18 flower users and 18 concentrate users) and a total of 143 samples (73 flower user samples and 70 concentrate user samples) was analyzed. For this proof-of-concept study, samples were not collected with the addition of stabilizing formic acid. It was the aim of this study to evaluate the endocannabinoid levels in the 2 study groups (flower users versus concentrate users) and to evaluate if the regular consumption of different potencies of Cannabis products affects the endocannabinoid levels.
Statistics
All statistics for the determination of validation parameters as well as distribution statistics, correlation analysis, and analysis of variance (with Tukey’s post hoc test) for the comparison of the different Cannabis user groups were carried out using IBM SPSS (SPSS Statistics for Windows, Version 26.0, IBM, Armonk, NY, USA). For correlation analysis, data were processed after and without log-transformation.
Results
Assay optimization
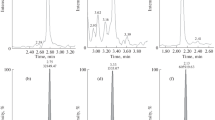
During the assay development, various chromatographic conditions and column materials were tested to achieve separation of 2-arachidonoylglycerol (the active endocannabinoid) from the naturally occurring structural isomer 1-arachidonoylgycerol (1-AG, not an endocannabinoid). This could be achieved using a superficially porous reversed phase column (4.6 · 50 mm Poroshell Eclipse C18, 2.7 μm) in combination with a stepwise gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile (for specifics, please see methods section). Figure 1 shows the chromatographic separation of 1-AG from 2-AG. Using the chromatographic parameters described in the methods section, baseline separation of these 2 structural isomers could be accomplished.

Representative extracted ion chromatograms of plasma and whole blood collected and stored under varying conditions: a K2EDTA plasma processed and stored with 0.5% formic acid final concentration (pH 4–5); b K2EDTA plasma with no other additives; c K2EDTA whole blood sample with additive of 0.5% formic acid final concentration; and d K2EDTA whole blood sample without addition of formic acid
Sample matrix evaluation
Our experiments also proved that isomerization from 2-AG to 1-AG was inhibited by lowering the pH of plasma or whole blood samples (see Fig. 1). Based on these results, it was determined that a pH range of 4–5 was sufficient to stabilize 2-AG. This could be accomplished if 9 parts K2EDTA whole blood or plasma was acidified with addition of 1 part of 5% formic acid in water. However, acidification of K2EDTA whole blood did not work out, since EDTA lost its ability to bind divalent ions and whole blood samples coagulated following freeze-thaw cycles. In addition, initial experiments showed that the amount of 1-AG formed from 2-AG is much lower than the amount of initial 2-AG in a stabilized sample. This becomes clear from Fig. 1a (stabilized plasma) compared to Fig. 1b (plasma without acidification) as well as Fig. 1c (stabilized whole blood) compared to Fig. 1d (whole blood without acidification), showing more than 2 orders of magnitude signal loss in the non-acidified samples.
Specificity, selectivity, recovery, matrix effect (ion suppression/enhancement), and carry-over effect
Since endocannabinoids are endogenous compounds that are naturally abundant in human blood, a matrix depleted of endocannabinoids is not available. The responses at the retention times of the 14 endocannabinoids in the ion chromatograms of extracted charcoal-stripped human EDTA plasma sample matrix was less than 20% of the detector response of that of the lowest calibrators or its internal standards with the exceptions of OLA, SEA, PEA, and LEA. However, this simply indicated that charcoal-stripped plasma was not completely depleted of said endogenous analytes. Additionally, enrichment of plasma with the study analytes and determination of retention times and signal comparison to buffer samples revealed that the method for analyzing the 14 endocannabinoids in human EDTA plasma was considered specific. Representative extracted ion chromatograms of the individual study compounds from blank (non-enriched) calibrator standards, standards at the corresponding lower limits of quantitation, and from representative study samples are shown in the ESM Fig. S2.
Extraction recovery was evaluated by comparison of QC samples that were enriched and then extracted to QC samples that extracted first and then enriched to the same concentrations. Six individual lots were tested at the 3 QC levels. All samples had initially to be corrected for the area of the endogenous endocannabinoid signal. All recoveries were above 70% and reproducible (%CV ≤20%) with the exceptions of OLA and SEA (see Table 1).
