
Abstract
A quick screening method of more than 200 pharmaceutical and other residues in aquatic foods based on ultrahigh-performance liquid chromatography–quadrupole-Orbitrap mass spectrometry (UHPLC-Q/Orbitrap MS) was established. In this method, after the addition of 200 μL of 1 M EDTA-Na2, 2 g of each sample homogenate was extracted successively with 10 mL of acetonitrile and 10 mL of ethyl acetate. The extracts were combined, dried under nitrogen flow, and redissolved in 0.1% formic acid in acetonitrile/water (4:6, v/v) for analysis. The prepared samples were analyzed by UHPLC- Q/Orbitrap MS system in Full MS/ddMS2 (full-scan data-dependent MS/MS) mode. Compound identification was performed through comparison of the sample data with the database for standard chemicals, including the retention time, precursor ion, product ions, and isotope pattern for all 206 compounds. Five different aquatic food matrices (carp, shrimp, crab, eel, and mussel) spiked with the analytes at 1, 10, and 50 ng/g were evaluated to assess recoveries, precision, matrix effects, stability, and detection limits using the method. UHPLC analyses required 25 min, and 178–200 analytes met identification criteria at 50 ng/g depending on the matrix. Furthermore, practical application of this method for real samples displayed strong screening capability.

A quick screening method of >200 pharmaceutical and other residues in aquatic foods based on ultrahighperformance liquid chromatography–quadrupole-Orbitrap mass spectrometer was established. Fivedifferent aquatic food matrices, including carp, shrimp, crab, eel and mussel, were studied to evaluatescreen limit at 1, 10 and 50 μg·kg-1 level. Results suggest the high reliability, high time-efficiency and goodsimplicity of the method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aquaculture provides a large part of the food from farming of freshwater or marine areas [1]. The frequent use of veterinary drugs is unavoidable and plays an important role in the prevention and treatment of aquaculture diseases, promoting growth and feed efficiency, or improvement of the cultural environment [2]. In addition, the aquaculture environment and the crop origin feed may induce the risk of exposure to unexpected contaminants passed from the water cycle [3] and residues in the crop component of feed [4]. Furthermore, the misuse or illegal use of veterinary drug in disease prevention and treatment as well as undesirable contamination from water or feed would lead to high risk associated with these component residues [5,6,7,8]. Therefore, a series of detection techniques must be developed to monitor and inspect the presence of these residues in aquatic foods [9,10,11,12,13,14,15,16,17,18]. To better monitor the safety of foods, it is especially preferable to develop generic methods which are capable of detecting as many different classes of concerned compounds as possible [19]. In the last few years, methods based on ultrahigh-performance liquid chromatography coupled to high-resolution mass spectrometry (UHPLC-HRMS) have been increasingly explored [20, 21].
There have been increasing reports on multiresidue screening in different food matrices based on UHPLC-HRMS. Initially, time-of-flight mass spectrometry (TOF-MS) was used in the development of screening methods for residues in various food matrices [22]. TOF-MS exhibits good performance for the monitoring and identification of a wide range of pharmaceuticals and contaminants [23]. However, TOF-MS cannot simultaneously perform positive and negative detection modes, thereby limiting its application for a large number of target residues when both ionization modes are required [24]. Compared to TOF-MS, the Orbitrap mass spectrometry shows excellent performance in terms of its much higher mass resolution and fast switching between positive-ion and negative-ion modes [25, 26]. Therefore, the capacity of Orbitrap-based mass spectrometry expands the scope and diversity of compounds suitable for screening.
Methods for screening pharmaceutical residues or contaminants in aquatic food are based on either TOF-MS or Orbitrap MS [27]. A qualitative LC-Q-TOF MS screening method for 143 veterinary drugs and pharmaceutical residues for fish tissues was investigated [28]. Using UHPLC- Q/Orbitrap MS, Jia et al. developed an untargeted screening method for 137 veterinary drugs and their metabolites (16 categories) in tilapia [29]. Turnipseed established a wide-scope screening method for 70 veterinary drugs in fish, shrimp, and eel using Q/Orbitrap MS [20]. In fish tissues, 133 pesticides and 24 polycyclic aromatic hydrocarbons were screened successfully through GC- Q/Orbitrap MS [30]. However, the scopes of the target compounds and matrices are specifically limited. Our study aims to develop a more generic screening method for a wider scope of residues and targeting more compounds simultaneously. In this research, a quick screening method in aquatic food, including 20 categories of pharmaceutical and other residues, was established.