Based on the analysis of enriched extracted blank plasma samples from 6 individuals that were enriched after extraction and compared to buffer samples (no matrix), the 14 compounds revealed matrix effects from mild ion suppression to mild ion enhancement. Matrix effects were determined as previously described by Matuszewski et al. [25]. All matrix effects were reproducible for the individual lots tested (%CV ≤ 20%) with the exception of OLA.
The analysis of blank and enriched human EDTA plasma samples from 6 individuals showed acceptable results for all analytes with the exception of SEA and OEA. More than 2/3 of the tested samples were required to be within 80–120%. The values for SEA and OEA were with 70–130% of the nominal concentration (data not shown). Thus, matrix interferences were not abundant for the majority of analytes.
Range of reliable response
Since endocannabinoids are endogenous samples, diluted blank samples were used to evaluate the LLOQ. The lower limit of quantification (LLOQ) and range of reliable response was determined by analysis of 6 sets (n = 6) of quality control samples. The assay was linear from 0.05–50, 0.25–50, 0.26–500, 2.1–500, 0.18–50, 0.55–50, 0.05–50, 0.10–50, 0.05–50, 74.8–250, 1–500, 1–50, 0.34–50, and 0.70–50 for AEA, O-AEA, 2-AG, 1-AG, DEA, 2-AGE, DH-g-LEA, NADA, ODA, OLA, PEA, SEA, LEA, and OEA, respectively. The acceptance criteria of more than 3/4 of calibrators were required to be within 80–120% of the nominal value with a %CV of less than 20%.
Intra- and inter-day accuracy and imprecision
Intra-day and inter-day accuracy and imprecision were established for over 4 different days. Six sets of QCs were analyzed per level (n = 6 each day). All compounds with the exceptions of ODA and OLA fulfilled the predefined acceptance criteria for intra- and inter-run accuracy and imprecision of 80–120% and ≤ 20%CV, respectively (see Table 2). In this context, one level for ODA one analysis day (intra-run on day 4 and inter-run at the LLOQ) failed the acceptance criteria, whereas the acceptance criteria for OLA could not be met on most days.
Stability assessment
Stability of extracted samples in the autosampler was evaluated for up to 48 h. QC samples were extracted, stored in the autosampler for 1 and 2 days. Each day, the QC samples were analyzed alongside a freshly prepared calibration curve. All endocannabinoids with the exception of OLA and ODA were stable in the autosampler. To determine the impact of freeze-thaw cycles, plasma samples were exposed to 0, 1, and 3 freeze-thaw cycles. All endocannabinoids except for 2-AG and DH-g-LEA were stable for up to 3 freeze-thaw cycles. In this context, 2-AG and DH-g-LEA showed 30% decay after one freeze-thaw cycle. To evaluate sample benchtop stability, plasma samples were stored on the benchtop at ambient temperature for 3, 6, and 24 h. Most endocannabinoids with the exception of O-AEA, 2-AG, 1-AG, 2-AGE, and ODA were stable for up to 3 h. For extended benchtop storage, most endocannabinoids showed instability. Short-term stability was evaluated in plasma samples, which were stored at 4 °C and − 20 °C for 1 and 7 days. All endocannabinoids were stable for up to 1 week when stored at −20 °C. Storage at 4 °C resulted in instability for most endocannabinoids. Further stability was established in plasma samples that were stored at −80 °C for 3 months. The results showed that most endocannabinoids with the exception of LEA and OEA were stable when stored under these conditions for 3 months.
Study sample stability assessment
Samples from 130 study subjects were collected. One sample from each individual was stored in plasma without formic acid and one in plasma supplemented with formic acid. The samples were collected in the time period between March 27, 2018, and December 18, 2018, and analyzed in October 2019 in collaboration with the Institute of Cognitive Science, University of Colorado Boulder, Boulder, CO, USA. Consequently, the samples were stored at −80 °C for the duration of 10 to 18 months prior to analysis. The results are shown in Table 3 and suggested that there were no significant differences observed for any endocannabinoids with the exception of 2-AG, 1-AG, 2-AGE, NADA, and OLA. The collection and storage in formic acid–treated containers resulted in 60-fold and 46-fold higher 2-AG and 1-AG levels compared to samples stored without formic acid, respectively.