Materials and methods
Chemicals and reagents
All the standards of pharmaceutical and other residues (high purity grade, >90%) were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany). Acetonitrile (MeCN) and methanol (MeOH) of HPLC grade were obtained from J.T. Baker (USA). Formic acid (FA, LC-MS grade, Fisher Scientific, Spain) was used. All the other reagents were purchased from common domestic suppliers.
Preparation of standard solutions
Stock standard solutions of individual compounds (around 100 mg mL−1) were prepared in MeOH and stored at −42 °C in brown glass volumetric flasks. FA (1.2 mg mL−1) was added to enhance solubility when the standard was not well dissolved. Standard solutions of different concentrations were prepared by mixing or diluting the stock solutions with MeOH and were also stored at −42 °C.
Instrumentation
An ultrahigh-performance liquid chromatography (UHPLC) system (DionexUltiMate 3000, Thermo Fisher Scientific) coupled to a quadrupole-Orbitrap mass spectrometer with electrospray ionization (Q-Exactive, Thermo Fisher Scientific) was used for data acquisition. An Accucore RP-MS C18 column (2.1 × 100 mm, 2.6 μm, Thermo Fisher Scientific, USA) was employed to perform sample separation with a thermostat at 30 °C. The binary mobile phases were 0.1% FA in MeCN (A) and 0.1% FA in water (B). Their gradient elution was started with 5% (A) 0.1% FA in MeCN for 3 min, then linearly increased to 100% in 19 min, and kept constant for 3 min. In the end, the eluent was restored to the initial conditions for 5 min to re-equilibrate the column for the next injection. The flow rate was kept at 0.3 mL min−1 for the whole elution process. The injection volume for analysis was 10 μL for each sample.
The electrospray ionization was operated in both positive and negative mode, with the following parameters: spray voltage, 3200 V (positive mode), 2800 V (negative mode), sheath gas flow rate at 8 L min−1, auxiliary gas flow rate at 10 L min−1, sweep gas flow rate at 1.5 L min−1, capillary temperature at 325 °C, S-lens RF level at 60 V. The HRMS was acquired under Full MS/ddMS2 (with inclusion list) mode over the mass range m/z 100–1000 (positive mode) and 150–1000 (negative mode). The Full MS/ddMS2 with inclusion list (full-scan data-dependent MS/MS) mode could simultaneously record the precursor mass and the MS/MS (fragmentation) spectra for the selected precursors. A full MS scan (resolution, 70,000) was conducted to search for the target ions in the inclusion list to perform the MS/MS acquisition (resolution, 15,000) for the selected ions (isolation window, 1.0 m/z). For each round of fragmentation acquisition, the top 5 (TopN, 5, loop count 1) most abundant precursors above the threshold 5e4 were sequentially transferred into the C-Trap (AGC, 5e4 Max IT, 80 ms) for collision at normalized energies (NCE, 20, 50, 80) in HCD multipole and transferred to Orbitrap for MS/MS acquisition. For compounds which cannot be well fragmented at the NCE, NCE was optimized and preset into the inclusion list.
All the parameters of the UHPLC- Q/Orbitrap MS system were controlled through the TraceFinder software.
Sample preparation
The brief procedure for sample preparation is shown in Fig. 1. Two grams of each properly homogenized aquatic food sample was weighed in a 50-mL centrifuge tube. Then 200 μL of EDTA-Na2 (0.1 M) and 10 mL of MeCN were added. The sample was mixed for 30 s in a grinder, vortexed for 5 min, and placed in an ultrasonic bath at 40 °C for 10 min. After being vortexed for another 5 min, the sample was centrifuged at 10,000 × g for 10 min. The supernatant was transferred into a glass tube. Afterwards, 10 mL of ethyl acetate (EtOAc) was added into the sample tube to repeat the above extraction procedure. The MeCN and EtOAc supernatant were combined and dried under nitrogen flow at 40 °C. Then, the resulting residues were dissolved in 1 mL of 0.1% FA in MeCN/water (2:3, v/v) and then filtered through a 0.22-μm nylon syringe filter for subsequent UHPLC- Q/Orbitrap MS analysis.