Endocannabinoid levels in flower users versus concentrate users
To determine if endocannabinoid levels differ depending on the preferred use of certain Cannabis products, 36 study subjects (18 flower users and 18 concentrate users) and a total of 143 samples (73 flower user samples and 70 concentrate users samples) were analyzed. Data were not corrected for specific timing of the Cannabis consumption, since it was the aim of this sub-study to investigate the differences in endocannabinoid levels based on the participants’ preferred Cannabis consumption habits. There were no statistically significant changes in endocannabinoid levels determined between the study groups with the exception of docosatetraenoyl ethanolamide (DEA, p = 0.006, please see Fig. 2). A similar trend was observed for AEA (Fig. 2) although statistical significance was not reached (p = 0.139). Fabritius et al. [28] determined that a level of THCCOOH higher than 40 μg/L is strongly correlated with a heavy consumption of Cannabis, whereas a concentration below 3 μg/L suggests occasional use. When these criteria were applied to both study groups, it was found that frequent users were in both groups and not just limited to the concentrate users. We used the free non-hydrolyzed THCCOOH concentrations as surrogate estimate for the frequency of Cannabis use independent of the study group and correlated AEA concentrations and DEA concentrations with the plasma THCCOOH concentrations. Using data without logarithmic normalization, AEA and DEA were significantly correlated (Spearman’s rho) with 11-Nor-9-carboxy-Δ9-tetrahydrocannabinol (THCCOOH) plasma levels independent of preferred consumption (flower or concentrate), with r = 0.255 (p = 0.002) and r = 0.388 (p = 0.000002), for AEA and DEA, respectively. Data that were logarithmic transformed revealed a significant correlation between THCCOOH and DEA only with r = 0.196 and p = 0.021.

Endocannabinoid levels in concentrate users (n = 18) versus flower users (n = 18). The closely related endocannabinoids anandamide (not significant, upper graph) and docosatetraenoylethanolamine (**p = 0.006, lower graph) are shown. Values are presented as mean ± standard error of the mean (SEM)
Discussion
In this study, we present a sensitive high-throughput online-trapping high-performance liquid chromatography–tandem mass spectrometry–based (LC/LC-MS/MS) assay using robust atmospheric pressure chemical ionization (APCI) for the quantitation of 14 endocannabinoids and congeners in human plasma. The internal validation proved successful for the quantitative analysis of 12 compounds and semi-quantitative for 2 analytes. We showed that endocannabinoid sample stability is problematic and was successfully minimized by the addition of formic acid to plasma as stabilizing additive. For the external validation, we applied this assay for the analysis of plasma samples from sub-study cohorts of Cannabis flower and concentrate users. In alignment with other studies, we showed that the levels of anandamide levels (non-significant) were increased within the high THC concentrate user group. In addition, we found the novel endocannabinoid docosatetraenoyl ethanolamide statistically significantly increased in concentrate users compared to the low THC flower consumers.
At the turn of the century, the endocannabinoid system (ECS) was defined as the ensemble of the two cannabinoid receptors CB1 and CB2, their 2 most studied endogenous ligands, the “endocannabinoids” N-arachidonoylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG), and a group of enzymes involved in their synthesis and breakdown [29]. The new definition of the “expanded endocannabinoid system” or “endocannabinoidome,” potentially includes hundreds of lipid mediators, more than 20 biosynthetic or inactivating enzymes, and more than 20 molecular targets, such as many previously identified nuclear receptors, ligand-activated ion channels, and orphan GPCRs [3, 29, 30]. To address the growing need of bioanalytics quantifying endocannabinoid levels to probe the ECS, we developed and validated a simple, reproducible high-throughput assay for the quantitative analysis of 14 endocannabinoids and congeners in human plasma.
The validation of a multi-analyte assay for endogenous compounds is more complex than the validation of a typical bioanalytical assay that quantifies xenobiotics and follows applicable regulatory guidelines [31]. In the case of the present biomarker validation study, the acceptance criteria were defined as accuracy within 80–120% (75–125 at the LLOQ) with an imprecision of ≤20%CV (≤25% at the LLOQ). The HPLC-MS/MS-based assay for the analysis of 14 endocannabinoids has shown to be specific and selective for all analytes with the exception of OLA (oleamide). It was possible to chromatographically separate 2-AG and 1-AG. Carry-over was not an issue (<0.2%) with the exception of OLA. The intra- and inter-run accuracy (80%–120%) and imprecision (≤20%) evaluation performed on 4 days showed acceptable results for all endocannabinoids with the exception of OLA and ODA. It has been shown by us and others that APCI minimizes ion suppression and matrix effects caused by phospholipids for cannabinoids [32]. Thus, we used the same approach for endocannabinoids and indeed no relevant matrix effects were detected. The matrix effects determined according to Matuszewski et al. [25] did not affect the reproducibility and reliability of the assay. Back-calculated enriched matrices from 6 individual donors were within the acceptance criteria of the assay with the exception of OLA. The recoveries for the 14 endocannabinoids were reliable, reproducible, and higher than 60%, again with the exception of OLA. In summary, the assay was successfully validated for the analysis of all endocannabinoid biomarkers in human plasma included in the present assay with the exception of the endocannabinoids ODA (N-oleoyl dopamine) and OLA (oleamide). The endocannabinoid levels for O-AEA, AGE, NADA, and ODA were below the lower limit of quantitation for more than 50% of human plasma samples analyzed.
Instability of several endocannabinoid biomarkers has been described in the literature [33]. This is especially the case for 2-AG. Stability of 2-AG was shown to be impacted by pH, with an optimum at pH 5 [34]. Thus, we tested a sampling protocol that included acidification of plasma immediately after sample collection using 10% of a 5% aqueous formic acid solution. The results of this study showed that sampling and storage conditions are essential to stabilize 2-AG and other endocannabinoids. Figure 1 and Table 3 show that the concentrations of 2-AG and 1-AG did significantly decrease, when sampling and storage conditions were not ideal. Isomerization from 2-AG to 1-AG cannot be the explanation for this observation. Thus, it can only be concluded that degradation/ hydrolysis of both 2-AG and 1-AG most likely resulting in arachidonic acid and glycerol is the main cause for the reduced 2-AG and 1-AG levels (Fig. 3). Whether this is enzyme-catalyzed or autocatalytic remains unclear. Many studies, during which samples were collected without addition of stabilizers, assumed that the sum of 1-AG and 2-AG reflects the endogenous levels of 2-AG (Zoerner et al. [33] list numerous publications which are based on the sum of 1-AG + 2-AG). One of the major findings of our results indicates that the separation of 2-AG from 1-AG and the sample treatment are essential for the correct interpretation of study data.

Isomerization and degradation of 2-arachidonoylglycerol (2-AG) to 1-arachidonoylglycerol (1-AG) and arachidonic acid. At physiological pH (ca. 7.4), 2-AG is rapidly transformed into 1-AG or degraded to arachidonic acid. At lower pH, the isomerization is inhibited and 2-AG stabilized
There have been several high-performance liquid chromatography–mass spectrometry–based assays recently published [15,16,17,18,19,20,21,22,23,24, 33]. These assays often used more extensive sample workup procedures. However, many of these assays reported the sum of 2-AG and 1-AG [20, 21, 35,36,37,38,39] for different reasons, including but not limited to the inability to separate the isomers and isomerization/ degradation happening during storage and extraction. Only few assays report 2-AG and 1-AG separated, which is in light of our data essential for precise interpretation of study data [33, 40, 41]. Only few assays have used simple sample workup with column switching technology or similar [15, 16, 24]. In comparison to these assays, the assay presented here has the advantage of using robust APCI in contrast to electrospray ionization. This technology has the advantage to be less impacted by matrix effects such as ion suppression, as previously shown by our group [32]. The disadvantage of APCI in this context is lower overall sensitivity. However, the use of column switching technology compensates for the loss of sensitivity since it enriches the analytes on the extraction column. Fanelli et al. [16] also utilized APCI in combination with a column switching approach, but the assay is limited to 2 endocannabinoids and 2 congeners. In summary, the assay presented here combines a simple sample workup with sensitivity increasing online-trapping technology and robust atmospheric pressure chemical ionization to measure 14 endocannabinoids and congeners. This becomes increasingly important, since the endocannabinoid system is comprised of many more endocannabinoids than just anandamide and 2-AG.