Schematic diagram of sample preparation for multiresidue screening in aquatic food
Establishment of database and identification criteria
All the standard solutions of analytes (100 ng mL−1) were analyzed through the UHPLC- Q/Orbitrap MS system, using aforementioned parameters. The m/z of the precursor ion, retention time (RT), and fragment ions (FI) were acquired through analysis, while the isotope pattern of the precursor was automatically calculated by TraceFinder software. Afterwards, a database containing m/z of precursor ion (peaks), RT, m/z of FI, and isotope pattern was built in TraceFinder. Screening of all the analytes was performed by TraceFinder with the self-built database. The identification criteria were established as follows: allowed m/z deviation of precursor ion was 3 ppm, allowed RT deviation was ±15 s, at least one fragment ion match with allowed m/z deviation at 20 ppm, and the fit threshold for precursor isotope pattern was 75% with allowed mass deviation at 10 ppm, and allowed intensity deviation less than 25%.
Matrix effect measurement
The matrix effect (ME%) was based on the ratio between the signal area in matrix-matched standard solution (A) and the signal area in standard solution (B) of identical concentration for each compound, calculated as the following equation:
It is apparent that ME% above 0 shows matrix enhancement and ME% below 0 stands for matrix suppression. In our experiment, ME% values between −20% and 20% are usually considered as acceptable for a non-matrix calibration, while ME% values outside of this range represent a strong matrix effect. In this research, six blank samples of each matrix were extracted and spiked with 100 ng mL−1 analytes separately. In combination with standard solutions of analytes, these samples were analyzed for matrix effect measurements. All the data was calculated through the average peak area of each analyte.
Method validation
Sensitivity, stability, and recovery in different matrices
To examine the method performance in screening of the concerned analytes, blank samples (carp, shrimp, crab, mussel, and eel) were spiked with all analytes at 1 μg kg−1, 10 μg kg−1, and 50 μg kg−1, respectively. All of these samples were analyzed by our method to investigate the profile of confirmable analytes in different matrices at each spiked level. The recoveries of analytes were calculated through the ratio between the blank samples spiked with equivalent analytes before and after pretreatment. Each sample was spiked with all analytes, well mixed, and kept at room temperature for 1 h to simulate the real positive samples. Each matrix at a spiked level was tested in six replicates. Sensitivity and recovery were evaluated on the basis of the average of six replicates, and the relative standards deviation (RSD) of the six replicates was used to evaluate the reproducibility of the method.
Practical application test
To further validate the feasibility of the method for wide-scope screening of real samples, 24 samples of aquatic food from different markets in China were collected and analyzed. The results were evaluated through the profile of positive analytes in different samples and their consistency with a specific detection method for single class compounds. The positive samples from screening were also roughly quantified through a single-point matrix-matched standard together with corresponding recovery calibration calculated at an appropriate spiking concentration. Firstly, the positive samples produce signal response (Ax) for analytes of interest. The positive samples were spiked with appropriate amounts of corresponding analytes (X0), and again analyzed through this method. The discrepancy (A0) between signal response before and after spiking in the samples accounts for the addition of analytes. On the basis of the ratio between Ax and A0, as well as the recoveries of analytes (Rx), the amounts of analytes of interest is calculated as \( \mathrm{X}=\frac{\left(\frac{A_x}{A_0}{X}_0\right)}{R_x} \).
Results and discussion
Working principle of full MS-ddMS2 scan mode for qualification and the importance of resolution
Full MS-ddMS2 collects accurate mass of the positive and negative precursor ions with fast alternate mode switching, which would be set as the first standard for identification of analytes. When the signals of the precursors reach a given intensity, the defined most abundant precursors are isolated through the quadrupole, fragmented in the HCD multipole, collected in the C-trap, and their high-resolution mass is recorded with the Orbitrap mass spectrometer. Therefore, it is the accurate mass of precursors that is continuously tracked instead of their fragmentation ions. Theoretically, each chromatographic peak of a precursor ion of an analyte would give the quantification data during the peak recording process. At the same time, the corresponding fragmentations for the precursors underwent identification and confirmation. The resolution for the acquisition of precursors and fragments affects the quality of identification. Higher resolution gives more accurate results for identification. However, higher resolution data acquisition takes a longer time, and compromises the time resolution of the chromatograph. Accordingly, we applied higher resolution (70,000) for precursor acquisition and lower resolution (15,000) for fragment acquisition, which can give 5–8 points of sampling per second for precursor acquisition and the speed for fragment acquisition would not have too much effect when the analytes are detected. Under these optimized conditions, the discrepancy for precursor and fragment ion acquisition can be less than 3 ppm and 20 ppm, respectively, which was validated when positive samples were tested each time.