It has been shown that cannabinoids (THC and CBD) alter the levels of endocannabinoids in the plasma or brain [42,43,44]. Chronic Cannabis abuse can lead to damaging effects in humans [45], and some authors examined whether components of the endocannabinoid system will undergo alterations as well. These studies reported an increase in serum 2-AG [44]. In contrast, anandamide levels were decreased in CSF and an alteration of serum anandamide was not observed in the same study [44]. Others reported increased levels of anandamide upon CBD treatment [43, 46, 47]. Muhl et al. [48] showed that in high-frequency users, the anandamide plasma levels were increased compared to controls. In the present study, we also observed increased levels of anandamide and the docosatetraenoyl ethanolamide (DEA) in concentrate users compared to users of the less potent flowers. DEA is an endocannabinoid with high similarity to anandamide, containing docosatetraenoic acid in place of the arachidonate moiety. It has been shown that DEA acts on CB1 receptors with potency and efficacy similar to that of AEA [49]. Hanus et al. [50] initially described the activity of DEA. However, its specific role and relative importance as a cannabinergic neurotransmitter have not yet been elucidated. Plasma levels of THCCOOH, and its O-ester glucuronide are considered to be increased in frequent users compared to infrequent users [28, 51]. The correlation between THCCOOH with AEA and DEA was significant indicating a change of these closely related endocannabinoids as a result of frequent use. Larger follow-up studies comparing frequent to non-frequent users might shed more light on the effect of frequency of use on endocannabinoid changes.
There were limitations to this study. Only subsets of samples were analyzed. The subsets used for the methodology development, sampling optimization, and feasibility were from the first volunteers participating in the larger observational studies. In these two broad observational Cannabis consumption studies, certain outcomes could be influenced by the time schedule and individual consumptions prior to sample collection. In this context, different frequencies of consumption could be considered, for instance regular versus intermittent versus “weekend binge weed smoking.” Another limitation is the fact that in this study no non-user controls were included.
In conclusion, a robust, sensitive high-throughput assay for the quantitation of 14 endocannabinoids and congeners was successfully validated. Endocannabinoid sample stability was an issue and could be successfully minimized by the addition of formic acid to plasma as stabilizing additive. Using this approach, large differences for some analytes including 2-AG between non-stabilized versus stabilized plasma samples were observed. Our study showed that it is mandatory to (A) appropriately stabilize samples and (B) separate and separately quantify 1-AG and 2-AG otherwise study results are unreliable. The analysis of study samples from Cannabis flower users versus Cannabis concentrate users revealed increased levels of AEA and DEA in concentrate users in accordance with other literature. Future studies with larger study cohorts and a non-user control cohort might give further insights into the effects of Cannabis consumption on circulating endocannabinoid levels.
References
Di Marzo V, Piscitelli F. The endocannabinoid system and its modulation by phytocannabinoids. Neurotherapeutics. 2015;12(4):692–8.
Zamberletti E, Gabaglio M, Parolaro D. The endocannabinoid system and autism spectrum disorders: insights from animal models. Int J Mol Sci. 2017;18(9).
Witkamp R. Fatty acids, endocannabinoids and inflammation. Eur J Pharmacol. 2016;785:96–107.
Alger BE. Endocannabinoids and their implications for epilepsy. Epilepsy Curr. 2004;4(5):169–73.
Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–77.
Di Marzo V, et al. Anandamide receptors. Prostaglandins Leukot Essent Fatty Acids. 2002;66(2–3):377–91.