Identification criteria
Guidelines for identification with HRMS have been taken into comprehensive consideration [31, 32]. Identification of the concerned analytes was performed through comparison of tested results with the database of chromatographic and mass spectroscopic information of all the standards. Besides the identical chromatographic information between sample and standards, their mass information should attain a given number of identification points (IPs) through comparison. As HRMS data was collected, 2 IPs are earned if the precursor ion matches, and 2.5 IPs are earned for each of their product ions [33, 34]. Usually, 4.5 IPs are enough for the identification of all compounds. In this experiment, comparison of isotope patterns of precursors within a given criterion leads to higher IPs for structure identification. As a result, the confirmation of analytes should give a strict and reliable result. The database of standard chemicals is displayed in the Electronic Supplementary Material (ESM, Table S1).
Optimization of extract solvent
It remains a significant challenge to find a generic solution for a wide scope of compounds with different physical and chemical properties. However, it is better to get as many compounds extracted as possible, where the preferable compounds are all included. MeCN and EtOAc are usually the most commonly used solvents for extracting analytes from samples with relatively good recovery and stability. In our method, we applied a two-step extraction using two generic solvents of different polarity, MeCN and EtOAc, which usually dissolve a wide range of compounds in various samples [35]. It is clearly shown in Fig. 2a that the sequence of solvents for extraction and the used solvent substantially affect the outcomes of extraction. In Fig. 2a, there is not too much difference in the number of compounds extracted from different solvents and steps between the combination of MeCN+MeCN and MeCN+EtOAc. The compounds extracted from the second step are completely overlapped with the first step. A total of 167 analytes were identified at the spiked level of 10 μg kg−1. The combination of EtOAc+MeCN also extracted 167 analytes; 53 and 20 of these analytes were identified in EtOAc and MeCN, respectively, which are not overlapped. It is shown in Fig. 2b that more compounds were extracted at recoveries of 70–120% and greater than 120% with the solvent combination of MeCN+EtOAc than the other two combinations. Therefore, MeCN+EtOAc was selected for the extraction of a wide range of residues in aquatic products for the subsequent in which more than 200 compounds belonging to 20 categories can be screened out qualitatively at 50 μg kg−1.

Number of compounds extracted and identified in separate steps for different solvent combinations (a), and the number of compounds with different recoveries for analyte-spiked samples (Carp) (b). Spiked level, 10 μg kg−1
We tried different solvents in combination with the commercially available Oasis PRiME HLB SPE or d-SPE of C18 and PSA to clean up the extract for screening. A total of 252 compounds were spiked and examined, and all three types of SPE could only extract around 130 compounds at 10 μg kg−1 spiking level. At each step of cleanup, it is likely that some of the targets were lost. Attempts to use SPE to extract and clean up samples for screening of such a wide range of compounds failed.
Effect of matrix component on recoveries of analytes during analysis
The component profile in the extract can be displayed through the total ion chromatogram, as shown in Fig. 3. For the matrix components extracted from all of the five samples (carp, shrimp, crab, eel, and mussel), most of the components were eluted in the first 0.5–5 min and the following 8–22 min. Matrix components from mussel seem to be more complicated in the range of 8–22 min compared to other samples. Most of the analytes were detected with a retention time between 5 and 15 min, when a comparable high matrix component was also eluted out. Nevertheless, these compounds could still be screened with a high portion of co-eluting components. Between 15 and 20 min when the most matrix components eluted out, analytes were also screened out (see ESM, Fig. S1). The matrix complexity can affect the co-extraction of analytes during pretreatment, and further result in different recoveries. These experimental results demonstrated the excellent performance of the UHPLC- Q/Orbitrap MS system regarding its tolerance to high matrix contents and resolution stability.

Total ion chromatogram (TIC) of matrix extract in five blank samples (carp, shrimp, crab, eel, and mussel) on the UHPLC- Q/Orbitrap MS system
Matrix effect evaluation
Matrix effects frequently occur in mass spectrometry analysis when electrospray ionization (ESI) is employed and happen when the matrix components co-eluted with the analytes either enhance or suppress the ionization of the analytes by a synergistic or antagonistic effect, and further affect the MS signal intensity. Besides, the Orbitrap mass spectrometer can also show signal suppression when an overabundance of ionized analytes and matrix components is present in the C-trap. As this research aims to develop a generic sample extract method and no specific cleanup method was utilized, the presence of large amounts of co-extracted matrix component is reasonable. It would give a matrix effect in the ESI process, and further affect the signal response of the mass analyzer, possibly inducing an inferior quality of screening.
As a result, the matrix effect should be evaluated and considered for subsequent qualification. A strong matrix effect occurred for 80 analytes in carp, 90 analytes in crab, 108 analytes in shrimp, 131 analytes in eel, and 150 compounds in mussel (see ESM, Fig. S2). Around half of the total confirmable analytes showed a weak matrix effect in carp, shrimp, and crab samples. The strongest matrix effect occurred in mussel samples. The corresponding TIC of the mussel samples (Fig. 3) shows more complicated and abundant matrix components. The linear response profile in different matrices also indicates the matrix effect (see ESM, linearity in different matrices). Figure 4 shows the matrix effect versus the retention time of analytes from different matrices. Most of the matrix components that can be ionized were eluted out between 8 and 22 min and the majority of the analytes were screened out between 5 and 15 min. There was no obvious ME% distribution on the specific range of retention times for carp and shrimp, implying less signal influence from matrix extracts. However, within this time range, more analytes showed strong matrix effects and broader variation for crab, eel, and mussel samples, which to some extent agrees with the matrix complexity profile from Fig. 3.

Matrix effect of different analytes detected in five different matrices (carp, shrimp, crab, eel, and mussel) versus their retention time on the UHPLC- Q/Orbitrap MS system; spiked level in the blank sample solution, 100 ng mL−1
Stability and recovery
To examine the profile of the stability and recovery of our method, a series of different samples were spiked with all of the concerned analytes, and analyzed through the screening method. Figure 5 displays the number of compounds with different recoveries (a) and relative standard deviation (b) of screened analytes spiked in different matrices. In carp samples, 200 analytes were successfully screened out at 50 μg kg−1. For these screened compounds, 160 of them can be recovered between 50% and 120%, 94% of which gave RSDs less than 15%. In shrimp samples, when spiked at 50 μg kg−1,176 compounds can be recovered between 50% and 120%, 98.3% of which had RSDs below 15%. The recoveries varied obviously for crab, eel, and mussel. This can be attributed to the complexity of these matrices. Nevertheless, more than 74% of the RSDs for analytes in these three matrices were still kept below 15%, which indicates a desirable stability of our method. The detailed information on RSD and recoveries for each analyte in different matrices is shown in the ESM (Table S2 and Fig. S3). As a screening method, the capability of accurate and stable qualification should be considered as a priority. The low RSD values for most of the screened analytes indicate the reliability of our method regarding the stable extraction efficiency and uniform signal response with complicated matrix components; these results help to definitely judge the screened residues. Therefore, the feasibility of our method in reliable screening of a wide scope of residues in various aquatic foods can be guaranteed.

Number of compounds with different recoveries (a) and relative standard deviation (b) of screened analytes spiked in different samples (carp, shrimp, crab, eel, and mussel) at 50 μg kg−1
Sensitivity of screened analytes in different matrices
To examine the sensitivity of the screening method for all the concerned analytes in different matrices, the numbers of compounds screened out at different spiked levels are displayed in Fig. 6. As different matrix components were extracted in these matrices, their effects on the screening of analytes also vary. The variation can be caused by the matrix disturbance on the analyte extraction and electrospray ionization for mass spectrometry analysis. In the five matrices, 200 analytes can be identified at 50 μg kg−1 for both carp and shrimp, and 181, 178, and 180 of the analytes were screened out for crab, eel, and mussel, respectively (Fig. 6). Furthermore, at the spiked level of 10 μg kg−1, 174, 170, 142, 124, and 135 of the compounds were detected for grass carp, shrimp, crab, eel, and mussel, respectively (Fig. 6). Moreover, 100, 95, 45, 54, and 60 of the analytes were confirmed at 1 μg kg−1 for carp, shrimp, crab, eel, and mussel, respectively (Fig. 6). The detailed screening profile for each analyte in different matrices is shown in the ESM (Table S3 and Fig. S4). Most of the identified compounds at 1 μg kg−1 distribute at the retention time between 5 and 15 min, where the co-eluted matrix components are relatively low. For retention time between 15 and 25 min, where a comparably high portion of matrix components was eluted, the percentage of analytes identified at 10 μg kg−1 or 50 μg kg−1 increased (Fig. S4). The number of detected compounds and their screening limit agreed well with the matrix complexity evaluated through the TIC profile. These verified results showed the strong capability of our method in screening for aquatic food safety. We did not calculate the limit of detection for each analyte according to the signal to noise ratio at a given spiked level, as it seems not achievable at all for such a low calculated concentration through the HRMS. Our verified screening limit for qualification should be a more practical reference for different analytes and matrices. Therefore, a more reliable screening result can be achieved for comprehensive evaluation of the residue profile of a wide range of compounds.