Woodhams SG, et al. The cannabinoid system and pain. Neuropharmacology. 2017;124:105–20.
Woodhams SG, et al. The role of the endocannabinoid system in pain. Handb Exp Pharmacol. 2015;227:119–43.
Pistis M, Melis M. From surface to nuclear receptors: the endocannabinoid family extends its assets. Curr Med Chem. 2010;17(14):1450–67.
Moriconi A, et al. GPR55: current knowledge and future perspectives of a purported “Type-3” cannabinoid receptor. Curr Med Chem. 2010;17(14):1411–29.
Alexander SP, Kendall DA. The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol. 2007;152(5):602–23.
Pertwee RG, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev. 2010;62(4):588–631.
Woodward DF, Liang Y, Krauss AH. Prostamides (prostaglandin-ethanolamides) and their pharmacology. Br J Pharmacol. 2008;153(3):410–9.
Maione S, Costa B, Di Marzo V. Endocannabinoids: a unique opportunity to develop multitarget analgesics. Pain. 2013;154(Suppl 1):S87–93.
Marchioni C, et al. A column switching ultrahigh-performance liquid chromatography-tandem mass spectrometry method to determine anandamide and 2-arachidonoylglycerol in plasma samples. Anal Bioanal Chem. 2017;409(14):3587–96.
Fanelli F, et al. Estimation of reference intervals of five endocannabinoids and endocannabinoid related compounds in human plasma by two dimensional-LC/MS/MS. J Lipid Res. 2012;53(3):481–93.
Bluher M, et al. Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes. 2006;55(11):3053–60.
Fernandez-Rodriguez CM, et al. Circulating endogenous cannabinoid anandamide and portal, systemic and renal hemodynamics in cirrhosis. Liver Int. 2004;24(5):477–83.
Balvers MG, Verhoeckx KC, Witkamp RF. Development and validation of a quantitative method for the determination of 12 endocannabinoids and related compounds in human plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(14–15):1583–90.
Di Marzo V, et al. Role of insulin as a negative regulator of plasma endocannabinoid levels in obese and nonobese subjects. Eur J Endocrinol. 2009;161(5):715–22.
Thomas A, et al. Quantitative and qualitative profiling of endocannabinoids in human plasma using a triple quadrupole linear ion trap mass spectrometer with liquid chromatography. Rapid Commun Mass Spectrom. 2009;23(5):629–38.
Acquaro Junior VR, et al. Analysis of endocannabinoids in plasma samples by biocompatible solid-phase microextraction devices coupled to mass spectrometry. Anal Chim Acta. 2019;1091:135–45.
Gurke R, et al. Determination of endocannabinoids and endocannabinoid-like substances in human K3EDTA plasma - LC-MS/MS method validation and pre-analytical characteristics. Talanta. 2019;204:386–94.
Souza ID, Hantao LW, Queiroz MEC. Polymeric ionic liquid open tubular capillary column for on-line in-tube SPME coupled with UHPLC-MS/MS to determine endocannabinoids in plasma samples. Anal Chim Acta. 2019;1045:108–16.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75(13):3019–30.
US Department of Health and Human Services, Guidance for Industry, Bioanalytical Method Validation. US FDA, Center for Drug Evaluation and Research, Center for Veterinary Medicine, MD, USA; 2018.
Bidwell LC, et al. Association of naturalistic administration of Cannabis flower and concentrates with intoxication and impairment. JAMA Psychiatry. 2020;77(8):787–96.
Fabritius M, et al. THCCOOH concentrations in whole blood: are they useful in discriminating occasional from heavy smokers? Drug Test Anal. 2014;6(1–2):155–63.
Lu HC, Mackie K. An introduction to the endogenous cannabinoid System. Biol Psychiatry. 2016;79(7):516–25.
Veilleux A, Di Marzo V, Silvestri C. The expanded endocannabinoid system/endocannabinoidome as a potential target for treating diabetes mellitus. Curr Diab Rep. 2019;19(11):117.
Chau CH, et al. Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res. 2008;14(19):5967–76.