Numbers of analytes of different screen limit in different matrices (carp, shrimp, crab, eel, and mussel)
Application to real samples
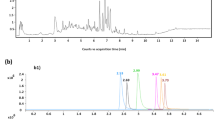
To evaluate the practicability of the method, 24 real aquatic food samples collected from markets in different provinces of China were analyzed. In all the real samples, the most frequently screened residues were ethoxyquin, enrofloxacin, ciprofloxacin, and trimethoprim, and their representative extracted ion chromatograms are shown in Fig. 7. Twelve samples were confirmed to be positive for ethoxyquin, which is an authenticated antioxidant in feedstuff. It might be transferred and accumulated during aquaculture by feedstuffs containing residues of this chemical. Enrofloxacin and ciprofloxacin were detected in 18 and 11 samples, respectively, which are approved wide-spectrum quinolone antibiotics in various animal culture industries. Ciprofloxacin is also the metabolite of enrofloxacin in animals, which means it can be derived from the enrofloxacin residues [36]. Trimethoprim was detected in five samples; it is used in combination with sulfonamides for the treatment of infection, and was also found co-existing with sulfonamides in some real samples [37].

Representative extracted ion chromatograms of compounds detected in real samples
Detailed information on positive analytes in samples is shown in the ESM (Table S4). The presence of residues of enrofloxacin and ciprofloxacin in all of the real samples was also confirmed through specific quantification methods based on triple-quadrupole mass spectrometry, which were proved to be consistent with our screening method. These practical results demonstrated the feasibility of our methods for wide-scope screening of 20 categories of 206 pharmaceutical and other residues in aquatic food.
Conclusions
We have established a quick screening method for 206 residues including 20 categories of pharmaceutical and other residues in aquatic food based on UHPLC- Q/Orbitrap MS. Identification was performed through comparison of the sample data with a database built from standard chemicals, which can be carried out with very strict tolerance error. Method evaluations validated the screening of compounds at spiked levels of 1, 10, and 50 μg kg−1. Practical application of this method to real samples highlighted the strong screening capability. These results suggested the desirable reliability, high time efficiency, and simplicity of the method when it was applied for routine screening of suspicious residues in aquatic food to meet the requirements of related regulations.
References
Broughton EI, Walker DG. Policies and practices for aquaculture food safety in China. Food Policy. 2010;35(5):471–8.
Masiá A, Suarez-Varela MM, Llopis-Gonzalez A, Picó Y. Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: a review. Anal Chim Acta. 2016;936(Supplement C):40–61.
Locatelli M, Sciascia F, Cifelli R, Malatesta L, Bruni P, Croce F. Analytical methods for the endocrine disruptor compounds determination in environmental water samples. J Chromatogr A. 2016;1434:1–18.
Gómez-Pérez ML, Romero-González R, Martínez Vidal JL, Garrido Frenich A. Analysis of veterinary drug and pesticide residues in animal feed by high-resolution mass spectrometry: comparison between time-of-flight and Orbitrap. Food Addit Contam Part A. 2015;32(10):1637–46.
Varol M, Sünbül MR. Organochlorine pesticide, antibiotic and heavy metal residues in mussel, crayfish and fish species from a reservoir on the Euphrates River, Turkey. Environ Pollut. 2017;230:311–9.
Guo J, Selby K, Boxall ABA. Assessment of the risks of mixtures of major use veterinary antibiotics in European surface waters. Environ Sci Technol. 2016;50(15):8282–9.
Anumol T, Lehotay SJ, Stevens J, Zweigenbaum J. Comparison of veterinary drug residue results in animal tissues by ultrahigh-performance liquid chromatography coupled to triple quadrupole or quadrupole–time-of-flight tandem mass spectrometry after different sample preparation methods, including use of a commercial lipid removal product. Anal Bioanal Chem. 2017;409(10):2639–53.
Baynes RE, Dedonder K, Kissell L, Mzyk D, Marmulak T, Smith G, et al. Health concerns and management of select veterinary drug residues. Food Chem Toxicol. 2016;88:112–22.
Wang Z, Beier RC, Shen J. Immunoassays for the detection of macrocyclic lactones in food matrices–a review. TrAC Trends Anal Chem. 2017;92:42–61.
Mainero Rocca L, Gentili A, Pérez-Fernández V, Tomai P. Veterinary drugs residues: a review of the latest analytical research on sample preparation and LC-MS based methods. Food Addit Contam Part A. 2017;34(5):766–84.
Zhang Y, Qiao H, Chen C, Wang Z, Xia X. Determination of nitrofurans metabolites residues in aquatic products by ultra-performance liquid chromatography–tandem mass spectrometry. Food Chem. 2016;192:612–7.
Lombardo-Agüí M, García-Campaña AM, Cruces-Blanco C, Gámiz-Gracia L. Determination of quinolones in fish by ultra-high performance liquid chromatography with fluorescence detection using QuEChERS as sample treatment. Food Control. 2015;50:864–8.
Zhu W-X, Yang J-Z, Wang Z-X, Wang C-J, Liu Y-F, Zhang L. Rapid determination of 88 veterinary drug residues in milk using automated TurborFlow online clean-up mode coupled to liquid chromatography-tandem mass spectrometry. Talanta. 2016;148:401–11.
Morris S, Allchin CR, Zegers BN, Haftka JJ, Boon JP, Belpaire C, et al. Distribution and fate of HBCD and TBBPA brominated flame retardants in North Sea estuaries and aquatic food webs. Environ Sci Technol. 2004;38(21):5497–504.
Nagata T, Saeki M. Simultaneous determination of thiamphenicol, florfenicol, and chloramphenicol residues in muscles of animals and cultured fish by liquid chromatography. J Liq Chromatogr Relat Technol. 1992;15(12):2045–56.
Zheng W, Park J-A, Zhang D, El-Aty AA, Kim S-K, Cho S-H, et al. Determination of fenobucarb residues in animal and aquatic food products using liquid chromatography-tandem mass spectrometry coupled with a QuEChERS extraction method. J Chromatogr B. 2017;1058:1–7.
Gan J, Lv L, Peng J, Li J, Xiong Z, Chen D, et al. Multi-residue method for the determination of organofluorine pesticides in fish tissue by liquid chromatography triple quadrupole tandem mass spectrometry. Food Chem. 2016;207:195–204.
Frenich AG, Romero-González R, del Mar Aguilera-Luiz M. Comprehensive analysis of toxics (pesticides, veterinary drugs and mycotoxins) in food by UHPLC-MS. TrAC Trends Anal Chem. 2014;63:158–69.
Dasenaki ME, Thomaidis NS. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography–tandem mass spectrometry. Anal Chim Acta. 2015;880:103–21.
Turnipseed SB, Storey JM, Lohne JJ, Andersen WC, Burger R, Johnson AS, et al. Wide-scope screening method for multiclass veterinary drug residues in fish, shrimp, and eel using liquid chromatography–quadrupole high-resolution mass spectrometry. J Agric Food Chem. 2017;65(34):7252–67.
Mol HG, Zomer P, De Koning M. Qualitative aspects and validation of a screening method for pesticides in vegetables and fruits based on liquid chromatography coupled to full scan high resolution (Orbitrap) mass spectrometry. Anal Bioanal Chem. 2012;403(10):2891–908.
Núñez O, Gallart-Ayala H, Martins CPB, Lucci P. New trends in fast liquid chromatography for food and environmental analysis. J Chromatogr A. 2012;1228(Supplement C):298–323.
Wang J, Leung D, Chow W, Chang J, Wong JW. Development and validation of a multiclass method for analysis of veterinary drug residues in milk using ultrahigh performance liquid chromatography electrospray ionization quadrupole orbitrap mass spectrometry. J Agric Food Chem. 2015;63(41):9175–87.