Klawitter J, et al. An atmospheric pressure chemical ionization MS/MS assay using online extraction for the analysis of 11 cannabinoids and metabolites in human plasma and urine. Ther Drug Monit. 2017;39(5):556–64.
Zoerner AA, et al. Quantification of endocannabinoids in biological systems by chromatography and mass spectrometry: a comprehensive review from an analytical and biological perspective. Biochim Biophys Acta. 2011;1811(11):706–23.
Rouzer CA, Ghebreselasie K, Marnett LJ. Chemical stability of 2-arachidonylglycerol under biological conditions. Chem Phys Lipids. 2002;119(1–2):69–82.
Engeli S, et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54(10):2838–43.
Bisogno T, et al. Brain regional distribution of endocannabinoids: implications for their biosynthesis and biological function. Biochem Biophys Res Commun. 1999;256(2):377–80.
Richardson D, et al. Quantitative profiling of endocannabinoids and related compounds in rat brain using liquid chromatography-tandem electrospray ionization mass spectrometry. Anal Biochem. 2007;360(2):216–26.
Valenti M, et al. The endocannabinoid system in the brain of Carassius auratus and its possible role in the control of food intake. J Neurochem. 2005;95(3):662–72.
Gonzalez S, et al. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002;954(1):73–81.
Kondo S, et al. 2-Arachidonoylglycerol, an endogenous cannabinoid receptor agonist: identification as one of the major species of monoacylglycerols in various rat tissues, and evidence for its generation through CA2+-dependent and -independent mechanisms. FEBS Lett. 1998;429(2):152–6.
Rohrig W, et al. Quantification of 24 circulating endocannabinoids, endocannabinoid-related compounds, and their phospholipid precursors in human plasma by UHPLC-MS/MS. J Lipid Res. 2019;60(8):1475–88.
Solinas M, et al. Dopaminergic augmentation of delta-9-tetrahydrocannabinol (THC) discrimination: possible involvement of D(2)-induced formation of anandamide. Psychopharmacology. 2010;209(2):191–202.
Leweke FM, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94.
Morgan CJ, et al. Cerebrospinal fluid anandamide levels, cannabis use and psychotic-like symptoms. Br J Psychiatry. 2013;202(5):381–2.
Hall W, Degenhardt L. Adverse health effects of non-medical cannabis use. Lancet. 2009;374(9698):1383–91.
Bisogno T, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134(4):845–52.
Gaston TE, Szaflarski JP. Cannabis for the treatment of epilepsy: an update. Curr Neurol Neurosci Rep. 2018;18(11):73.
Muhl D, et al. Increased CB2 mRNA and anandamide in human blood after cessation of cannabis abuse. Naunyn Schmiedeberg's Arch Pharmacol. 2014;387(7):691–5.
Felder CC, et al. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc Natl Acad Sci U S A. 1993;90(16):7656–60.
Hanus L, et al. Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J Med Chem. 1993;36(20):3032–4.
Kelly P, Jones RT. Metabolism of tetrahydrocannabinol in frequent and infrequent marijuana users. J Anal Toxicol. 1992;16(4):228–35.
Funding
Funding for this study was provided by grants from the National Institutes of Health (DA039707 to Dr. Hutchison) and Colorado Department of Public Health and Environment (96947 to Dr. Bidwell). The funding organizations had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Clinical samples
The clinical study was approved by the University of Colorado Boulder Institutional Review Board and the primary outcomes have been published previously [27]. The study was registered as an observational study in ClinicalTrials.gov (NCT03522103).
Source of biological material
Blank human K2EDTA plasma was received from Bioreclamation IVT (Westbury, NY, USA) and charcoal-stripped human plasma was received from BioCheMed (Winchester, VA, USA).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 1.94 mb)
Rights and permissions
About this article

Cite this article
Sempio, C., Klawitter, J., Jackson, M. et al. Analysis of 14 endocannabinoids and endocannabinoid congeners in human plasma using column switching high-performance atmospheric pressure chemical ionization liquid chromatography–mass spectrometry. Anal Bioanal Chem 413, 3381–3392 (2021). https://doi.org/10.1007/s00216-021-03280-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03280-0