Kaufmann A, Teale P. Capabilities and limitations of high-resolution mass spectrometry (HRMS): time-of-flight and Orbitrap™. In: Kay JF, MacNeil JD, Wang J, editors. Chemical analysis of non-antimicrobial veterinary drug residues in food. New York: Wiley; 2016. p. 93–139.
Cheng Q, Shou L, Chen C, Shi S, Zhou M. Application of ultra-high-performance liquid chromatography coupled with LTQ-Orbitrap mass spectrometry for identification, confirmation and quantitation of illegal adulterated weight-loss drugs in plant dietary supplements. J Chromatogr B. 2017;1064:92–9.
Williamson JC, Edwards AV, Verano-Braga T, Schwämmle V, Kjeldsen F, Jensen ON, et al. High-performance hybrid Orbitrap mass spectrometers for quantitative proteome analysis: observations and implications. Proteomics. 2016;16(6):907–14.
Turnipseed SB, Lohne JJ, Boison JO. Application of high resolution mass spectrometry to monitor veterinary drug residues in aquacultured products. J AOAC Int. 2015;98(3):550–8.
Dasenaki ME, Bletsou AA, Koulis GA, Thomaidis NS. Qualitative multiresidue screening method for 143 veterinary drugs and pharmaceuticals in milk and fish tissue using liquid chromatography quadrupole-time-of-flight mass spectrometry. J Agric Food Chem. 2015;63(18):4493–508.
Jia W, Chu X, Chang J, Wang PG, Chen Y, Zhang F. High-throughput untargeted screening of veterinary drug residues and metabolites in tilapia using high resolution orbitrap mass spectrometry. Anal Chim Acta. 2017;957:29–39.
Nácher-Mestre J, Serrano R, Portolés T, Berntssen MH, Pérez-Sánchez J, Hernández FL. Screening of pesticides and polycyclic aromatic hydrocarbons in feeds and fish tissues by gas chromatography coupled to high-resolution mass spectrometry using atmospheric pressure chemical ionization. J Agric Food Chem. 2014;62(10):2165–74.
Martinello M, Borin A, Stella R, Bovo D, Biancotto G, Gallina A, et al. Development and validation of a QuEChERS method coupled to liquid chromatography and high resolution mass spectrometry to determine pyrrolizidine and tropane alkaloids in honey. Food Chem. 2017;234:295–302.
European Commission Directorate-General for Health and Food Safety. SANTE/11945/2015:Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed. Brussels, European Commission; 2015.
Rochat B. Proposed confidence scale and ID score in the identification of known-unknown compounds using high resolution MS data. J Am Soc Mass Spectrom. 2017;28(4):709–23.
Schymanski EL, Singer HP, Slobodnik J, Ipolyi IM, Oswald P, Krauss M, et al. Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem. 2015;407(21):6237–55.
Gómez-Ramos M, Ferrer C, Malato O, Agüera A, Fernández-Alba A. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: screening and quantitative studies. J Chromatogr A. 2013;1287:24–37.
Justino CI, Duarte KR, Freitas AC, Panteleitchouk TS, Duarte AC, Rocha-Santos TA. Contaminants in aquaculture: overview of analytical techniques for their determination. TrAC Trends Anal Chem. 2016;80:293–310.
Phu TM, Scippo M-L, Phuong NT, Tien CTK, Son CH, Dalsgaard A. Withdrawal time for sulfamethoxazole and trimethoprim following treatment of striped catfish (Pangasianodon hypophthalmus) and hybrid red tilapia (Oreochromis mossambicus× Oreochromis niloticus). Aquaculture. 2015;437:256–62.
Acknowledgments
The authors would like to acknowledge the financial support from the Shanghai Municipal Agricultural Commission, the Yangfan project (14YF1408100) from Science and Technology Commission of Shanghai Municipality – PR China and the special research fund for the national non-profit institutes (East China Sea Fisheries Research Institute) (No. 2014T05).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This research did not involve human participants or animals.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Published in the topical collection Food Safety Analysis with guest editor Steven J. Lehotay.
Electronic supplementary material
ESM 1
(PDF 3.29 MB)
Rights and permissions
About this article

Cite this article
Kong, C., Wang, Y., Huang, Y. et al. Multiclass screening of >200 pharmaceutical and other residues in aquatic foods by ultrahigh-performance liquid chromatography–quadrupole-Orbitrap mass spectrometry. Anal Bioanal Chem 410, 5545–5553 (2018). https://doi.org/10.1007/s00216-018-1124-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-1124-5